 Jie Yin
Jie Yin Kai Wu
Kai Wu Qingyang Ma
Qingyang Ma Hang Dong
Hang Dong- State Key Laboratory of Medical Genomics, Institute of Health Sciences, Shanghai Jiao Tong University School of Medicine and Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
Through linkage and candidate gene screening, many breast cancer (BC) predisposition genes have been identified in the past 20 years. However, the majority of genetic risks that contribute to familial BC remains undetermined. In this study, we revisited whole exome sequencing datasets from non-BRCA1/2 familial BC patients, to search for novel BC predisposition genes. Based on the infinite mutation model, we supposed that rare non-silent variants that cooccurred between familial and TCGA-germline datasets, might play a predisposition contributing role. In our analysis, we not only identified novel potential pathogenic variants from known cancer predisposition genes, such as MRE11, CTR9 but also identified novel candidate predisposition genes, such as NCK1. According to the TCGA mRNA expression dataset of BC, NCK1 was significantly upregulated in basal-like subtypes and downregulated in luminal subtypes. In vitro, NCK1 mutants (D73H and R42Q) transfected MCF7 cell lines, which attributed to the luminal subtype, were much more viable and invasive than the wild type. On the other side, our results also showed that overall survival and disease-free survival of patients with NCK1 variations might be dependent on the genomic context. In conclusion, genetic heterogeneity exists among non-BRCA1/2 BC pedigrees and NCK1 could be a novel BC predisposition gene.
Introduction
Breast cancer (BC) is the most malignant cancer type, affecting women worldwide (30%) and is the secondary cause of death in women (14%) (Siegel et al., 2018). Although most BC patients are sporadic, about 10–15% of BC s show familial aggregation (Kiiski et al., 2014; Lynch et al., 2015). High penetrance genes, such as BRCA1 and BRCA2, contribute about 20% to the etiology of familial BC (Mavaddat et al., 2010; Rizzolo et al., 2011; Melchor and Benitez, 2013). While linkage analyses failed to identify any compelling evident region of linkage in non-BRCA1/2 BC pedigrees (Antoniou and Easton, 2006). According to candidate gene screening, other high or moderate penetrance genes, such as TP53, PALB2, STK11, ATM, and CHEK2 have been identified (Stratton and Rahman, 2008; Melchor and Benitez, 2013). With the application of Whole Exome Sequencing (WES), several novel BC predisposition genes have been identified from BC pedigrees, which further confirms that non-BRCA1/2 familial BC is highly heterogeneous.
An evaluation of potential predisposition roles of germline variants is challenging. First, to distinguish disease-causative variants from the non-pathogenic ones during WES analysis usually involves a series of filtering steps, including in silico prediction; however, such filtering steps might cause over-filtering or be misleading (Bamshad et al., 2011). For instance, on one hand, in silico predictions might not be sensitive enough to detect all deleterious or damaging variants; on the other hand, the in silico predicted damaging variants might not be clinically pathogenic (Rahman, 2014). Second, to identify predisposition factors usually starts with an inspection of familial aggregation datasets, followed by a case-cohort confirmation (Kiiski et al., 2014); however, variants may be misclassified as having a uncertain significance due to their extreme rarity and heterogeneity. The efficiency of predisposition gene identification cannot be promoted significantly by simply increasing sample size. Third, incidental findings, which are not related to the observed phenotype of the patient, also complicate the analysis of the WES result (Kohane et al., 2006).
The American College of Medical Genetics and Genomics-Association for Molecular Pathology (ACMG-AMP) based guidelines have been widely used in variant classification (Hampel et al., 2015). Recently, ACMG-AMP-based variant classification rules have also been used in familial BC (Maxwell et al., 2016) and pan-cancer datasets (Huang et al., 2018). Of note, a co-segregation status of a germline variant is also important for variant classification (Jarvik and Browning, 2016). Pan-cancer studies have provided valuable sources to inspect tumor initiation and progression (Weinstein et al., 2013). An integrative analysis of germline and somatic variants could help to decipher tumor progression (Kanchi et al., 2014). We supposed that the co-occurrence between non-silent familial co-segregation variants and TCGA derived germline datasets could provide supporting evidence for a predisposition. Furthermore, pan-cancer datasets would also provide additional clues and evidence. Given that, we reanalyzed the WES datasets including 10 familial non-BRCA1/BRCA2 BC pedigrees (Gracia-Aznarez et al., 2013; Hilbers et al., 2013), manually evaluated variants as recommended (Hampel et al., 2015), and performed data mining on pan-cancer datasets.
In our analysis, some recently published BC predisposition genes, including MRE11 (Bartkova et al., 2008), CTR9 (Hanks et al., 2014), were recalibrated in our results, but were missed in the original publication. In addition, we identified novel cancer predisposition genes, such as NCK1. NCK1 encodes the cytoplasmic adaptor protein NCK1, which contains Src homolog2 and 3 (SH2 and SH3) domains. As an adaptor, NCK1 mediates multiple signals from receptors, including EGFR, PDGFR, to downstream effectors and the overexpression of Nck in the NIH 3T3 cell line showed oncogenic features (Li et al., 1992). In mammalians, most Nck1 effectors are involved in cytoskeletal dynamics (Li et al., 2001). For instance, Nck1 is involved in actin cytoskeletal remodeling via the WASp/Arp2/3 complex, which in turn causes the polarization and directional migration of the cell (Lapetina et al., 2009). Interestingly, the mutation NCK1 (p.D73H) identified from the BC pedigree (F2887) is located in an N-WASP activation motif (Okrut et al., 2015). Therefore, we supposed that NCK1 (p.D73H) might impact cell invasion. MCF7 cell lines, which are non-invasive, transfected with NCK1 mutants and were much more viable and invasive, in vitro. In conclusion, our results support that NCK1 could be a candidate cancer predisposition gene.
Materials and Methods
Whole Exome Sequencing Datasets
In this study, we reanalyzed WES data of non-BRCA1/BRCA2 BC pedigrees (Gracia-Aznarez et al., 2013; Hilbers et al., 2013). Ten pedigrees with at least two independent patients applied to whole exome sequencing were involved in this study. The raw data of pedigrees (2887, 3311, RUL36, and RUL153) are available at National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (Project ID: PRJEB3235). The raw data of pedigrees (NIJM6, NIJM8, RUL39, RUL70, RUL79, and RUL154) were transmitted with permission. The authority of the datasets about those pedigrees belongs to the original authors.
Variant Calling, Annotation, and Evaluation
We mapped the WES reads against the human reference genome (hg19) using BWA mem mode, with parameters set as default (Li and Durbin, 2009) and preprocessed as recommended (McKenna et al., 2010). Mindful that highly quality off-target variants could be identified from WES (Guo et al., 2012), we generated all exon regions with flanking 100 bp via UCSC Table browser supplied to GATK for variant calling. We combined VQSR (Variant Quality Score Recalibration) and a hard filters to filter out potential false positive variants. The parameters are summarized in the Supplementary Table S1. The variants were then annotated with ANNOVAR (Wang et al., 2010) and classified as recommended (Hampel et al., 2015). The databases involved in annotation and the variant classification methods are summarized in the Supplementary Table S1.
Vector Construction, Cell Culture, and Transfection
Full-length NCK1 was cloned from pLX304 to MSCV-5′HA (3×). We generated point mutants of NCK1 (p.D73H and p.R42Q) via site-directed mutagenesis with primers designed by Primer X1. All the vectors were confirmed via Sanger sequencing. For lentivirus production, the NCK1 mutants containing MSCV vectors were co-transfected with pCMV-VSVg and GAG/pol plasmids into 293FT cells by Lipo2000. Cell lines were cultured at 37°C under 5% CO2 in DMEM, high glucose medium (Gibco) with 10% (v/v) fetal bovine serum (FBS; Gibco) and penicillin G (100 U/ml, Gibco) and streptomycin (100 ug/ml, Gibco).
Cell Viability Assay and Transwell Invasion Assay
Cell viability was assessed with MTT colorimetric assay (Ameresco), at time periods of 6 days. The optical absorbance was measured at 562 nm on a spectrophotometer (Biotek), and the reference wavelength at 630 nm. All the experiments were performed in triplicate and repeated three times. Cell invasion assays were performed using 24-well transwell (8 μm pore, Corning) that were coated with 1:10 diluted Matrigel Matrix (BD Biosciences). A total of 2 × 104 cells, in 200 μL of serum-free DMEM medium, were added into the upper transwell chamber, and 500 μL of 10% FBS DMEM medium containing 1 μg/mL EGF was added into the lower chamber. After incubation for 48 h, the cells were fixed in 4% paraformaldehyde and stained with 0.1% crystal violet. The cell images were taken at five random microscopic fields (Olympus, 10×). All experiments were repeated three times. The Student’s t-test was used to test whether the difference was significant.
NCK1 Mutation Analysis
TCGA-germline variants were retrieved by subtracting the non-TCGA variants (ExAC-non-TCGA) from the whole dataset (ExAC) (Lek et al., 2016). Pan-cancer somatic mutations of NCK1 were retrieved from cBioportal (Cerami et al., 2012). We performed a hotspot analysis on NCK1 somatic mutations via the R package DominoEffect (Buljan et al., 2018). The flanking regions were determined after normalizing the gene length and impaired residues by function calculate boundary (Buljan et al., 2018). In order to evaluate substitution tolerance of NCK1 mutations, position specific score matrix (PSSM) was generated by PSI-BLAST (Altschul et al., 1997). For a given missense mutation, we obtained the score difference between the mutation and wild type residue: ΔS = S mutation – S wild–type. We generated 10,000 sets of three random mutations of NCK1 and evaluated the mean score for each set.
NCK1 Mutation Burden Analysis
To perform mutation burden analysis of NCK1 germline mutations in a cancer-cohort and normal controls, we retrieved the allele count and allele number of corresponding NCK1 mutations from the general cohort, control-cohort, non-cancer cohort collected from the Genome Aggregation Database (genomAD) (Karczewski et al., 2019). The cancer-cohort specific allele count and allele number of NCK1 mutations was obtained by deducting the non-cancer cohort from the general cohort. A Fisher test was used to test the occurrence of non-silent mutations in NCK1 across the cohorts mentioned above.
NCK1 Expression Analysis
As described before (Chen et al., 2016), the mRNA expression level in NCK1 (RNA-seq V2) of 99 tumor-normal matched BC samples were retrieved from the Cancer Genome Atlas database (Weinstein et al., 2013) and the RSEM normalized result were applied to the downstream analysis. Among them, 95 patients owned inferred PAM50 subtypes (Netanely et al., 2016).
Results
Re-evaluation Variants Identified From Familial Breast Cancer Patients
We reanalyzed published Whole Exome Sequencing datasets from 10 non-BRCA1/2 BC pedigrees (Gracia-Aznarez et al., 2013; Hilbers et al., 2013). Two samples per pedigree were applied to whole exome sequencing, and the kinship of the samples varied from 0.016 to 0.25 (Table 1). We set those rare non-silent variants, shared between patients per pedigree, as candidate co-segregated ones. To reduce incidental findings, we first focused on the genes that had been assigned with pathogenic supporting evidence (Supplementary Table S1), especially the known cancer predisposition genes (Rahman, 2014). Second, we filtered for variants with uncertain clinical significance, which must show in both the familial and TCGA germline dataset. The detailed variant filtering and classification parameters are summarized in Supplementary Table S1.
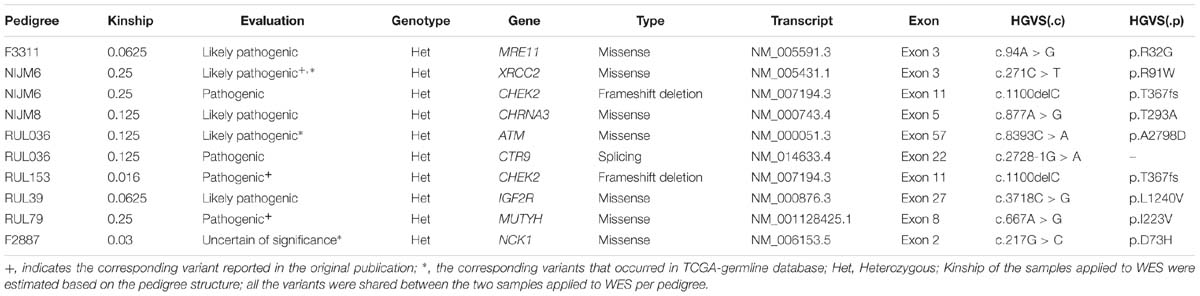
Table 1. Candidate predisposition genes identified from non-BRCA1/2 pedigrees.
In our analysis, we found that seven out of 10 pedigrees had potential co-segregated pathogenic variants in known cancer-associated genes (Table 1 and Supplementary Table S1), including CHEK2, ATM, MRE11, and CTR9, and some other cancer-associated genes, such as IGF2R and CHRNA3 (Table 1 and Supplementary Table S1). Interestingly, we found that XRCC2 (p.R91W) and ATM (p.A2798D) co-occurred in the ExAC TCGA-germline dataset (Table 1 and Supplementary Table S1). Furthermore, the XRCC2 (p.R91W) was also reported in the original publication (Hilbers et al., 2012) and an independent pedigree (Park et al., 2012), which further confirmed our approach was effective. Finally, we identified a novel candidate gene, NCK1, from pedigree F2887 (Table 1 and Supplementary Table S1). NCK1 (p.D73H) occurred once in about 7000 TCGA samples, but did not show up in more than 60,000 control samples (Supplementary Table S2). Generally, we succeeded in identifying potential cancer predisposition variants from eight in 10 pedigrees in the evaluation.
Most of the Somatic and Germline Mutations in NCK1 Were Intolerant
So far, few publications have reported the cancer predisposition role of NCK1. First, we inspected the NCK1 variants in the genome aggregation database (genomAD), which contained the cancer patient cohort and provided detailed cohort information, such as non-cancer, control (Supplementary Table S2). We could therefore retrieve the allele counts and allele numbers of the corresponding variants recorded in genomAD for enrichment analysis (Supplementary Table S2). Additionally, we only focused on the high-quality variants, which were marked as a pass in both the exome and genome datasets. The NCK1 mutations were significantly enriched in the cancer cohort, non-cancer cohort, and general cohort in comparison to the control cohort (Fisher-test; P < 0.001) (Supplementary Table S2).
Second, we inspected the occurrence of somatic mutations in NCK1 among pan-cancer datasets since the somatic event is another important factor involved in cancer progression. According to pan-cancer datasets, 0.3% of patients had NCK1 somatic mutations, including 102 non-silent mutations from 97 patients, and four fusion variants impaired NCK1 in four patients (Figure 1A). NCK1 mutations were enriched in some cancer types, including uterine endometrioid carcinoma (P = 1e−16), stomach adenocarcinoma (P = 6.679e−06), cutaneous melanoma (P = 2.63e−05), but not BC (P > 0.05) (Supplementary Figure S1; Binominal test). Among these mutations, residue 42 is the most frequent in somatic, and residue 73 mutated in both the BC pedigree and the cancer cohort (Figures 1A,B). Given the rarity of the NCK1 germline and somatic mutations, we supposed that mutations in NCK1 might be intolerant.
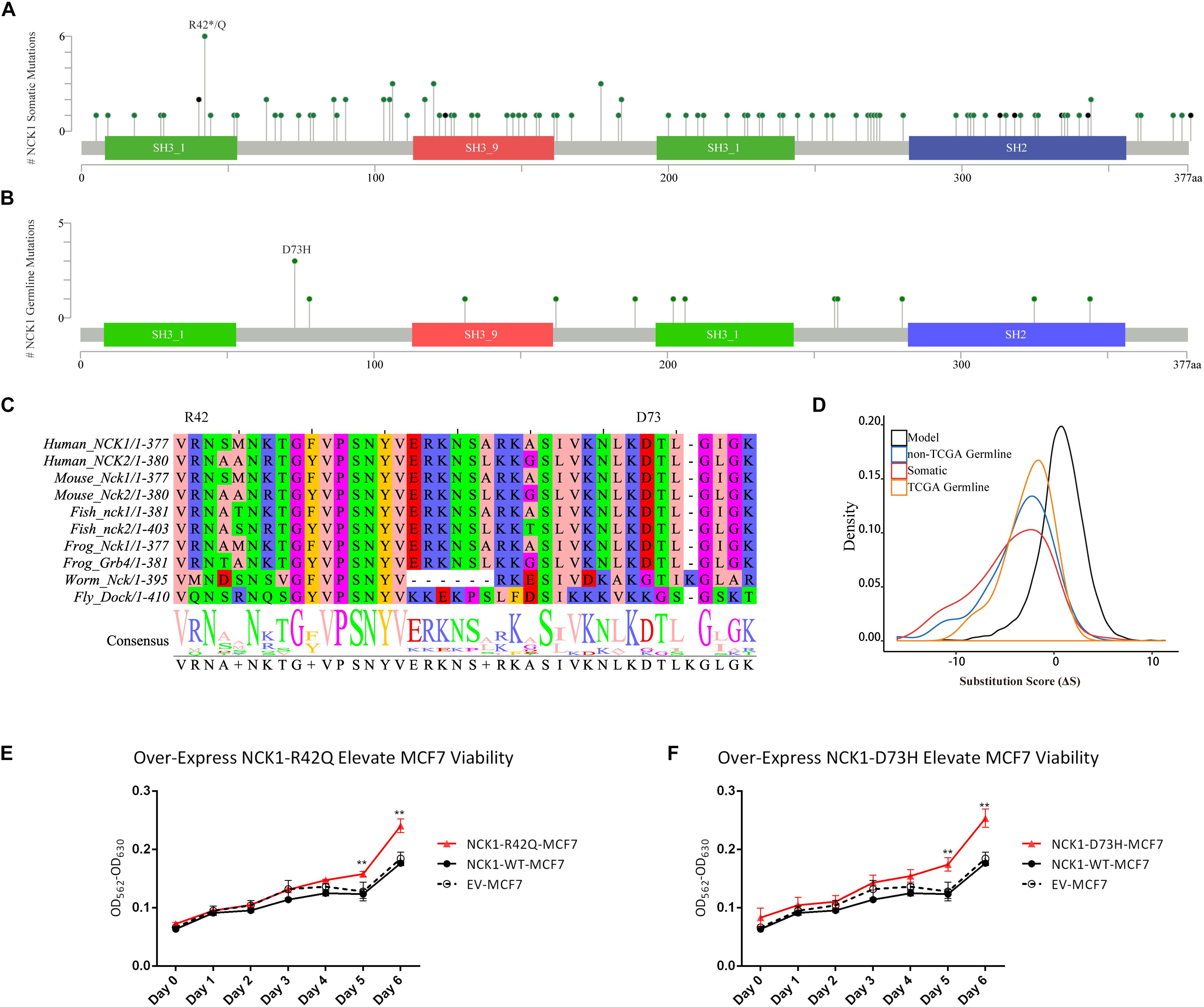
Figure 1. NCK1 mutation diagram and potential functional effect. (A) Mutation diagram of NCK1 collected in cBioportal (Pan-Cancer). (B) Mutation diagram of germline mutations in NCK1, including all TCGA-germline variants and NCK1 D73H, identified in familial breast cancer pedigree (F2887). (C) Multiple sequence alignment of sequence flanking NCK1 D73 residue. (D) Distribution of substitution score (ΔS) of NCK1 based on Position Specific Score Matrix. (E and F) The cell viabilities in all groups of mutant over-expression assay about R42Q (E) and D73H (F) at different time points (0, 1, 2, 3, 4, 5, and 6 days). Data were expressed as mean ± standard deviation (SD) of experiments with triplicates. Asterisks indicate significant increasing of cell viability in mutant (R42Q and D73H) transfected MCF7 cells compared with wild type transfected MCF7 cells (Student’s t-test; P < 0.01). Model: random mutations generated by in silico, non-TCGA germline: variants collected in ExAC non-TCGA dataset; TCGA-germline: variants collected in ExAC, but not in the ExAC non-TCGA dataset; Somatic: somatic variants collected in cBioportal.
To confirm that supposition, we generated a position specific score matrix (PSSM) via PSI-BLAST (Altschul et al., 1997) and predicted the damaging effect with SIFT (Ng and Henikoff, 2003) and PolyPhen2 (Adzhubei et al., 2010). In silico, PolyPhen2 predicted that those two were possibly damaging, and SIFT predicted that those two mutations were tolerant. Paradoxically, the residue D73 and R42 are conserved among 100 vertebrates according to MultiZ alignment (Supplementary Figure S2; Rosenbloom et al., 2015), and the residue R42 and D73 are both conserved in NCK1 and NCK2, which is the paralog of NCK1, but not conserved in the orthologs in Caenorhabditis elegans and Drosophila melanogaster (Figure 1C). According to PSSM, both germline and somatic mutations of NCK1 were more intolerant than randomly modeling mutations (Figure 1D), and substitution score of NCK1 D73H (ΔS = −3) and NCK1 R42Q (ΔS = −1) both are negative. In vitro, we found that both the mutants could increase cell viability (Figures 1E,F); therefore, both the NCK1 mutations should be deleterious.
Role of NCK1 Variations in Tumor Progression
Based on the “20/20” rule (Vogelstein et al., 2013), which means that more than 20 percent missense were located in recurred residues (Figures 1A,B), we supposed that NCK1 might have an oncogenic role. According to hotspot analysis of NCK1 somatic mutations, we found that the residue 42 turned to be a hotspot site (P < 0.001) (Supplementary Table S3). Indeed, NCK1-D73H and NCK1-R42Q transfected MCF7 cell lines showed significantly increased cell viability in comparison with wild type (Figures 1E,F). In addition, NCK1 contains an N-WASP activation motif (Okrut et al., 2015), where the residue D73 locates. Given this, we supposed that NCK1 might involve in tumor invasion.
To further prove that, we assessed the NCK1 mRNA expression level among 99 tumor-normal matched samples from TCGA-BRCA. However, the expression of NCK1 mRNA in tumor samples was significantly lower than the matched normal samples (Figure 2A), which was also observed across different tumor stages (Figures 2B–D). Mindful that BC is a molecular heterogenous cancer type, we retrieved PAM50 subtypes of the corresponding samples (Netanely et al., 2016). We found that NCK1 was significantly upregulated in the basal-like subtype (Figure 2E). No significant difference was observed in the Her2 subtype (Figure 2F), but the expression of NCK1 was still significantly downregulated in the Luminal A (Figure 2G) and Luminal B subtype (Figure 2H), especially in Luminal A. In this study, both NCK1-R42Q and NCK1-D73H transfected MCF cell lines, which are luminal subtypes, and showed a significantly increased invasion ability (Figures 3A,B). Recently, Morris et al. (2017) reported that the deficiency of Nck in MDA-MB-231, which is a basal-like subtype, could delay BC progression and metastasis, which was consistent with our results - given that NCK1 also plays a vital role in tumor invasion. Finally, we inspected the survival status of the patients with NCK1 variations, including CNVs, somatic mutations, and a Z-score normalized mRNA expression level, via cBioPortal (Gao et al., 2013). We found that the patients with both NCK1 variations and TP53 mutations had poorer overall survival (P < 0.05) and disease-free survival (P < 0.05) (Figures 3C,D). In general, the roles of NCK1 in tumor progression could be genomic context dependent and differentiated in cancer types.
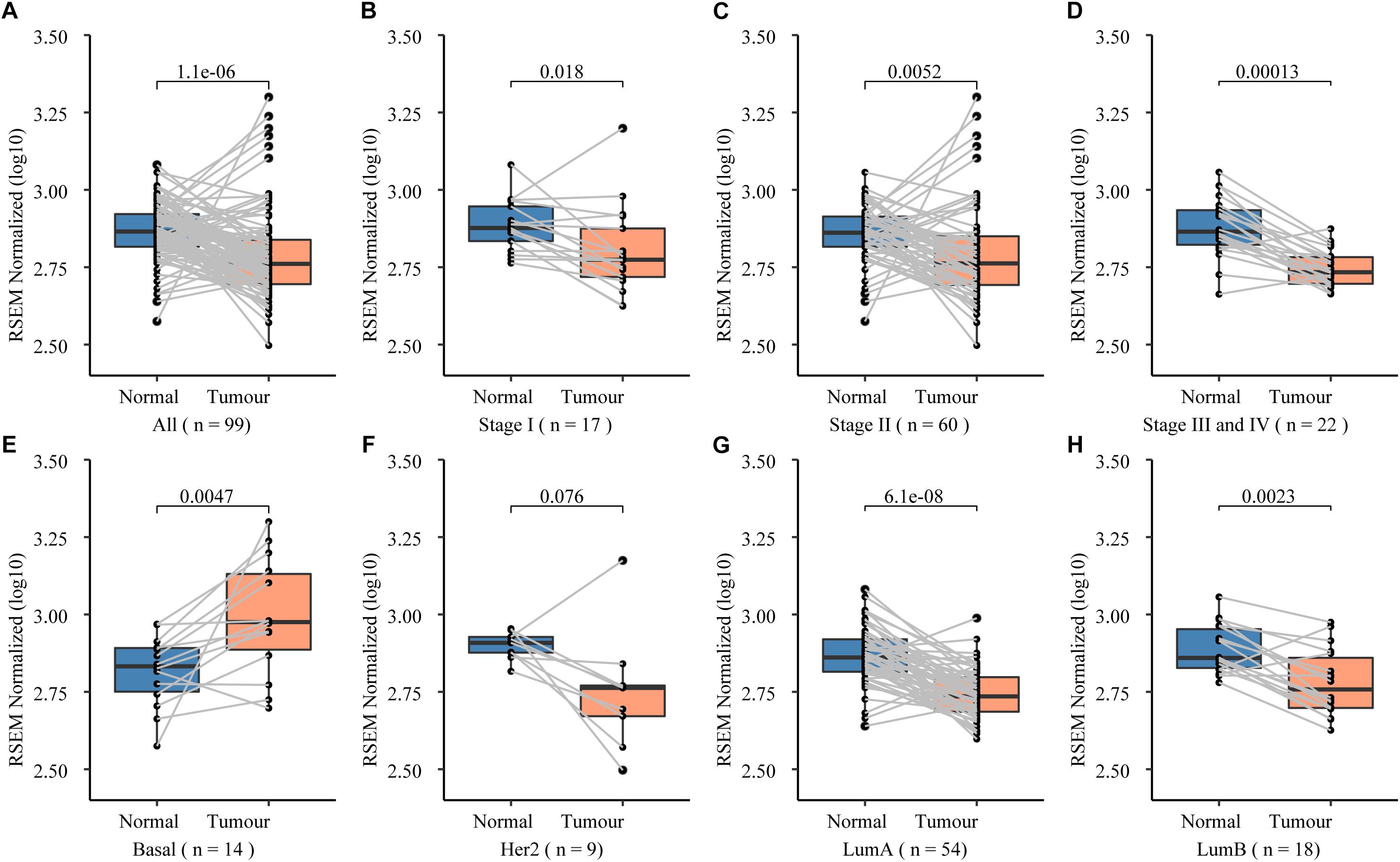
Figure 2. Expression spectrum of NCK1 in 99 tumor-normal paired samples across different stage and subtypes. (A) All; (B) Stage I; (C) Stage II; (D) Stage III and IV; (E) basal-like; (F) Her2; (G) LumA (LuminalA); and (H) LumB (LuminalB).
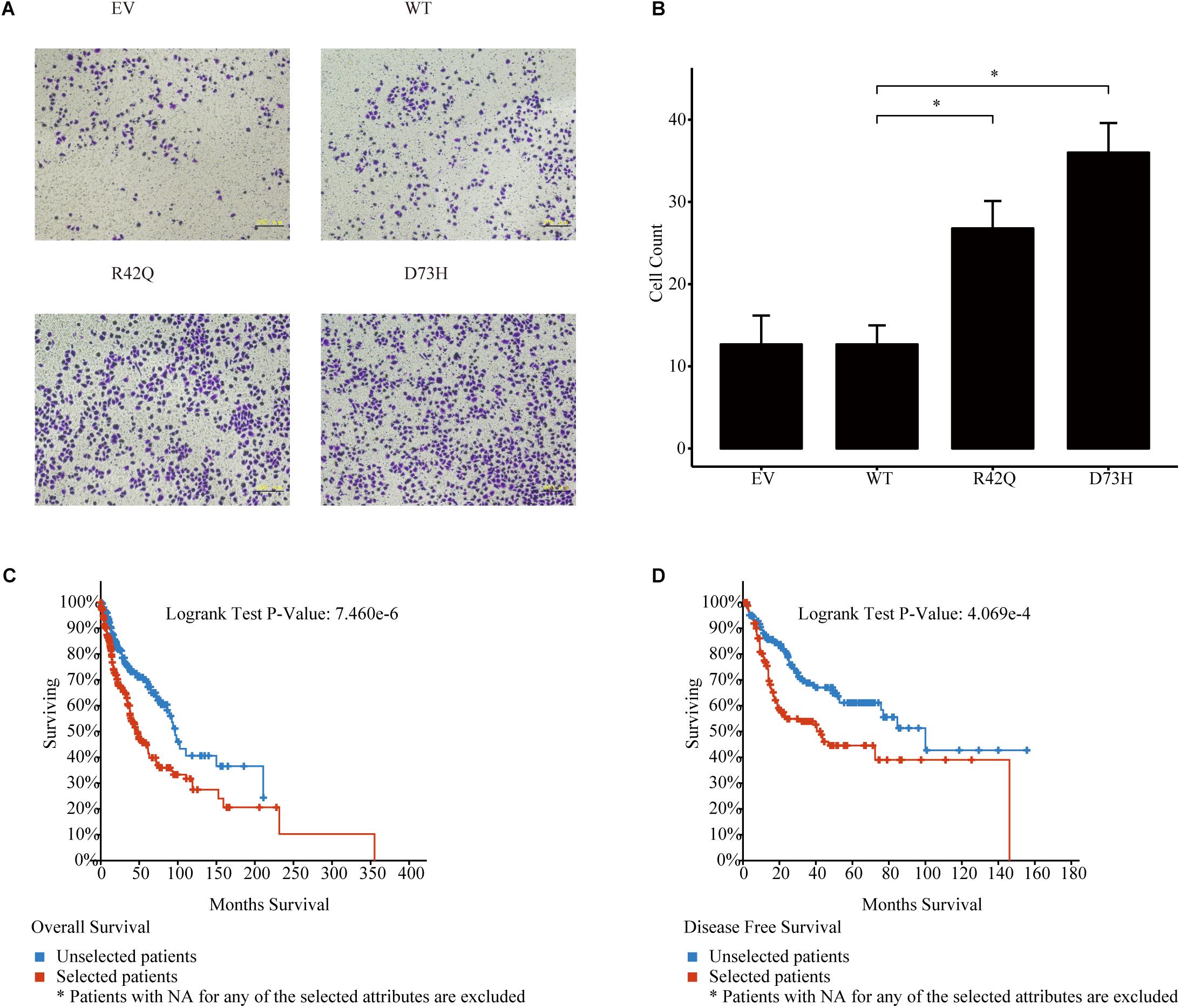
Figure 3. Roles of NCK1 in tumor progression might be context dependent. (A) Images of MCF7 cells migrated from transwell membrane (B) Cell count and quantitative analysis of the migrated MCF7 cells. Patients with both NCK1 aberrations and TP53 mutations showed a much poorer overall survival (C) and disease-free survival (D). Selected patients: patients with both NCK1 aberrations and TP53 mutation. Unselected patients: patients with only NCK1 aberrations. Scale bar: 200 μm. Data are depicted as mean ± standard deviation (SD).
Discussion
Intense efforts have been dedicated to identifying BC genes; however, more than 50% of familial BC heritability is still undetermined (Melchor and Benitez, 2013). Furthermore, non-BRCA1/2 familial BC patients are highly heterogeneous. For instance, we found CHEK2 mutations from four pedigrees, including pedigree RUL153, NIJM6, NIJM8 and RUL70 (Supplementary Table S4). The CHEK2 (p.T367fs) in pedigree NIJM8 appears to be homozygous but was only identified in one patient. Two separate CHEK2 variants were identified from members of pedigree NIJM8 (Supplementary Table S4). In RUL70, we also identified a CHEK2 mutation from only one patient. However, the confident predisposition variant in XRCC2 (Table 1) identified from another CHEK2 positive pedigree (NIJM6) further complicate the evaluation. CHEK2 (p.T367fs) was not co-segregated across all patients in RUL153, which was explained as a phenocopy (Gracia-Aznarez et al., 2013). Although CHEK2 (p.T367fs) is a well-known BC predisposition gene (Meijers-Heijboer et al., 2002), the co-segregation status of the variant has turned out to be negative among those pedigrees. Due to the patients in RUL70 and NIJM6, NIJM8 has been reported with a chromosome 22 gain like profile (Hilbers et al., 2013), where CHEK2 locates, and we therefore suppose that structural variants might also contribute.
During our analysis, we also identified some likely pathogenic variants in recently established cancer predisposition genes, such as MRE11 (Bartkova et al., 2008; Damiola et al., 2014) and CTR9 (Hanks et al., 2014). MRE11A, encoded by MRE11, acting as a component of the MRN (MRE11A-RAD50-NBN) complex, which plays a vital role in DNA double-strand break repair (Yuan et al., 2012). Dysfunction of the MRN complex could promote BC invasion and metastasis (Gupta et al., 2013). In pedigree RUL036, we identified two candidate predisposition genes, including ATM and CTR9. Although the ATM variant occurred in the TCGA-germline dataset, multiple in silico tools predicted it to be benign or tolerant. CTR9 was first reported as a Wilms tumor predisposition gene, and the mutations are almost truncated (Hanks et al., 2014). As it occurs in the Wilms tumor, we also identified a splicing site mutation in CTR9. Interestingly, evidence indicates that CTR9 plays an import role in regulating the estrogen signaling pathway, which promotes estrogen receptor α (ERα) positive BC progression (Zeng and Xu, 2015). In addition, we found a rare non-silent mutation in IGF2R. IGF2R is a polymorphic imprinting locus in humans (Xu et al., 1993), which indicates that individuals with IGF2R imprinted, might have increased cancer susceptibility (Feinberg, 1993). CHRNA3 encodes an α type subunit of the nicotinic acetylcholine receptor. Polymorphisms in CHRNA3 have been associated with increased smoking initiation risk and increases susceptibility to lung cancer (Hung et al., 2008). Given the heterogeneity in BC, the predisposition genes might have different disease-causative mechanisms and predisposition factors of non-BRCA1/2 pedigrees might be multifactorial, such as gene-environment interaction.
In our study, we mainly focused on gene NCK1, because few reports suggest the underlying predisposition role of NCK1 mutations. As an adaptor, NCK1 mediated multiple signaling pathways, especially actin dynamic and organization involved in invadopodia formation and maturation (Stylli et al., 2009; Oser et al., 2010). The SH2 domain of NCK1 involves the recognition of cell surface receptors and transduces signals to downstream effectors (Li et al., 2001). The SH3 domain of NCK1 usually interacts with downstream effectors, most of which involves the actin cytoskeletal dynamic. For instance, NCK1 is required for EGFR-mediated cell migration and tumor metastasis (Huang et al., 2012). And the metastasis-promoting role of NCK1 has been reported in multiple cancer types, such as colorectal cancer (Zhang et al., 2017) and BC (Morris et al., 2017). Interestingly, NCK1 also have connections to the hotspot mutation of PIK3CA. Wu et al. reported that oncogenic mutations of PIK3CA mediate tumor cell invasion through cortactin (Wu et al., 2014), which is a partner of NCK1 in invadopodia maturation (Oser et al., 2010). Therefore, NCK1 might be an invisible participant in tumor progression, because NCK1 mutations rarely occur in cancer patients.
On the one hand, overexpression of NCK1 shows oncogenic roles (Li et al., 1992), and the high expression of NCK1, at least in basal-like BC, contributes to tumor proliferation and metastasis (Morris et al., 2017). In our study, we identified a mutation in a motif that is involved in N-WASP activation, which is involved in invadopodia maturation (Okrut et al., 2015). Our results showed that both the NCK1 mutants (D73H and R42Q) indeed promote cell proliferation and invasion in vitro. We propose that NCK1 not only contributes to cancer predisposition but is also involved in cancer progression and prognosis. In addition, our results also suggest that the tumor-promoting role of NCK1 might be a cancer subtype dependent. On the other hand, downregulation of NCK1 might also be pathogenic, but in different mechanisms. For instance, Nck degradation could prevent cancer cells from apoptosis (Li et al., 2013) and regulate actin dynamics (Buvall et al., 2013). Furthermore, NCK1 played important roles in angiogenesis (Zhang et al., 2017; Xia et al., 2018) and even has an unexpected link to CHEK2 activation (Kremer et al., 2007).
Traditional approaches to identify underlying predisposition genes usually involves allele frequency filtering and in silico prediction and the sequences involved in the comparative analysis could also impact the final accuracy. Although we identified some novel candidate cancer predisposition variants, the power to confirm the predisposition role of those variants was limited. Because most of candidate cancer predisposition variants identified in our analysis turn out to be familial specific, which indicates that the power to establish a novel predisposition variant depends on an extremely large sample size (Guo et al., 2016). For instance, the variant NCK1 (p.D73H), identified from the pedigree F2887, occurred once in about 7,000 cancer samples, but not in about 60,000 controls according to the genomAD datasets. The predisposition role of NCK1 mutations was ignored probably because of its rare occurrence. In general, our results support NCK1 as a candidate cancer gene; however, the underlying mechanisms requirefurther investigation. In addition, we imagine that many more cancer genes like NCK1 might exist.
Author Contributions
JY, LH, and XK aided in data collection and developed the concepts. JY performed the data analysis, variants evaluation, vector construction, table, created the figures, and drafted and revised the manuscript. KW aided in variants evaluation, vector construction, in vitro experiments, and manuscript revision. QM and HD aided in experiments design and manuscript revision. YZ and LH aided in data collection and manuscript revision. XK supervised all the study. All the authors read and approved the final manuscript.
Funding
This work was supported by the Key Programs of the Chinese Academy of Sciences (Grant No. QYZDJ-SSW-SMC01), the National Natural Science Foundation of China (Grant Nos. 31371499, 31471224, and 81570827), and the National Basic Research Program of China (Grant No. 2011CB510102).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all patients and families for their participation in the original study. We thank Florentine S. Hilbers for sharing the Whole Exome Sequencing data of the non-BRCA1/2 breast cancer pedigrees. The pan-cancer analysis result here are in whole based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The pan-cancer data mining analysis are mainly based on cBioportal: https://www.cbioportal.org. The large cohort analysis result about germline variants here was based on data generated by both the Exome Aggregation Consortium (ExAC): http://exac.broadinstitute.org and the Genome Aggregation Database (genomAD): https://gnomad.broadinstitute.org.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00527/full#supplementary-material
FIGURE S1 | Summary of NCK1 variations across TCGA studies.
FIGURE S2 | Multiple sequence alignment of residue flanking R42 and D73 across 100 vertebrates.
TABLE S1 | Variant filtering and classification parameters and the whole list of candidate genes.
TABLE S2 | The allele count and allele number of the non-silent NCK1 mutations retrieved from genomAD and the burden test result.
TABLE S3 | Hotspot analysis of NCK1 somatic mutations.
TABLE S4 | All CHEK2 variants identified from the pedigrees.
Footnotes
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Antoniou, A. C., and Easton, D. F. (2006). Models of genetic susceptibility to breast cancer. Oncogene 25, 5898–5905. doi: 10.1038/sj.onc.1209879
Bamshad, M. J., Ng, S. B., Bigham, A. W., Tabor, H. K., Emond, M. J., Nickerson, D. A., et al. (2011). Exome sequencing as a tool for mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755. doi: 10.1038/nrg3031
Bartkova, J., Tommiska, J., Oplustilova, L., Aaltonen, K., Tamminen, A., Heikkinen, T., et al. (2008). Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol. Oncol. 2, 296–316. doi: 10.1016/j.molonc.2008.09.007
Buljan, M., Blattmann, P., Aebersold, R., and Boutros, M. (2018). Systematic characterization of pan-cancer mutation clusters. Mol. Syst. Biol. 14:e7974. doi: 10.15252/msb.20177974
Buvall, L., Rashmi, P., Lopez-Rivera, E., Andreeva, S., Weins, A., Wallentin, H., et al. (2013). Proteasomal degradation of Nck1 but not Nck2 regulates RhoA activation and actin dynamics. Nat. Commun. 4:2863. doi: 10.1038/ncomms3863
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. doi: 10.1158/2159-8290.cd-12-0095
Chen, X., Cao, X., Sun, X., Lei, R., Chen, P., Zhao, Y., et al. (2016). Bcl-3 regulates TGFbeta signaling by stabilizing Smad3 during breast cancer pulmonary metastasis. Cell Death Dis. 7:e2508. doi: 10.1038/cddis.2016.405
Damiola, F., Pertesi, M., Oliver, J., Le Calvez-Kelm, F., Voegele, C., Young, E. L., et al. (2014). Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 16:R58. doi: 10.1186/bcr3669
Feinberg, A. P. (1993). Genomic imprinting and gene activation in cancer. Nat. Genet. 4, 110–113. doi: 10.1038/ng0693-110
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6:l1. doi: 10.1126/scisignal.2004088
Gracia-Aznarez, F. J., Fernandez, V., Pita, G., Peterlongo, P., Dominguez, O., de la Hoya, M., et al. (2013). Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS One 8:e55681. doi: 10.1371/journal.pone.0055681
Guo, M. H., Dauber, A., Lippincott, M. F., Chan, Y. M., Salem, R. M., and Hirschhorn, J. N. (2016). Determinants of power in gene-based burden testing for monogenic disorders. Am J Hum. Genet. 99, 527–539. doi: 10.1016/j.ajhg.2016.06.031
Guo, Y., Long, J., He, J., Li, C. I., Cai, Q., Shu, X. O., et al. (2012). Exome sequencing generates high quality data in non-target regions. BMC Genomics 13:194. doi: 10.1186/1471-2164-13-194
Gupta, G. P., Vanness, K., Barlas, A., Manova-Todorova, K. O., Wen, Y. H., and Petrini, J. H. (2013). The Mre11 complex suppresses oncogene-driven breast tumorigenesis and metastasis. Mol. Cell 52, 353–365. doi: 10.1016/j.molcel.2013.09.001
Hampel, H., Bennett, R. L., Buchanan, A., Pearlman, R., and Wiesner, G. L. (2015). A practice guideline from the american college of medical genetics and genomics and the national society of genetic counselors: referral indications for cancer predisposition assessment. Genet. Med. 17, 70–87. doi: 10.1038/gim.2014.147
Hanks, S., Perdeaux, E. R., Seal, S., Ruark, E., Mahamdallie, S. S., Murray, A., et al. (2014). Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat. Commun. 5:4398. doi: 10.1038/ncomms5398
Hilbers, F. S., Meijers, C. M., Laros, J. F., van Galen, M., Hoogerbrugge, N., Vasen, H. F., et al. (2013). Exome sequencing of germline DNA from non-BRCA1/2 familial breast cancer cases selected on the basis of aCGH tumor profiling. PLoS One 8:e55734. doi: 10.1371/journal.pone.0055734
Hilbers, F. S., Wijnen, J. T., Hoogerbrugge, N., Oosterwijk, J. C., Collee, M. J., Peterlongo, P., et al. (2012). Rare variants in XRCC2 as breast cancer susceptibility alleles. J. Med. Genet. 49, 618–620. doi: 10.1136/jmedgenet-2012-101191
Huang, K. L., Mashl, R. J., Wu, Y., Ritter, D. I., Wang, J., Oh, C., et al. (2018). Pathogenic germline variants in 10,389 adult cancers. Cell 173, 355.e–370.e. doi: 10.1016/j.cell.2018.03.039
Huang, M., Anand, S., Murphy, E. A., Desgrosellier, J. S., Stupack, D. G., Shattil, S. J., et al. (2012). EGFR-dependent pancreatic carcinoma cell metastasis through Rap1 activation. Oncogene 31, 2783–2793. doi: 10.1038/onc.2011.450
Hung, R. J., McKay, J. D., Gaborieau, V., Boffetta, P., Hashibe, M., Zaridze, D., et al. (2008). A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452, 633–637. doi: 10.1038/nature06885
Jarvik, G. P., and Browning, B. L. (2016). Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 98, 1077–1081. doi: 10.1016/j.ajhg.2016.04.003
Kanchi, K. L., Johnson, K. J., Lu, C., McLellan, M. D., Leiserson, M. D., Wendl, M. C., et al. (2014). Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 5:3156. doi: 10.1038/ncomms4156
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv
Kiiski, J. I., Pelttari, L. M., Khan, S., Freysteinsdottir, E. S., Reynisdottir, I., Hart, S. N., et al. (2014). Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc Natl Acad Sci U.S.A. 111, 15172–15177. doi: 10.1073/pnas.1407909111
Kohane, I. S., Masys, D. R., and Altman, R. B. (2006). The incidentalome: a threat to genomic medicine. Jama 296, 212–215. doi: 10.1001/jama.296.2.212
Kremer, B. E., Adang, L. A., and Macara, I. G. (2007). Septins regulate actin organization and cell-cycle arrest through nuclear accumulation of NCK mediated by SOCS7. Cell 130, 837–850. doi: 10.1016/j.cell.2007.06.053
Lapetina, S., Mader, C., Machida, K., Mayer, B., and Koleske, A. (2009). Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J. Cell Biol. 185, 503–519. doi: 10.1083/jcb.200809085
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Li, B., Pi, Z., Liu, L., Zhang, B., Huang, X., Hu, P., et al. (2013). FGF-2 prevents cancer cells from ER stress-mediated apoptosis via enhancing proteasome-mediated Nck degradation. Biochem. J. 452, 139–145. doi: 10.1042/bj20121671
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, W., Fan, J., and Woodley, D. T. (2001). Nck/Dock: an adapter between cell surface receptors and the actin cytoskeleton. Oncogene 20, 6403–6417. doi: 10.1038/sj.onc.1204782
Li, W., Hu, P., Skolnik, E. Y., Ullrich, A., and Schlessinger, J. (1992). The SH2 and SH3 domain-containing Nck protein is oncogenic and a common target for phosphorylation by different surface receptors. Mol. Cell Biol. 12, 5824–5833. doi: 10.1128/mcb.12.12.5824
Lynch, H., Synder, C., and Wang, S. M. (2015). Considerations for comprehensive assessment of genetic predisposition in familial breast cancer. Breast J. 21, 67–75. doi: 10.1111/tbj.12358
Mavaddat, N., Antoniou, A. C., Easton, D. F., and Garcia-Closas, M. (2010). Genetic susceptibility to breast cancer. Mol. Oncol. 4, 174–191. doi: 10.1016/j.molonc.2010.04.011
Maxwell, K. N., Hart, S. N., Vijai, J., Schrader, K. A., Slavin, T. P., Thomas, T., et al. (2016). Evaluation of ACMG-guideline-based variant classification of cancer susceptibility and non-cancer-associated genes in families affected by breast cancer. Am. J. Hum. Genet. 98, 801–817. doi: 10.1016/j.ajhg.2016.02.024
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meijers-Heijboer, H., van den Ouweland, A., Klijn, J., Wasielewski, M., de Snoo, A., Oldenburg, R., et al. (2002). Low-penetrance susceptibility to breast cancer due to CHEK2(∗)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 31, 55–59. doi: 10.1038/ng879
Melchor, L., and Benitez, J. (2013). The complex genetic landscape of familial breast cancer. Hum. Genet. 132, 845–863. doi: 10.1007/s00439-013-1299-y
Morris, D. C., Popp, J. L., Tang, L. K., Gibbs, H. C., Schmitt, E., Chaki, S. P., et al. (2017). Nck deficiency is associated with delayed breast carcinoma progression and reduced metastasis. Mol. Biol. Cell 28, 3500–3516. doi: 10.1091/mbc.E17-02-0106
Netanely, D., Avraham, A., Ben-Baruch, A., Evron, E., and Shamir, R. (2016). Expression and methylation patterns partition luminal-A breast tumors into distinct prognostic subgroups. Breast Cancer Res. 18:74. doi: 10.1186/s13058-016-0724-722
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Okrut, J., Prakash, S., Wu, Q., Kelly, M. J., and Taunton, J. (2015). Allosteric N-WASP activation by an inter-SH3 domain linker in Nck. Proc. Natl. Acad. Sci. U.S.A. 112, E6436–E6445. doi: 10.1073/pnas.1510876112
Oser, M., Mader, C. C., Gil-Henn, H., Magalhaes, M., Bravo-Cordero, J. J., Koleske, A. J., et al. (2010). Specific tyrosine phosphorylation sites on cortactin regulate Nck1-dependent actin polymerization in invadopodia. J. Cell. Sci. 123(Pt 21), 3662–3673. doi: 10.1242/jcs.068163
Park, D. J., Lesueur, F., Nguyen-Dumont, T., Pertesi, M., Odefrey, F., Hammet, F., et al. (2012). Rare mutations in XRCC2 increase the risk of breast cancer. Am. J. Hum. Genet. 90, 734–739. doi: 10.1016/j.ajhg.2012.02.027
Rahman, N. (2014). Realizing the promise of cancer predisposition genes. Nature 505, 302–308. doi: 10.1038/nature12981
Rizzolo, P., Silvestri, V., Falchetti, M., and Ottini, L. (2011). Inherited and acquired alterations in development of breast cancer. Appl. Clin. Genet. 4, 145–158. doi: 10.2147/tacg.s13226
Rosenbloom, K. R., Armstrong, J., Barber, G. P., Casper, J., Clawson, H., Diekhans, M., et al. (2015). The UCSC genome browser database: 2015 update. Nucleic Acids Res. 43, D670–D681. doi: 10.1093/nar/gku1177
Siegel, R. L., Miller, K. D., and Jemal, A. (2018). Cancer statistics, 2018. Can. J. Clin. 68, 7–30. doi: 10.3322/caac.21442
Stratton, M. R., and Rahman, N. (2008). The emerging landscape of breast cancer susceptibility. Nat. Genet. 40, 17–22. doi: 10.1038/ng.2007.53
Stylli, S. S., Stacey, T. T., Verhagen, A. M., Xu, S. S., Pass, I., Courtneidge, S. A., et al. (2009). Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J. Cell Sci. 122(Pt 15), 2727–2740. doi: 10.1242/jcs.046680
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A. Jr., Kinzler, K. W., et al. (2013). Cancer genome landscapes. Science 339, 1546–1558. doi: 10.1126/science.1235122
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi: 10.1093/nar/gkq603
Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R. M., Ozenberger, B. A., Ellrott, K., et al. (2013). The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. doi: 10.1038/ng.2764
Wu, X., Renuse, S., Sahasrabuddhe, N. A., Zahari, M. S., Chaerkady, R., Kim, M. S., et al. (2014). Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat. Commun. 5:4961. doi: 10.1038/ncomms5961
Xia, P., Huang, M., Zhang, Y., Xiong, X., Yan, M., Xiong, X., et al. (2018). NCK1 promotes the angiogenesis of cervical squamous carcinoma via Rac1/PAK1/MMP2 signal pathway. Gynecol. Oncol. 152, 387–395. doi: 10.1016/j.ygyno.2018.11.013
Xu, Y., Goodyer, C. G., Deal, C., and Polychronakos, C. (1993). Functional polymorphism in the parental imprinting of the human IGF2R gene. Biochem. Biophys. Res. Commun. 197, 747–754. doi: 10.1006/bbrc.1993.2542
Yuan, S. S., Hou, M. F., Hsieh, Y. C., Huang, C. Y., Lee, Y. C., Chen, Y. J., et al. (2012). Role of MRE11 in cell proliferation, tumor invasion, and DNA repair in breast cancer. J. Natl. Cancer Inst. 104, 1485–1502. doi: 10.1093/jnci/djs355
Zeng, H., and Xu, W. (2015). Ctr9, a key subunit of PAFc, affects global estrogen signaling and drives ERalpha-positive breast tumorigenesis. Genes Dev. 29, 2153–2167. doi: 10.1101/gad.268722.115
Keywords: breast cancer, non-BRCA1/2, NCK1, predisposition gene, invasion
Citation: Yin J, Wu K, Ma Q, Dong H, Zhu Y, Hu L and Kong X (2019) Revisiting Non-BRCA1/2 Familial Whole Exome Sequencing Datasets Implicates NCK1 as a Cancer Gene. Front. Genet. 10:527. doi: 10.3389/fgene.2019.00527
Received: 03 February 2019; Accepted: 14 May 2019;
Published: 04 June 2019.
Edited by:
Jialiang Yang, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Quan Zou, University of Electronic Science and Technology of China, ChinaIan Campbell, Peter MacCallum Cancer Centre, Australia
Guang Wu, Guangxi Academy of Sciences, China
Copyright © 2019 Yin, Wu, Ma, Dong, Zhu, Hu and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangyin Kong, eHlrb25nQHNpYnMuYWMuY24=
†These authors have contributed equally to this work