 Christian S. Jensen1*
Christian S. Jensen1* Charles J. Norsigian2
Charles J. Norsigian2 Xin Fang2
Xin Fang2 Xiaohui C. Nielsen1
Xiaohui C. Nielsen1 Jens Jørgen Christensen1,3
Jens Jørgen Christensen1,3 Bernhard O. Palsson2,4
Bernhard O. Palsson2,4 Jonathan M. Monk2
Jonathan M. Monk2- 1The Regional Department of Clinical Microbiology, Region Zealand, Slagelse, Denmark
- 2Department of Bioengineering, University of California, San Diego, San Diego, CA, United States
- 3Institute of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark
- 4Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Lyngby, Denmark
The mitis group of streptococci (MGS) is a member of the healthy human microbiome in the oral cavity and upper respiratory tract. Troublingly, some MGS are able to escape this niche and cause infective endocarditis, a severe and devastating disease. Genome-scale models have been shown to be valuable in investigating metabolism of bacteria. Here we present the first genome-scale model, iCJ415, for Streptococcus oralis SK141. We validated the model using gene essentiality and amino acid auxotrophy data from closely related species. iCJ415 has 71-76% accuracy in predicting gene essentiality and 85% accuracy in predicting amino acid auxotrophy. Further, the phenotype of S. oralis was tested using the Biolog Phenotype microarrays, giving iCJ415 a 82% accuracy in predicting carbon sources. iCJ415 can be used to explore the metabolic differences within the MGS, and to explore the complicated metabolic interactions between different species in the human oral cavity.
Introduction
The mitis group of streptococci (MGS) consists of 20 different species, all gram positive cocci arranged as pairs or in chains (Spellerberg and Brandt, 2008). MGS is a part of the lactic acid bacteria, which along with the human pathogenic genera Streptococcus, Enterococcus, and Aerococcus is composed of a wide variety of bacteria important for the food industry (Wu et al., 2017).
Most species in the MGS are considered commensal inhabitants of the oral cavity. These bacteria exist in a complex metabolic relationship with other oral streptococci, as well as other species residing in the oral cavity. These interactions are thought to have an influence on the development of certain oral diseases in humans, e.g., caries (Kreth et al., 2009; Nascimento et al., 2009; Abranches et al., 2018). Sometimes these MGS are able to escape their oral niche and cause an infection of the heart valves, infective endocarditis (IE), a severe disease with a high mortality even with proper treatment. Of all the IE causing species in the MGS, Streptococcus oralis is the most common (Rasmussen et al., 2016).
Genome-scale models (GEMs) of metabolism have proven to be valuable in investigating the metabolism in a single bacterial strain (McCloskey et al., 2013). GEMs have been used to compare metabolism between different strains of the same species and between different species (Monk et al., 2013; Ong et al., 2014; Bosi et al., 2016; Monk et al., 2017; Norsigian et al., 2018). When a GEM is used to compare different strains, it is possible to compare the metabolic capabilities with known phenotypic or virulence characteristics, enabling us to identify the metabolic capabilities that either enhance the virulence of a strain or is necessary for a species to be pathogenic (Monk et al., 2013). This provides an unprecedented opportunity to investigate complex mechanisms behind pathogenesis.
Given the implications of Lactococcus, Lactobacillus, and Streptococcus for the food industry, GEMs have been created and published (Oliveira et al., 2005; Teusink et al., 2006; Pastink et al., 2009; Thiele and Palsson, 2010). However, only two GEMs for human pathogenic streptococci have been published so far. One is for Streptococcus pyogenes, a distant relative of the MGS with a very different disease spectrum (Jensen and Kilian, 2012; Levering et al., 2016). The other published GEM is for a closer relative, Streptococcus pneumoniae (Dias et al., 2019). Although phylogenetically closely related, S. oralis and S. pneumoniae have several differences. S. pneumoniae is a part of the throat microbiome, while S. oralis is commensal in the oral microbiome. Furthermore, S. pneumoniae is a common cause of pneumonia, otitis media and meningitis. These diseases are only very rarely caused by S. oralis.
In this article we present the first GEM for a S. oralis strain, named iCJ415. This GEM is based on the genome sequence of S. oralis SK141 combined with experimental data using the Biolog phenotypic array or retrieved in published literature. The model is validated with experimental data obtained using either S. oralis SK141 or other closely related species in the MGS. This model is intended as a starting point for exploring the complicated metabolic interactions between different bacterial species in the human oral microbiome and for exploring the differences between pathogenic and non-pathogenic bacteria being a part of the human microbiome.
Materials and Methods
Strains
GEMs are built using an annotated genome sequence combined with experimental data. We used the strain S. oralis SK141, isolated from human dental biofilm (Kilian et al., 2014). The SK141 genome sequence was downloaded from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/, accession number: JPGA00000000.1). The genome sequence was annotated using the RAST server on default settings (Aziz et al., 2008).
Reconstruction
The reconstruction was made using the protocol made by Thiele and Palsson (2010). In short, we used the genome annotation to generate a draft reconstruction using Modelseed (Henry et al., 2010). This draft reconstruction was intensively manually curated using the databases BiGG (King et al., 2016), KeGG (Ogata et al., 1999), and MetaNetx (Ganter et al., 2013). If possible, reactions were verified with experimental data. If there was no available S. oralis specific data, we used data from closely related species (e.g. Streptococcus mitis or S. pneumoniae). For gapfilling, pathways were visualized using Escher (King et al., 2015). To identify missing genes, BLASTn homology searches were done using the NCBI webserver with default settings (Boratyn et al., 2013). To ensure a standardized nomenclature and to make comparison between other GEMs easier, all reactions and metabolites in the model were named with BiGG IDs if possible. BiGG Models is a knowledge base now incorporating more than 70 published GEMs (King et al., 2016).
The reconstruction was converted to a mathematical model and all simulations were done, using Constraint-Based Metabolic Modeling in Python (COBRApy) version 0.13.4 (Ebrahim et al., 2013).
Biolog
Biolog experiments were performed using Biolog’s phenotype microarrays (Biolog, Hayward, CA, USA). All experiments were carried out according to the phenotype microarray protocol for S. agalactiae and other Streptococcus species (version: 16-May-09) provided by the manufacturer with the following exception: Due to a high degree of background noise in the negative control well, phenotype microarrays 3 was tested using only 1/10 of the carbon sources reported.
The Biolog results were analyzed with the “OPM” package in R, and statistical analysis was done with the “Dunnett-type comparison: one-against-all” function (Vaas et al., 2013). All wells with a P-value < 0.05 when compared with the negative control were considered as having growth.
Biomass Reaction
The biomass reaction should include all essential constituents, and their fraction, in the biomass composition. Due to the lack of a detailed biomass composition of S. oralis we used the macromolecular composition of a Lactobacillus plantarum previously published (Teusink et al., 2006). We used the Python package BOFdat (Lachance et al., 2019) to calculate coefficients for DNA, RNA, proteins, lipids, coenzymes and ions. BOFdat divides the biomass calculations into different steps. Step 1 calculates biomass coefficients for DNA, RNA, proteins, and lipids. For determining coefficients for the nucleotides in DNA, it calculates the relative abundance of each nucleotide based on the genome provided. For RNA and amino acid calculations BOFdat uses transcriptomics and proteomics data, respectively. BOFdat uses these data to compare the frequency of RNA and proteins found in these experimental data to calculate the coefficients for nucleotides and amino acids present in RNA and proteins. The data used to RNA and amino acid calculations were obtained from S. pneumoniae strain D39 (Aprianto et al., 2018; Sun et al., 2011). To calculate lipids, BOFdat uses the relative abundance, found in experimental data, and the molecular weight of these lipids in the model. We used the lipid composition data from a previously published Lactococcus lactis model iAO358 (Oliveira et al., 2005). BOFdat step 2 adds coenzymes and inorganic ions to the model.
Since Gram positive bacteria, in contrast to Gram negative, contains a large amount of peptidoglycan, teichoic acid and lipoteichoic acid, we calculated the coefficients for these structures using data from a L. plantarum (Teusink et al., 2006). Further we used data from Xavier et al. to include the following coenzymes considered essential when constructing a metabolic model: Nad, nadp, S-Adenosyl-L-methionine, fad, pyridoxal 5′-phosphate, poenzyme A, tetrahydrofolate, methyltetrahydrofolate, formyltetrahydrofolate, thiamine diphosphate, and riboflavin-5-phosphate (Xavier et al., 2017). The individual contribution of these coenzymes was evenly distributed among the remaining part of the biomass.
All data used for BOFdat and the calculations for metabolites not found by BOFdat are summarized in Supplementary Table S1.
Validation of iCJ415
Gene Essentiality
No gene essentiality data exists for S. oralis, therefore comparison between iCJ415 and experimental gene essentiality data was done using data from the closely related species S. pneumoniae (Strain TIGR4) (van Opijnen et al., 2009) and Streptococcus sanguinis (Strain SK36) (Xu et al., 2011). Orthologous genes were found using NCBI Bidirectional Blast. COBRApy contains a function, which allows all genes in the model to be deleted individually, and for each gene deletion growth is assessed. This function is similar to in vivo experiments of tn-seq and transposon mutagenesis experiments (van Opijnen et al., 2009; Kleckner et al., 1977). Since all gene essentiality experiments were carried out in rich media (e.g. Broth), the simulations were done with all exchange reactions open.
Amino Acid Auxotrophy
Amino acid auxotrophies were found using data from S. pneumoniae strain D39 (Härtel et al., 2012). To compare amino acid auxotrophies found in the experimental data and iCJ415 we opened the exchange reactions corresponding to all metabolites present in the CDM media used to obtain the experimental data. When simulating amino acid omissions we sequentially closed the exchange reactions for the amino acid omitted and tested for growth of the model.
Carbon and Nitrogen Sources
When testing for carbon sources we found a minimal media in the literature, which did not support in silico growth of S. oralis without an added carbon source (Homer et al., 1993; Paixão et al., 2015). We used this media and sequentially opened the exchange reaction for the carbon sources and tested for growth of iCJ415. Only Biolog metabolites readily mapped to metabolites in iCJ415 or positive in the Biolog phenotypic system were included.
Nitrogen sources were tested with the same media used to test carbon sources. We used glucose (Opened the EX_glc__D_e exchange reaction) as a carbon source. We sequentially opened and closed the exchange for all nitrogens sources tested. If the opening resulted in an increase in Biomass yield, it was considered a positive result.
All simulations on gene essentiality, amino acid auxotrophy and carbon and nitrogen sources were done using Flux Balance Analysis (FBA) while optimizing for the Biomass reaction. For all metabolites present in the media, the upper and lower bounds were set to -1000 and 1000 mmol/g/h, respectively. When doing Biolog validations the exchange reactions for the carbon- and nitrogen sources tested were also set to -1000 and 1000 mmol/g/h for lower and upper bounds, respectively. Supplementary Table S2 shows the media content used in the simulations for carbon- and nitrogen sources and amino acid auxotrophies.
For all FBA simulations we used a value of 10-8 as a growth/no growth cutoff.
Biomass Yield
To test the biomass yield and byproduct production, we compared with previously published experimental data obtained in the S. pneumoniae strain D39 (Paixão et al., 2015). The D39 strain was grown in a chemically defined media, using four different carbon sources, and the byproducts were measured. These data have been used to validate a recently published S. pneumoniae GEM, iDS372 (Dias et al., 2019). We used the same media conditions to simulate growth of iCJ415 (see Supplementary Table S2). To simulate the repression of genes by the carbon catabolite repressor A (CcpA) present in MGS, we added additional constraints of the reactions catalyzed by genes repressed by the CcpA, This was done by testing the flux through the reactions catalyzed by the genes repressed by the CcpA using only the constraints imposed by the media. Fluxes through the reactions were then set to 0% (LACOX) or a maximum of 10% of the unconstrained value (ACKr, ALCD2x, PTAr). Due to alleviation of the repression when grown on galactose ACKr, ALCD2x and PTAr were set to a maximum of 100% of the unconstrained value. For the pfl gene, and associated reaction, Dias et al. simulated an underexpression by testing the growth rate and byproduct formation with fluxes from 0-100% of the unconstrained value. For comparison with iDS371, we tested only the value of pfl underexpression giving the best match with experimental data.
These calculations were visualized and calculated using the Escher FBA webapplication (Rowe et al., 2018).
Memote Test
iCJ415 was tested using memote, which provides a platform to test the performance of a metabolic model (Lieven et al., 2018).
Results and Discussion
The iCJ415 S. oralis metabolic model contains 604 reactions, 504 metabolites, and 415 genes. All reactions and metabolites in iCJ415 are detailed in Supplementary Table S3 and can be found in json and sbml format in Supplementary Data Sheets 1 and 2. Of those 604 reactions, 475 are included in the BiGG database, 52 are not included in the BiGG database, and the remaining 76 (excluding the biomass reaction) are exchange reactions. Figure 1 shows the characteristics of the reactions included in iCJ415. Of the non-gene associated reactions, six are spontaneous and four are added as sink or demand reactions due to lack of available data (e.g., the reaction “Skgcald”) or due to the need for a non-metabolic metabolite in the model, e.g., a sink reaction for DNA. Three reactions were added as gapfilling and 37 reactions were added based solely on biochemical information retrieved either from the literature or Biolog experiments, with no information on the associated gene. We were not able to find the genes responsible for making unsaturated fatty acids, and we added these based only on data present in literature. The same was the case for the reaction “UDPACGALRIBGALUDPAATGALS,” which produces one of the last reactions in the teichoic acid synthesis. The gene responsible for this reaction was not found, but we added the reaction based on experimental data (Denapaite et al., 2012). These non-gene associated reactions are placed in a separate sheet in Supplementary Table S3.
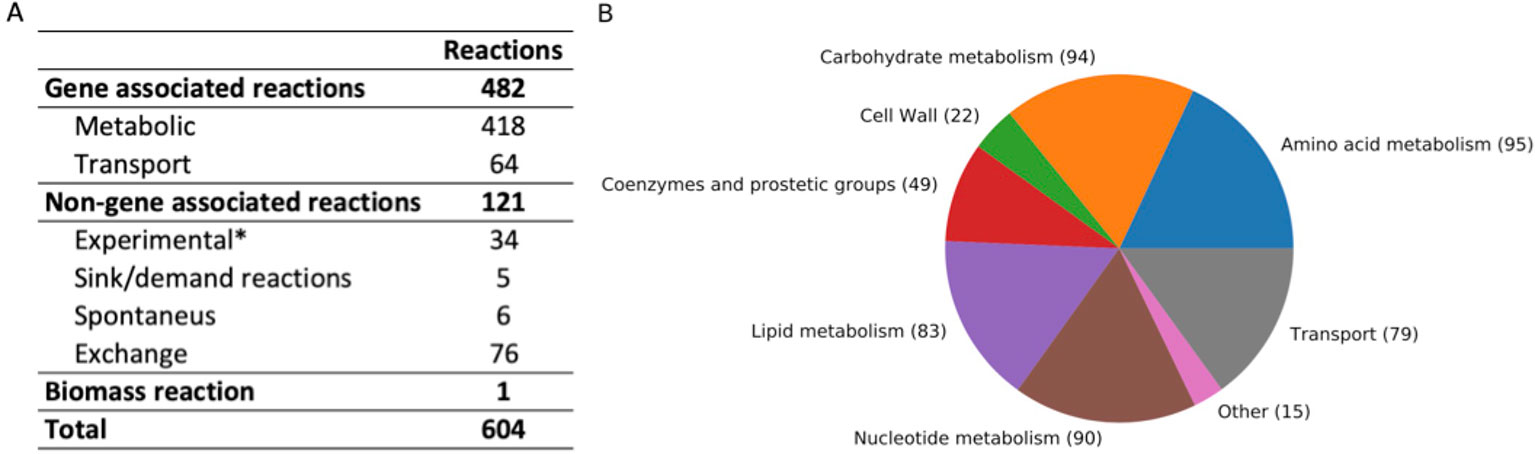
Figure 1 (A) Summary of reaction characteristics in iCJ415. (B) Reactions in iCJ415 sorted by pathway. Exchange reactions are left out. The reaction pathways are merged together according based on pathways and according to https://www.kegg.jp/kegg/pathway.html. *Based on experimental data, either found in literature or found in Biolog experiments.
Biomass Reaction
Using the software BOFdat we constructed a biomass reaction using genomic, transcriptomic, proteomic and lipidomic data from either S. oralis SK141 or closely related species. When comparing with another GEM from a closely related species, S. pneumoniae, iDS372, the biomass reactions have many similarities, though there are differences. In contrast to iDS372 we decided not to include polyamines into the Biomass reaction of iCJ415, despite polyamines has been shown to be import in nasopharyngeal carriage, and virulence of S. pneumoniae. This is because S. oralis does not colonize the nasopharynx and is usually not considered a pneumonia-causing agent like S. pneumoniae (Shah et al., 2011). The biomass reaction of iCJ415 is also similar to that of a S. pyogenes GEM (Levering et al., 2016). However, S. pyogenes, contains a capsule while S. oralis SK141 does not. Therefore we did not include a capsule in the biomass reaction of S. oralis SK141 (Levering et al., 2016).
Verification of iCJ415 Using Experimental Essentiality Data
Gene essentiality analysis is an important measure for validating the model, especially when considering new possible antimicrobial drug targets. We predicted a list of genes that are essential for growth using COBRApy to do a single gene knockout simulation in iCJ415. By using data from the closely related bacteria S. sanguinis and S. pneumoniae, Bidirectional Blast Hits found 356 and 375 orthologous genes in S. oralis SK141, respectively. The gene essentiality predictions in iCJ415 matched the S. sanguinis and S. pneumoniae gene essentiality datasets in 76% and 71% of the cases, respectively (See Figure 2, Supplementary Table S4).
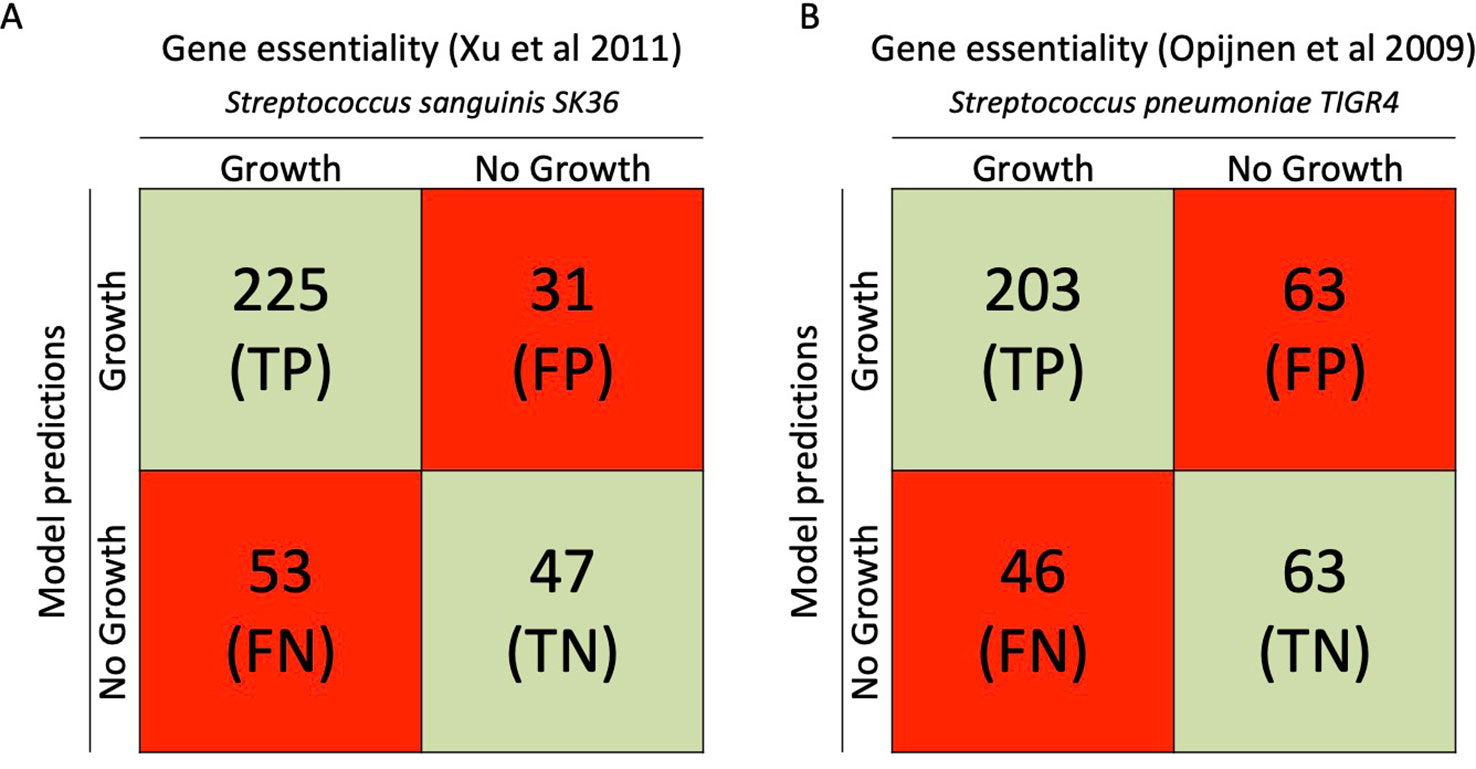
Figure 2 Gene essentiality comparison between iCJ415 and experimental data. (A) When iCJ415 is compared with S. sanguinis SK36 there is a 76% (272/356) concordance in gene essentiality. (B) When comparing iCJ415 with S. pneumoniae TIGR4 tn seq data there is a 71% (266/375) concordance. Gene orthologs in S. sanguinis SK36 and S. pneumoniae TIGR4 were found using bidirectional blast.
When comparing the gene essentiality datasets from S. sanguinis and S. pneumoniae, 336 of the genes in iCJ415 were found in both essentiality datasets and 79 were found in only one, or in none of the datasets. Discrepancy between the two essentiality datasets were observed with 62 genes. These discrepancies can partly be explained by how the experimental data were obtained. The S. pneumoniae essentiality data were from a tn-seq experiment and the S. sanguinis data were from gene knockout mutants.
Since we are using two gene essentiality dataset from two closely related bacteria, we only look at those genes where the experimental gene essentiality data agrees. Genes are termed “concordant,” when both gene essentiality datasets and iCJ415 results are the same and “discordant” when the gene essentiality datasets are the same, but they are different from gene essentiality predictions in iCJ415. The terms discordant and concordant are also used in Supplementary Table S4 to describe the two situations. Table 1 shows all the concordant and discordant genes categorized on pathway. Most pathways are almost equally represented in the concordant and discordant group, though amino acid metabolism has a higher representation in the discordant group and transport has a higher representation in the concordant group. It is worth noticing that both peptidoglycan and terpenoid backbone synthesis are highly represented in the concordant group. They are both simple synthetic pathways with only one gene responsible for each reaction, and the end product is essential for growth of the bacteria. This is in opposition to the amino acid metabolism where several reactions can lead to the same product.
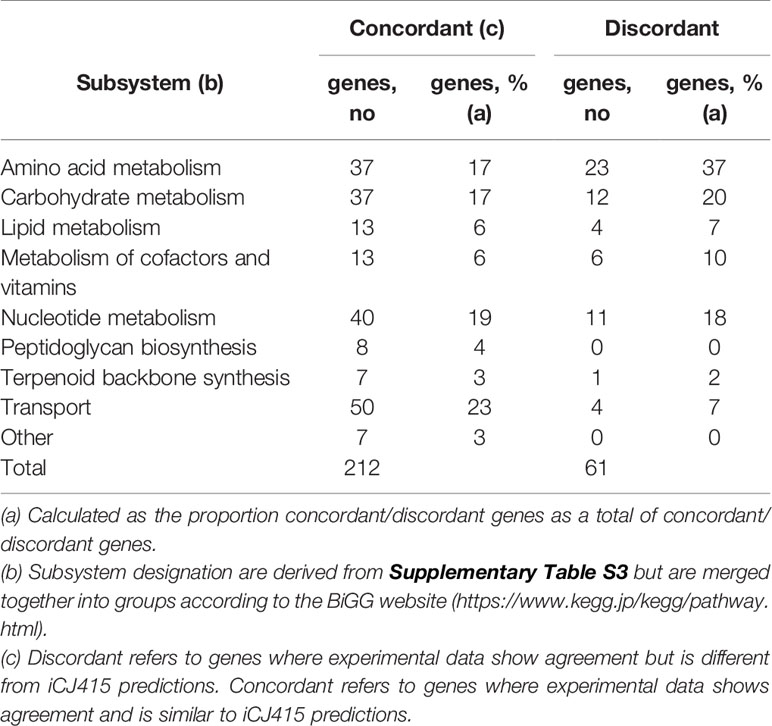
Table 1 Concordant and discordant genes in iCJ415 categorized according to subsystem.
There are 61 genes where there are discordant results. In 36 of those genes, iCJ415 shows essentiality while the essentiality datasets show non-essentiality. An explanation is that not much S. oralis specific data is available, which causes a lack of alternative genes or pathways in the model. This leads to genes being essential in silico, while they are not essential in vitro. This is the case with all but one of the amino acid genes (SK141_RS08355) categorized as discordant.
In the remaining 25 genes where there are discordant results, iCJ415 shows genes to be non-essential, while the essentiality data shows the genes to be essential. This can be explained in part by the use of rich medium in both gene essentiality datasets. Both of these studies use a broth-based, non-defined media, where it is not possible to get the exact composition of the different metabolites. When we simulate gene deletions in rich media we open all exchange reactions in the model, which corresponds to an experimental setup where all metabolites are present in the growth media. But if a metabolite is absent from the experimental growth media, a gene could be deemed essential, however the gene would not be essential if the metabolite was present.
The two gene essentiality studies use either Todd Hewitt or BHI media for the experiments (van Opijnen et al., 2009; Xu et al., 2011). Both of these media contain ingredients such as digests of beef-heart, neopeptones, brain, or gelatin. These ingredients make the exact composition of the media unknown, and they will probably differ between batches and manufacturer. Despite the unspecific media conditions, both Todd Hewitt and BHI have added glucose as a carbon source, which probably make it the primary carbon source available (Atlas and Snyder, 2019). Therefore we tried to do the gene essentiality calculations for all discordant genes from Supplementary Table S4, leaving out all carbon sources except glucose. See Table 2 for discordant genes that are non-essential in iCJ415, but essential in the experimental essentiality data. Furthermore, Table 2 shows differences in essentiality and growth rate when grown in full media, compared with glucose as the sole carbon source. The growth calculations are percent of growth when gene is not knocked out.
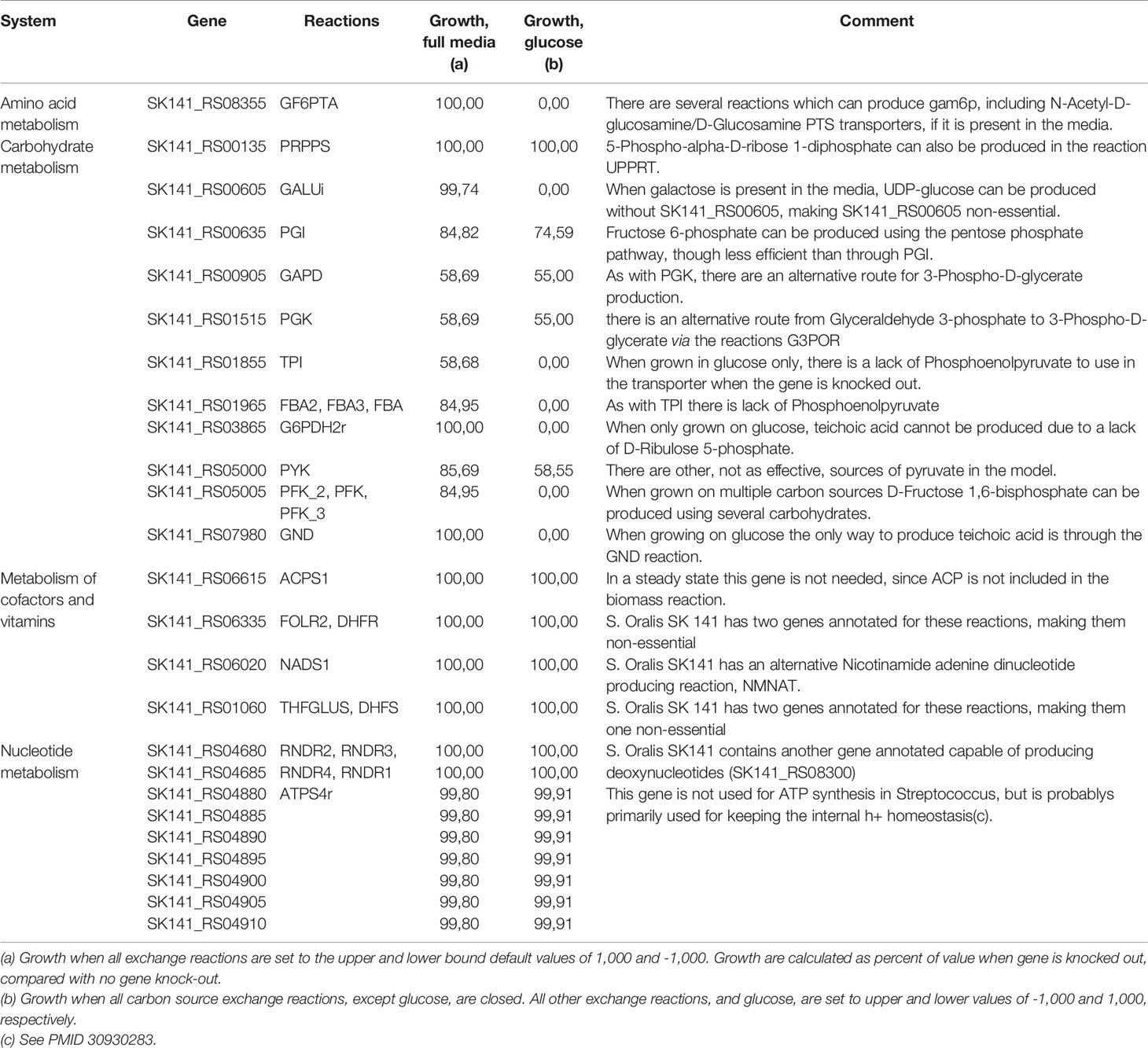
Table 2 Discordant genes that shows non-essentiality in iCJ415 in-silico, while they were shown to be essential in both experimental datasets.
When using glucose as a sole carbon source, seven “discordant” genes become essential; six of those are part of the carbon metabolism. These genes are becoming essential due to the lack of essential metabolite synthesis when only glucose is used as a carbon source, e.g. teichoic acid cannot be produced when the gene SK141_RS03865 is knocked out due to the inability of the model to synthesize ribulose 5-phosphate. When retesting all genes that were “concordant” in the full media, only one of the 169 genes were essential when testing with glucose as the sole carbon source. Furthermore, only 10 gene deletions had a lower growth rate when glucose was the only carbon source, and no deletion gave more than a 20% reduction in growth rate.
When publishing iDS372, the authors did gene essentiality testing in iDS372 and compared with essentiality data in the Online Gene Essentiality Database using a chemically defined media, despite the experimental essentiality data was generated in a complex media (Chen et al., 2012; Dias et al., 2019). We tested the gene essentiality of iDS372 by opening all exchanges (Simulating rich media conditions) and did gene knockout using COBRApy. Thirteen genes, which showed concordance between iCJ415 and the experimental data, did not show concordance in iDS372. Thirty-three genes that showed discordance between iCJ415 and experimental data showed concordance in iDS372. These differences can be explained, by that none of the essentiality datasets is from a S. oralis. Further, S. oralis has one of the smallest genomes in the MGS, having a genome size of 1.9 mio basepairs (Rasmussen et al., 2016). In contrast, both S. pneumoniae and S. sanguinis have larger genome sizes of around 2.1 and 2.4 mio basepairs, respectively, giving a relative larger amount of genes (Tettelin et al., 2001; Rasmussen et al., 2016). This can lead to alternative pathways, or multiple genes catalyzing the same reaction. The latter is the case in at least nine of the genes showing discordance in iCJ415 but concordance in iDS372.
These results indicate that most gene deletions in iCJ415, give the expected result, but there are still discrepancy between iCJ415 and the experimental data. However, much of the difference can be explained by how the essentiality data were obtained and the use of different species.
The essential genes found in iCJ415, can be used to find potential drug targets. Especially when iCJ415 can be applied to a larger set of strains, essential genes found in all strains could be possible drug targets. To further validate iCJ415, species-specific data would be helpful, preferably obtained in a chemically defined medium.
Experimental Verification of Carbon- and Nitrogen Utilization in iCJ415
To investigate S. oralis SK141 growth on different Carbon- and nitrogen sources we used the Biolog microarray system. For model simulations we used a minimal media found in the literature (Homer et al., 1993; Paixão et al., 2015). All metabolites mapped to iCJ415 or found to be positive in the Biolog system were tested for growth in iCJ415 (see Figure 3, Supplementary Table S4). The Biolog results for S. oralis SK141 can be found in Supplementary Data Sheet S3.
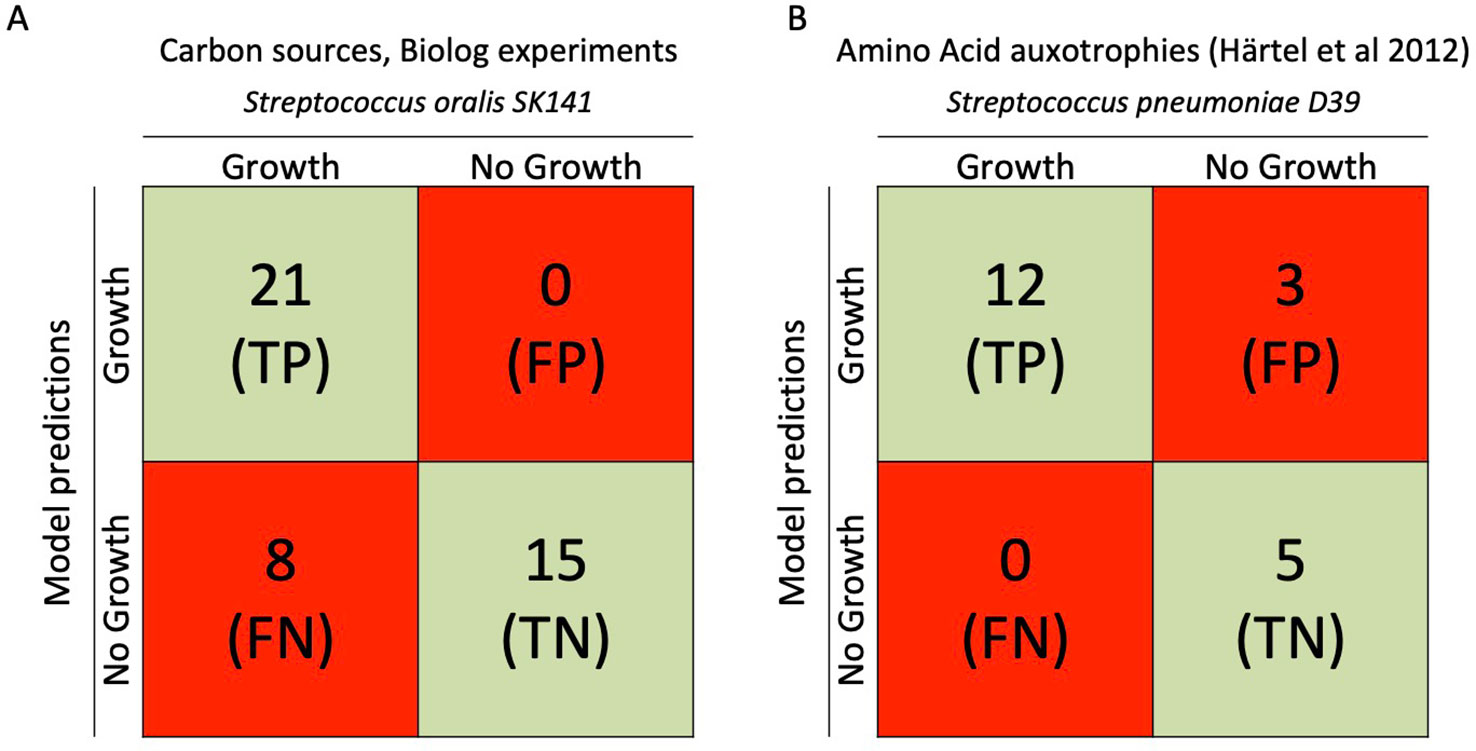
Figure 3 Comparison between iCJ415 and experimental data. (A) Comparison between carbon utilization in iCJ415 and data obtained using S. oralis SK141 and the Biolog phenotypic array. There is an 84% (36/43) concordance between iCJ415 and Biolog phenotypic array using different carbon sources. (B) Amino acid auxotrophy comparison between iCJ415 and experimental data obtained using S. pneumoniae D39. There is an 85% (17/20) concordance between iCJ415 and the experimental data.
The Biolog microarray system tests 190 different carbon sources. Of those 190 carbon sources, 44 where either mapped to iCJ415 or were positive in the Biolog microarray system. Though, dextrin and pectin were positive in the Biolog microarray system, they were not included due to the heterogeneity of the two carbon sources.
Knowledge about the differences in carbon source utilization within the MGS is limited. The published data is mainly about S. pneumoniae (Leonard and Lalk, 2018; Marion et al., 2012). Therefore it is interesting to compare the differences between S. oralis and S. pneumoniae. Though, phylogenetically closely related, S. oralis and S. pneumoniae are part of two different microbiomes in the human body, the oral cavity and upper respiratory airways respectively. Furthermore, their spectrum of disease is very different. S. pneumoniae being the etiologic agent of pneumonia, meningitis and otitis media, while S. oralis being etiologic agent of endocarditis. Of the 43 carbon sources mapped to iCJ415, eight (18%) were not able to promote growth in iCJ415. Four of them had transport reactions in iCJ415 but were still not able to use the metabolites as carbon sources (glycerol, dihydroxyacetone, deoxyribose, and tagatose). The remaining four metabolites were not present in the model and we could not find any information on the transport or metabolism in the genome annotation or in the literature (propionic acid, 5-keto-D-gluconic acid, oxalomalic acid, sorbic acid).
These metabolic associated genes are increasingly considered crucial to the pathogenesis since they determine in which ecological niche an organism can grow, both during asymptomatic carriage and in disease (Shelburne et al., 2008). Therefore, the variety of the carbon sources a strain is capable of utilizing is thought to be important for the pathogenesis, and can be valuable for understanding which pathogenetic traits are important for specific bacteria under certain conditions. When S. oralis and S. pneumoniae resides in two different ecological niches, different nutritional sources are available. While the oral cavity contains multiple different carbon sources, partly degraded by oral enzymes, pharynx has a low content of readily available carbon sources (Shelburne et al., 2008; Philips et al., 2003). When comparing genome annotations for S. pneumoniae R6 and S. oralis SK141 this is reflected by the content of transport systems specific for breakdown products of human tissue. S. pneumoniae R6 contains a transporter for hyaluronate, which is not present in S. oralis SK141, and together with lyases this transporter can make part of the human extracellular matrix available as carbon sources (Marion et al., 2012). S. pneumoniae mutants deficient of these lyases are attenuated in their ability to colonization and infection making them important virulence factors (Manco et al., 2006; King, 2010).
The S. pneumoniae and S. pyogenes GEMs previously published all contains several carbohydrate transporters, but the hyaluronate transporter was not included in any of them. The GEM for S. pneumoniae R6, iDS372, contains transporters for nine carbon sources also present in iCJ415. While iDS371 can grow on six of these carbon sources, iCJ415 can grow on 8. Neither of the models can use glycerol as a carbon despite having a transporter. In contrast to iCJ415, iDS372 cannot grow on either stachyose or raffinose, which is due to lack of a raffinose degrading reaction (Stachyose is degraded to raffinose). Raffinose utilization by S. pneumoniae has been shown to be an independent predictor for disease spectrum (Minhas et al., 2019).
Though, Biolog data showed that S. oralis SK141 was able to use some nitrogen sources which could be mapped to iCJ415 (D-glucosamine, N-acetyl-D-glucosamine, and N-acetyl-D-mannosamine), we were not able to distinguish single nitrogen capabilities in iCJ415 with the used in silico media, revealing that there are still knowledge gaps in iCJ415.
iCJ415 reflects that S. oralis is able to grow on multiple carbon sources, but the transport and metabolism of some of these metabolites are unknown or only poorly understood.
Verification of Amino Acid Auxotrophies in iCJ415 Using Experimental S. pneumoniae Data
To evaluate iCJ415 we compared amino acid auxotrophies in iCJ415 with experimental data. By omitting each one of the 20 amino acids in a synthetic chemically defined medium, Härtel et al., found the amino acid auxotrophies for S. pneumoniae D39. We opened the exchange reactions for all the amino acids in iCJ415 and all the metabolites present in the media used by Härtel et al. To test amino acid auxotrophy we consecutively closed each of the amino acid exchange reactions in iCJ415 (Figure 3, Supplementary Table S4). Three amino acids had discrepant results between experimental data and iCJ415.
Isoleucine, valine, and leucine are not auxotrophic in iCJ415 but show auxotrophy in the experimental omission data. In iCJ415 all three amino acids can be synthesized from pyruvate and threonine. All the reactions in these pathways are added based solely on the genome annotations. According to Härtel et al., the genes for the synthesis of the three amino acids are also present in S. pneumoniae D39, but are apparently not active under the conditions tested. Whether the same applies for S. oralis SK141 is not known.
Like the ability to grow on different carbon sources, amino acid auxotrophies can be an important pathogenetic trait. The ability to grow in the absence of a specific amino acid has been shown to be important for which metabolic niche a bacterium can cause disease. This can be important when a commensal bacteria, like S. oralis, causes disease. If the bacterium is auxotrophic for an amino acid not present in the environment, it cannot grow, and hence not cause disease. To exemplify this, it has been shown that the human commensal Haemophilus influenza, is more likely to have a histidin synthesizing machinery when isolated from the middle ear in otitis media patients, than when isolated from the throat in healthy humans (Juliao et al., 2007).
Validating Growth Rate of iCJ415 Using S. pneumoniae Data
For validating Biomass yield and byproduct formation of iCJ415 we used previously published experimental growth data from S. pneumoniae D39 (Paixão et al., 2015). The catabolite control protein A (CcpA) is used in many bacteria, including streptococci, to organize the carbohydrate catabolism for obtaining maximal growth. These data have previously been used to validate a Streptococcus pneumoniae R6 GEM, iDS372 (Dias et al., 2019). To simulate the impact of the carbon catabolite repressor on the genes in the model, fluxes in reactions catalyzed by repressed genes were set to a maximum flux defined by the flux in the unconstrained media, see materials and methods.
Table 3 shows the results from iCJ415, the results from iDS372 and experimentally obtained data from S. pneumoniae D39. The experimental data was obtained when grown in glucose, galactose, N-acetyl-D-glucosamine, and mannose. When compared with iDS372, iCJ415 has a lower growth rate than iDS372 except when N-acetyl-D-glucosamine is used as a carbon source. Especially when using glucose as a carbon source, the growth rate is only 0.44 h-1 in iCJ415, with growth rates in iDS372 and in the experimental of 0.85 and 0.82 h-1 respectively. When grown on glucose the 0% activity of pfl causes severe growth retardation in iCJ415, but only a small increase to 1% pfl activity causes an increase in growth rate to 0.76 h-1. The reason for decrease in growth rate is due to lack of formate production when the pfl gene is knocked out. Formate is used for the synthesis of folates in the reaction “FTHLFI.” This reveals differences in the synthesis of folates, which in part can be explained by a folate transporter in iDS372. A folate transporter has earlier been identified in Streptococcus mutans, but we could not find any evidence for the folate transporter being present in the annotation of S. oralis or S. pneumoniae and it has been postulated earlier that such a transporter is not present in S. pneumoniae (Eudes et al., 2008; Burghout et al., 2013). Due to the lack of evidence, we choose not to include a formate transporter in the model.
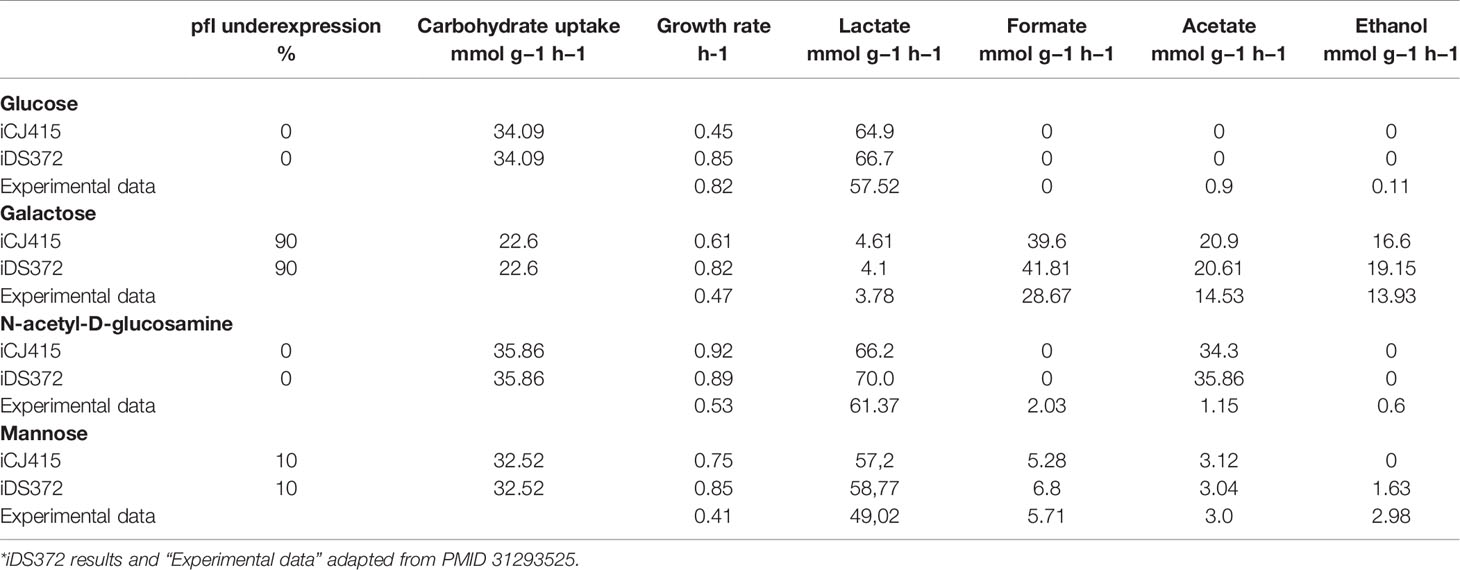
Table 3 Growth rate and byproduct formation in iCJ415 and an S. Pneumoniae GEM (iDS372). The simulations were done with additional constraints trying to simulate the action of the Carbon Catabolite repressor.
Interestingly, iCJ415, like iDS372, has a much higher rate of acetate production, than the experimental data, when N-acetyl-D-glucosamine is used as a carbon source. The high acetate production I iCJ415 is explained by a production of acetate in the breakdown of N-acetyl-D-glucosamine 6-phosphate. This anomaly reveals a knowledge gap in the metabolism of N-acetyl-glucosamine in S. oralis.
Memote Testing
iCJ415 was tested using the Memote web application with all exchanges open. iCJ415 scores >99% on all consistency scores except “unbounded flux in default medium” where it scores 95% (Lieven et al., 2018).
Conclusions
The aim of this study was to construct a GEM of a S. oralis, the first GEM of a bacterium that is a major constituent of the human oral microbiome and an IE causing bacterium. We used an annotated genome together with strain, species or genus specific data obtained from the literature. To get information in carbon source utilization in S. oralis SK141 we used the Biolog phenotypic microarray.
When comparing gene essentiality between iCJ415 and published experimental data, we found concordance in 71–76% of the genes tested. When testing carbon sources and amino acid auxotrophies we found concordance with experimental data in 82% and 85% of metabolites tested, respectively. Especially the carbon source utilization data obtained in this study are valuable. It is, beside the genomic data, the only strain specific data used. Furthermore, carbon source utilization capacities are increasingly being considered as possible virulence factors in bacteria (Leonard and Lalk, 2018; McAllister et al., 2012; Juliao et al., 2007). We found that S. oralis are able to grow on at least 28 carbon sources, including commonly known carbohydrates as glucose and galactose, but also rarely encountered ones like lyxose. iCJ415 were able to grow on 21 of the 28 carbon sources tested. In the remaining seven carbon sources we did not have enough information to simulate growth on these carbon sources.
iCJ415 should be used as a starting point for exploring the complicated metabolic interactions between different strains of the same species and between different species in the human mouth. Further it can be used to compare metabolic capabilities between non-pathogenic and known pathogenic strains, finding metabolic traits important for virulence.
iCJ415 was tested using memote and scored >99% on all consistency scores except “unbounded flux in default medium” where it scores 95%.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
BP, XN, JC, and CJ conceived the project. XF, CN, JM, and CJ performed the metabolic network reconstruction. CN and CJ performed the model simulations. CJ prepared the draft manuscript. All authors have read, commented and approved the final manuscript.
Funding
This work was supported by the Region Zealand Foundation for Health Research, The Knud Højgård Foundation (Grant no: 18-02-0767), and the Ovesen Foundation. None of the funders had any influence on the study or the conclusions.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank JC Lachance for help with BOFdat. The Biolog experiments were done by Ado Van Assche at the Laboratory for Process Microbial Ecology and Bioinspirational Management, Sint-Katelijne-Waver, Belgium.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00116/full#supplementary-material
Table S1 | Biomass calculations.
Table S2 | in-silico media contents.
Table S3 | Reactions and metabolites in iCJ415.
Table S4 | Gene essentiality, amino acid auxotrophies and carbon source testing of iCJ415.
Data Sheet S1 | iCJ415 in JSON format.
Data Sheet S2 | iCJ415 in SBML format
Data Sheet S3 | Biolog results for S. oralis SK141.
References
Abranches, J., Zeng, L., Kajfasz, J. K., Palmer, S. R., Chakraborty, B., Wen, Z. T., et al. (2018). Biology of oral streptococci. Microbiol. Spectr. 6. doi: 10.1128/microbiolspec.GPP3-0042-2018
Aprianto, R., Slager, J., Holsappel, S., Veening, J.-W. (2018). High-Resolution Analysis of the Pneumococcal Transcriptome under a Wide Range of Infection-Relevant Conditions. Nucleic Acids Res. 46 (19), 9990–10006.
Atlas, R., Snyder, J. (2019). In Manual of Clinical Microbiology, 2 Volume Set, edited by Karen C. Carroll, Michael A. Pfaller, Marie Louise Landry, Alexander J. McAdam, Robin Patel, Sandra S. Richter, and David W. Warnock. (Wiley), 316–349.
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi: 10.1186/1471-2164-9-75
Boratyn, G. M., Camacho, C., Cooper, P. S., Coulouris, G., Fong, A., Ma, N., et al. (2013). BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–W33. doi: 10.1093/nar/gkt282
Bosi, E., Monk, J. M., Aziz, R. K., Fondi, M., Nizet, V., Palsson, B. Ø. (2016). Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 113, E3801–E3809. doi: 10.1073/pnas.1523199113
Burghout, P., Zomer, A., van der Gaast-de Jongh, C. E., Janssen-Megens, E. M., Françoijs, K.-J., Stunnenberg, H. G., et al. (2013). Streptococcus Pneumoniae Folate biosynthesis responds to environmental CO2 levels. J. Bacteriol. 195 (7), 1573–1582.
Chen, W.-H., Minguez, P., Lercher, M. J., Bork, P. (2012). OGEE: An Online Gene Essentiality Database. Nucleic Acids Res. 40, D901–D906.
Dias, O., Saraiva, J., Faria, C., Ramirez, M., Pinto, F., Rocha, I. (2019). iDS372, a phenotypically reconciled model for the metabolism of streptococcus Pneumoniae Strain R6. Front. Microbiol. 10, 1283.
Denapaite, D., Reinhold, B., Hakenbeck, R., Vollmer, W. (2012). Biosynthesis of Teichoic Acids in Streptococcus Pneumoniae and Closely Related Species: Lessons from Genomes. Microbial Drug Resistance. 18 (3), 344–358.
Ebrahim, A., Lerman, J. A., Palsson, B. O., Hyduke, D. R. (2013). COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 7, 74. doi: 10.1186/1752-0509-7-74
Eudes, A., Erkens, G. B., Slotboom, D.J., Rodionov, D. A., Naponelli, V., Hanson, A. D. (2008). Identification of genes encoding the Folate- and Thiamine-binding membrane proteins in Firmicutes.” J. Bacteriol. 190 (22), 7590–7594.
Ganter, M., Bernard, T., Moretti, S., Stelling, J., Pagni, M. (2013). MetaNetX.org: a website and repository for accessing, analysing and manipulating metabolic networks. Bioinformatics 29, 815–816. doi: 10.1093/bioinformatics/btt036
Härtel, T., Eylert, E., Schulz, C., Petruschka, L., Gierok, P., Grubmüller, S., et al. (2012). Characterization of central carbon metabolism of Streptococcus pneumoniae by isotopologue profiling. J. Biol. Chem. 287, 4260–4274. doi: 10.1074/jbc.M111.304311
Henry, C. S., DeJongh, M., Best, A. A., Frybarger, P. M., Linsay, B., Stevens, R. L. (2010). High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982. doi: 10.1038/nbt.1672
Homer, K. A., Patel, R., Beighton, D. (1993). Effects of N-acetylglucosamine on carbohydrate fermentation by Streptococcus mutans NCTC 10449 and Streptococcus sobrinus SL-1. Infect. Immun. 61, 295–302. doi: 10.1128/IAI.61.1.295-302.1993
Jensen, A., Kilian, M. (2012). Delineation of Streptococcus dysgalactiae, its subspecies, and its clinical and phylogenetic relationship to Streptococcus pyogenes. J. Clin. Microbiol. 50, 113–126. doi: 10.1128/JCM.05900-11
Juliao, P. C., Marrs, C. F., Xie, J., Gilsdorf, J. R. (2007). Histidine auxotrophy in commensal and disease-causing nontypeable Haemophilus Influenzae.” J. Bacteriol. 189 (14), –5001.
Kilian, M., Riley, D. R., Jensen, A., Brüggemann, H., Tettelin, H. (2014). Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. MBio 5, e01490–e01414. doi: 10.1128/mBio.01490-14
King, Z. A., Dräger, A., Ebrahim, A., Sonnenschein, N., Lewis, N. E., Palsson, B. O. (2015). Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 11, e1004321. doi: 10.1371/journal.pcbi.1004321
King, Z. A., Lu, J., Dräger, A., Miller, P., Federowicz, S., Lerman, J. A., et al. (2016). BiGG Models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 44, D515–D522. doi: 10.1093/nar/gkv1049
King, S. J. (2010). Pneumococcal modification of host sugars: a major contributor to colonization of the human airway? Mol. Microbiol. 25, 15–24. doi: 10.1111/j.2041-1014.2009.00564.x
Kleckner, N., Roth, J., Bonstein, D. (1977). Genetic Engineering in Vivo Using Translocatable Drug-Resistance Elements. New Methods in Bacterial Genetics. J. Mol. Biol. 116 (1), 125–159.
Kreth, J., Merritt, J., Qi, F. (2009). Bacterial and host interactions of oral streptococci. DNA Cell Biol. 28, 397–403. doi: 10.1089/dna.2009.0868
Lachance, J.-C., Lloyd, C. J., Monk, J. M., Yang, L., Sastry, A. V., Seif, Y., et al. (2019). BOFdat: Generating biomass objective functions for genome-scale metabolic models from experimental data. PLoS Comput. Biol. 15, e1006971. doi: 10.1371/journal.pcbi.1006971
Leonard, A., Lalk, M. (2018). Infection and metabolism - Streptococcus Pneumoniae metabolism facing the host environment. Cytokine 112, 75–86.
Lieven, C., Beber, M. E., Olivier, B. G., Bergmann, F. T., Ataman, M., Babei, P., et al. (2016). Memote: A Community Driven Effort towards a Standardized Genome-Scale Metabolic Model Test Suite. doi: 10.1101/350991
Levering, J., Fiedler, T., Sieg, A., van Grinsven, K. W. A., Hering, S., Veith, N., et al. (2016). Genome-scale reconstruction of the Streptococcus pyogenes M49 metabolic network reveals growth requirements and indicates potential drug targets. J. Biotechnol. 232, 25–37. doi: 10.1016/j.jbiotec.2016.01.035
Manco, S., Hernon, F.,, Yesilkaya, H., Paton, J. C., Andrew, P. W., Kadioglu, A. (2006). Pneumococcal neuraminidases A and B both have essential roles during infection of the respiratory tract and sepsis. Infect. Immunity 74 (7), 4014–4020.
Marion, C., Stewart, J. M., Tazi, M. F., Burnaugh, A. M., Linke, C. M., Woodiga, S. A., et al. (2012). Streptococcus Pneumoniae can utilize multiple sources of hyaluronic acid for growth. Infect. Immunity 80 (4), 1390–1398.
McAllister, L. J., Ogunniyi, A. D., Stroeher, U. H., Paton, J. C. (2012). Contribution of a genomic accessory region encoding a putative Cellobiose Phosphotransferase system to Virulence of Streptococcus Pneumoniae. PloS One 7 (2) .
McCloskey, D., Palsson, B. Ø., Feist, A. M. (2013). Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol. Syst. Biol. 9, 661. doi: 10.1038/msb.2013.18
Minhas, V., Harvey, R. M., McAllister, L. J., Seemann, T., Syme, A. E., Baines, S. L., et al. (2019). Capacity to utilize raffinose dictates Pneumococcal disease phenotype. mBio 10 (1). doi: mBio.02596-18/mBio.02596-18
Monk, J. M., Charusanti, P., Aziz, R. K., Lerman, J. A., Premyodhin, N., Orth, J. D., et al. (2013). Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc. Natl. Acad. Sci. U. S. A. 110, 20338–20343. doi: 10.1073/pnas.1307797110
Monk, J. M., Lloyd, C. J., Brunk, E., Mih, N., Sastry, A., King, Z., et al. (2017). iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 35, 904–908. doi: 10.1038/nbt.3956
Nascimento, M. M., Gordan, V. V., Garvan, C. W., Browngardt, C. M., Burne, R. A. (2009). Correlations of oral bacterial arginine and urea catabolism with caries experience. Microbiol. Immunol. 24, 89–95. doi: 10.1111/j.1399-302X.2008.00477.x
Norsigian, C. J., Kavvas, E., Seif, Y., Palsson, B. O., Monk, J. M. (2018). iCN718, an Updated and Improved Genome-Scale Metabolic Network Reconstruction of AYE. Front. Genet. 9, 121. doi: 10.3389/fgene.2018.00121
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., Kanehisa, M. (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34. doi: 10.1093/nar/27.1.29
Oliveira, A. P., Nielsen, J., Förster, J. (2005). Modeling Lactococcus lactis using a genome-scale flux model. BMC Microbiol. 5, 39. doi: 10.1186/1471-2180-5-39
Ong, W. K., Vu, T. T., Lovendahl, K. N., Llull, J. M., Serres, M. H., Romine, M. F., et al. (2014). Comparisons of Shewanella strains based on genome annotations, modeling, and experiments. BMC Syst. Biol. 8, 31. doi: 10.1186/1752-0509-8-31
Paixão, L., Oliveira, J., Veríssimo, A., Vinga, S., Lourenço, E. C., Ventura, M. R., et al. (2015). Host glycan sugar-specific pathways in Streptococcus pneumoniae: galactose as a key sugar in colonisation and infection [corrected]. PLoS One 10, e0121042. doi: 10.1371/journal.pone.0121042
Pastink, M. I., Teusink, B., Hols, P., Visser, S., de Vos, W. M., Hugenholtz, J. (2009). Genome-scale model of Streptococcus thermophilus LMG18311 for metabolic comparison of lactic acid bacteria. Appl. Environ. Microbiol. 75, 3627–3633. doi: mBio.02596-18/AEM.00138-09
Philips, B. J., Meguer, J.-X., Redman, J., Baker, E. H.(2003). Factors determining the appearance of glucose in upper and lower respiratory tract secretions. Intensive Care Med.29 (12), 2204–2010
Rasmussen, L. H., Dargis, R., Højholt, K., Christensen, J. J., Skovgaard, O., Justesen, U. S., et al. (2016). Whole genome sequencing as a tool for phylogenetic analysis of clinical strains of Mitis group streptococci. Eur. J. Clin. Microbiol. Infect. Dis. 35, 1615–1625. doi: 10.1007/s10096-016-2700-2
Rowe, E., Palsson, B. O., King, Z. A. (2018). Escher-FBA: a Web Application for Interactive Flux Balance Analysis. BMC Syst. Biol. 12 (1), 84.
Shah, P., Nanduri, B., Swiatlo, E., Ma, Y., Pendarvis, K. (2011). Polyamine Biosynthesis and Transport Mechanisms Are Crucial for Fitness and Pathogenesis of Streptococcus Pneumoniae. Microbiol. 157 (2), 504–515.
Shelburne, S. A., Davenport, M. T., Keith, D. B., Musser, J. M. (2008). The role of complex carbohydrate catabolism in the pathogenesis of invasive streptococci. Trends Microbiol. 16 (7), 318–25 .
Spellerberg, B., Brandt, C. (2008). “Streptococci,” in Manual of Clinical Microbiology, vol. 412–430. Eds. Patrick, M., Baron, E. J., James, H., J., Marie Louise, L., Michael, A. ,. P. (Washington DC: ASM Press).
Sun, X., Jia, H.-L., Xiao, C.-L., Yin, X.-F., Yang, X.-.Y, Lu, J., et al. (2011). Bacterial Proteome of Streptococcus Pneumoniae through Multidimensional Separations Coupled with LC-MS/MS. Omics. 15 (7–8), 447–482.
Tettelin, H., Nelson, K. E., Paulsen, I. T., Eisen, J. A., Read, T. D., Peterson, S., et al (2001). Complete Genome Sequence of a Virulent Isolate of Streptococcus Pneumoniae. Sci. 293 (5529), 498–506.
Teusink, B., Wiersma, A., Molenaar, D., Francke, C., de Vos, W. M., Siezen, R. J., et al. (2006). Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. J. Biol. Chem. 281, 40041–40048. doi: 10.1074/jbc.M606263200
Thiele, I., Palsson, B. Ø. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi: 10.1038/nprot.2009.203
Vaas, L. A. I., Sikorski, J., Hofner, B., Fiebig, A., Buddruhs, N., Klenk, H.-P., et al. (2013). opm: an R package for analysing OmniLog(R) phenotype microarray data. Bioinformatics 29, 1823–1824. doi: 10.1093/bioinformatics/btt291
van Opijnen, T., Bodi, K. L., Camilli, A. (2009). Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772. doi: 10.1038/nmeth.1377
Wu, C., Huang, J., Zhou, R. (2017). Genomics of lactic acid bacteria: Current status and potential applications. Crit. Rev. Microbiol. 43, 393–404. doi: 10.1080/1040841X.2016.1179623
Xavier, J. C., Patil, K. R., Rocha, I. (2017). Integration of Biomass Formulations of Genome-Scale Metabolic Models with Experimental Data Reveals Universally Essential Cofactors in Prokaryotes. Metabolic Engineering 39, 200–208.
Keywords: Streptococcus oralis, mitis group of streptococci, genome-scale reconstruction, constraint-based modeling, Biolog phenotypic profiling
Citation: Jensen CS, Norsigian CJ, Fang X, Nielsen XC, Christensen JJ, Palsson BO and Monk JM (2020) Reconstruction and Validation of a Genome-Scale Metabolic Model of Streptococcus oralis (iCJ415), a Human Commensal and Opportunistic Pathogen. Front. Genet. 11:116. doi: 10.3389/fgene.2020.00116
Received: 16 August 2019; Accepted: 31 January 2020;
Published: 03 March 2020.
Edited by:
Alessio Mengoni, University of Florence, ItalyReviewed by:
Luana Presta, University of Luxembourg, LuxembourgSabina Zoledowska, Institute of Biotechnology and Molecular Medicine (IBMM), Poland
Copyright © 2020 Jensen, Norsigian, Fang, Nielsen, Christensen, Palsson and Monk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian S. Jensen, Q1NKQGRhZGxuZXQuZGs=