 Fei Su1†
Fei Su1† Rui Tian2†Yi Yang3†Hexin Li1Gaoyuan Sun1Ying Li4
Rui Tian2†Yi Yang3†Hexin Li1Gaoyuan Sun1Ying Li4 Bingqing Han4Xiaomao Xu4Xue Chen5Gang Zhao6
Bingqing Han4Xiaomao Xu4Xue Chen5Gang Zhao6 Hongyuan Cui6*Hongtao Xu5*
Hongyuan Cui6*Hongtao Xu5*- 1Clinical Biobank, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Cardiovascular Disease, Beijing Anzhen Hospital, Capital Medical University, Beijing, China
- 3Department of Otorhinolaryngology, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
- 4The Key Laboratory of Geriatrics, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
- 5Department of Laboratory Medicine, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
- 6Department of General Surgery, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China
Staphylococcus epidermidis is one of the most commonly isolated species from human skin and the second leading cause of bloodstream infections. Here, we performed a large-scale comparative study without any pre-assigned reference to identify genomic determinants associated with the diversity and adaptation of S. epidermidis strains to various environments. Pan-genome of S. epidermidis was open with 435 core proteins and had a pan-genome size of 8,034 proteins. Genome-wide phylogenetic tree showed high heterogeneity and suggested that routine whole genome sequencing was a powerful tool for analyzing the complex evolution of S. epidermidis and for investigating the infection sources. Comparative genome analyses demonstrated a range of antimicrobial resistance (AMR) genes, especially those within mobile genetic elements. The complicated host-bacterium and bacterium-bacterium relationships help S. epidermidis to play a vital role in balancing the epithelial microflora. The highly variable and dynamic nature of the S. epidermidis genome may contribute to its success in adapting to broad habitats. Genes related to biofilm formation and cell toxicity were significantly enriched in the blood and skin, demonstrating their potentials in identifying risk genotypes. This study gave a general landscape of S. epidermidis pan-genome and provided valuable insights into mechanisms for genome evolution and lifestyle adaptation of this ecologically flexible species.
Introduction
The coagulase-negative Staphylococcus epidermidis is a common human skin commensal bacterium that can be cultured from the body surface of almost all healthy individuals. It also plays a central role in the skin microbiome (Otto, 2009; Oh et al., 2016; Zhou et al., 2020), especially in keeping the ecological balance of human skin microflora (Schommer and Gallo, 2013). S. epidermidis can produce various bacteriocins, which directly kill other microorganisms and may enhance survival of the producer strains in a competitive fashion (Jetten and Vogels, 1972; Jack et al., 1995). Especially, serine protease Esp, secreted by S. epidermidis, can inhibit the biofilm formation of Staphylococcus aureus and destroy the pre-existing S. aureus biofilms (Iwase et al., 2010).
However, S. epidermidis is the second most common cause of nosocomial infections, which in most cases are antibiotic-resistant (Otto, 2009; Joo and Otto, 2015). Antibiotic resistance remarkably complicates the treatment and increases the medical expenses (Foster, 2017; Lee et al., 2018). The large gene pool of antibiotic resistance in S. epidermidis is shared with many other pathogenic species (e.g., S. aureus) through rare horizontal gene transfer events (Diep et al., 2006). Mobile genetic elements such as multidrug-resistant conjugative plasmids, arginine catabolic mobile element (ACME; Diep et al., 2006), and staphylococcal chromosome cassette mec (SCCmec) elements (Mcmanus et al., 2015) conferring β-lactam resistance are transferred frequently, enabling rapid evolution and adaptation of S. epidermidis against antibiotic selection pressure (Miragaia et al., 2007; Bloemendaal et al., 2010). When the protective layer of the human epithelium is breached and the host immunity fails, staphylococcal infections can become extremely dangerous and even fatal (Yao et al., 2005). S. epidermidis is particularly associated with the increased use of indwelling medical devices such as artificial heart valves, prosthetic joints, and vascular catheters, which provide a substrate for biofilm formation. On the other hand, during the long-time coevolution with various pathogens, human beings have developed versatile immunity system with antimicrobial peptides (AMPs) as the first line of innate immune defense on the human skin (Zhou et al., 2020); meanwhile, S. epidermidis also owns multiple mechanisms such as surface charge alteration, extracellular proteases, exopolymers, and efflux pump proteins to fight against AMPs (Joo and Otto, 2015). The complex host-bacterium and bacterium-bacterium relationships make it necessary to investigate the genetic diversity, genome evolution, and lifestyle adaptation of S. epidermidis.
Much attention has been focused on understanding the evolution and spread of S. epidermidis by different methods (Miragaia et al., 2007; Meric et al., 2015, 2018; Zhou et al., 2020). As high-throughput sequencing has yielded a large amount of genomic data on S. epidermidis, it is essential to perform more comprehensive comparative and evolutionary studies on ecologically diverse strains of S. epidermidis. Here, we compared the genomic features of S. epidermidis isolates of clinical and non-clinical relevance by using a pan-genome analysis of 198 publicly available S. epidermidis strains at the GenBank database of National Center for Biotechnology Information (NCBI) with isolation or clinical information when the analysis was performed. We assembled the consensus “pan-chromosome” without any pre-assigned genome reference and identified both core and variable regions within the chromosome. Second, we utilized a comparative genomics approach on 198 genomes to analyze the diversity of antibiotic resistance of S. epidermidis. Our results revealed that S. epidermidis isolates encoded a vast collection of genetic determinants and mechanisms to confer antibiotic resistance, AMPs resistance, and survival adaptations. These analyses were expected to provide insight into the coevolution of S. epidermidis as a nosocomial pathogen and directly aid the future efforts for large-scale epidemiological studies of this continuously evolving multi-drug resistant organism.
Materials and Methods
Strains
A total of 198 S. epidermidis isolates were selected to represent known diversity within geographical position, isolation source, and host tissue sampled when this analysis was performed, including reference genome of strain RP62A (Gill et al., 2005). All the available genome sequence of S. epidermidis strains and related annotation data were downloaded through the GenBank database (Benson et al., 2013) of NCBI (see Supplementary Table S1 in the Supplementary Material). Totally 121 strains were isolated from different human tissues and the others were from environments.
SCCmec and ACME Typing
An SCCmec sequence cassette database was prepared with the following accession numbers downloaded from NCBI: AB033763.2 (Type I), AB433542.1 (Type I.2), D86934.2 (Type II), AB261975.1 (Type II.4), AJ810123 (Type II-B), AB127982.1 (Type II-B), AM983545.1 (Type II-D), HE858191.1 (Type II-E), AB037671.1 (Type III), HM030721.1 (Type IV), HM030720.1 (Type IV), AM292304.1 (ZH47 mobile elements), AB425824.1 (Type IV), EU437549.2 (Type IV-A), AB063172.2 (Type IV-A), AB063173 (Type IV-B), AY271717.1 (Type IV-C), AB096217 (Type IV-C), AB245470.1 (Type IV-C), AB097677.1 (Type IV-D), AJ810121.1 (Type IV-E), DQ106887.1 (Type IV-G), AB633329.1 (Type IV-I), AB425823.1 Type IV), AB121219.1 (Type V), AB478780.1 (Type V), AB512767.1 (Type V), AF411935.3 (Type VI), AB462393.1 (Type VII), AB373032.1 (Type V-C1), FJ670542.1 (Type VIII), FJ390057.1 (Type VIII), AB505628.1 (Type IX), AB505630.1 (Type X), and FR821779.1 (Type XI; Ugolotti et al., 2016).
The ACME-arcA and ACME-opp3AB genes were used as markers of the ACME-arc cluster and the ACME-opp3 cluster, respectively. ACME was classified as type I (contains the ACME-arcA and ACME-opp3AB gene clusters), type II (carries only the ACME-arcA locus), and type III (carries only the ACME-opp3AB locus; Barbier et al., 2011). ACME-arcA and ACME-opp3AB identified in this study were compared with the reference sequences of ACME-arcA (USA300_FPR3757) and ACME-opp3AB (USA300_FPR3757).
kSNP S. epidermidis Trees
Because microbial genomes are subject to massive gene gains and losses, insertion, deletions and rearrangements, alignment of whole microbial genome sequences has proven to be computationally intensive. Here, we used kSNP (version: 3.01), a validated method to build the phylogenetic tree without alignment (Gardner et al., 2015) by using a k-mer length 19 nucleotides and based on a requirement that at least 80% of the genomes have a nucleotide at a given Single-nucleotide polymorphisms (SNPs) position in order for the SNP to be considered to be a core and included in tree building. A total of 1,832 core SNP positions were identified. jModelTest2 was used to carry out statistical selection of best-fitmodels of nucleotide substitution by implementing different model selection strategies (Ehsan Vafadarnejad et al., 2020) and maximum-likelihood tree was built with RAxML (Stamatakis, 2014) with generalized time reversible (GTR) model. Branch support values were inferred from a rapid bootstrap method applied with 100 replications. Visualization and annotation of the phylogenetic tree was performed by ggtree (Yu et al., 2018).
Pan-Genome Analysis
Cluster of orthologous proteins were generated with version 3.24 of PanOCT2) as previously described (Fouts et al., 2012). Briefly, PanOCT dealt with recently diverging paralogs by using neighborhood gene information. All the parameters were set to default values except for the length ratio to discard shorter protein fragments when a protein was split due to a frameshift or other mechanisms was set to 1.33 as recommended by the authors. Orthologous clusters were stringently defined as all sequences in a cluster having shared sequence identity ≥ 70% and coverage ≥ 75%. Plots and calculations of pan-genome sizes, new genes discovered, and pan-genome status were also determined as described previously (Tettelin et al., 2005). Core genes were defined as the genes present in all of the S. epidermidis genomes and accessory genes as genes present in at least one isolate.
Characterization of Strains
In silico multilocus sequence typing of 198 strains was performed with the MLST 1.8 online server (Larsen et al., 2012). The antimicrobial resistance (AMR) genes in the sequenced isolates were identified by BLASTp (Altschul et al., 1990) searching against the databases of ARDB (Liu and Pop, 2009), CARD(Alcock et al., 2020), and ResFinder (Zankari et al., 2012), with 10–5 as expect value threshold. Genes conferring virulence factors were identified using BLASTp with VFDB (Chen et al., 2012). Given that many virulence factors for S. epidermidis that are not contained in the VFDB, we used the orthologous proteins and virulence factors from RP62a (Assembly: GCF_000011925.1; Gill et al., 2005) and ATCC1228 (Assembly: GCA_000007645.1; Zhang et al., 2003) to make up the missing information.
Functional Analysis
All genes were BLASTed against the database of KOBAS 2.03 (Xie et al., 2011). The cutoffs were BLASTp E-value < 10–5 and BLAST subject coverage > 70%. We used the genes from the same genome as the default background distribution and considered the only pathways for which there were at least two genes mapped. For the enrichment analysis, Fisher’s exact test was performed, and Bonferroni correction was used to reduce the high overall Type-I error with p.adjust from R package.
Statistical Analyses
The differences in the prevalence of AMR genes and phenotypes among isolates were analyzed by using two-tailed Fisher’s exact test, and Bonferroni correction was also performed as mentioned above. All the statistical analyses were carried out using R package (version: 3.3). A P-value of < 0.05 was regarded as statistically significant.
Results
Core Pan-Genome of S. epidermidis
Despite the intensive effort to characterize S. epidermidis and the sizable number of whole genome comparisons in literature (Conlan et al., 2012; Meric et al., 2018), more and more genome data is rapid accumulated and could easily obtained from public database, such as NCBI. Based on PanOCT, a total of 8,034 orthologous protein clusters were identified from a collection of all S. epidiermidis genomes publicly available at the time of the analysis (Supplementary Table S1). PanOCT only includes non-paralogs in clusters and uses conserved gene neighborhood to separate duplicated genes. This means that insertion sequence elements that are in novel contexts will often form singleton clusters even though they are identical in sequence to other IS elements within or between genomes analyzed. When the “core” pan-genome was defined to be present at all 198 genomes analyzed, there were 435 (5.4%) core protein clusters and 2,915 (36.3 %) novel clusters (groups with a single member from a single genome; Figure 1A). To predict the theoretical maximum pan-genome size (i.e., the total number of genes, including core, unique, and accessory genes), a pan-genome model was implemented using medians and an exponential decay function (Figure 1B). The maximum pan-genome size was estimated to be 12,554 ± 65 genes. To determine whether the S. epidiermidis pan-genome had an unlimited large gene repertoire (open) or seemed to be limited by a maximum number of genes in their gene pool (closed), the number of new genes identified (i.e., unique or strain-specific genes) for each genome added was determined and fit to a power law function (n = κ N–α) as described previously (Tettelin et al., 2005). According to the result, we found the pan-genome of S. epidiermidis appeared to be open (α = 0.226 ± 0.002; Figure 1B). For each genome added, the number of new genes was extrapolated by calculating tg(θ), which was determined to be 7.7 ± 0.4 (Figure 1C).
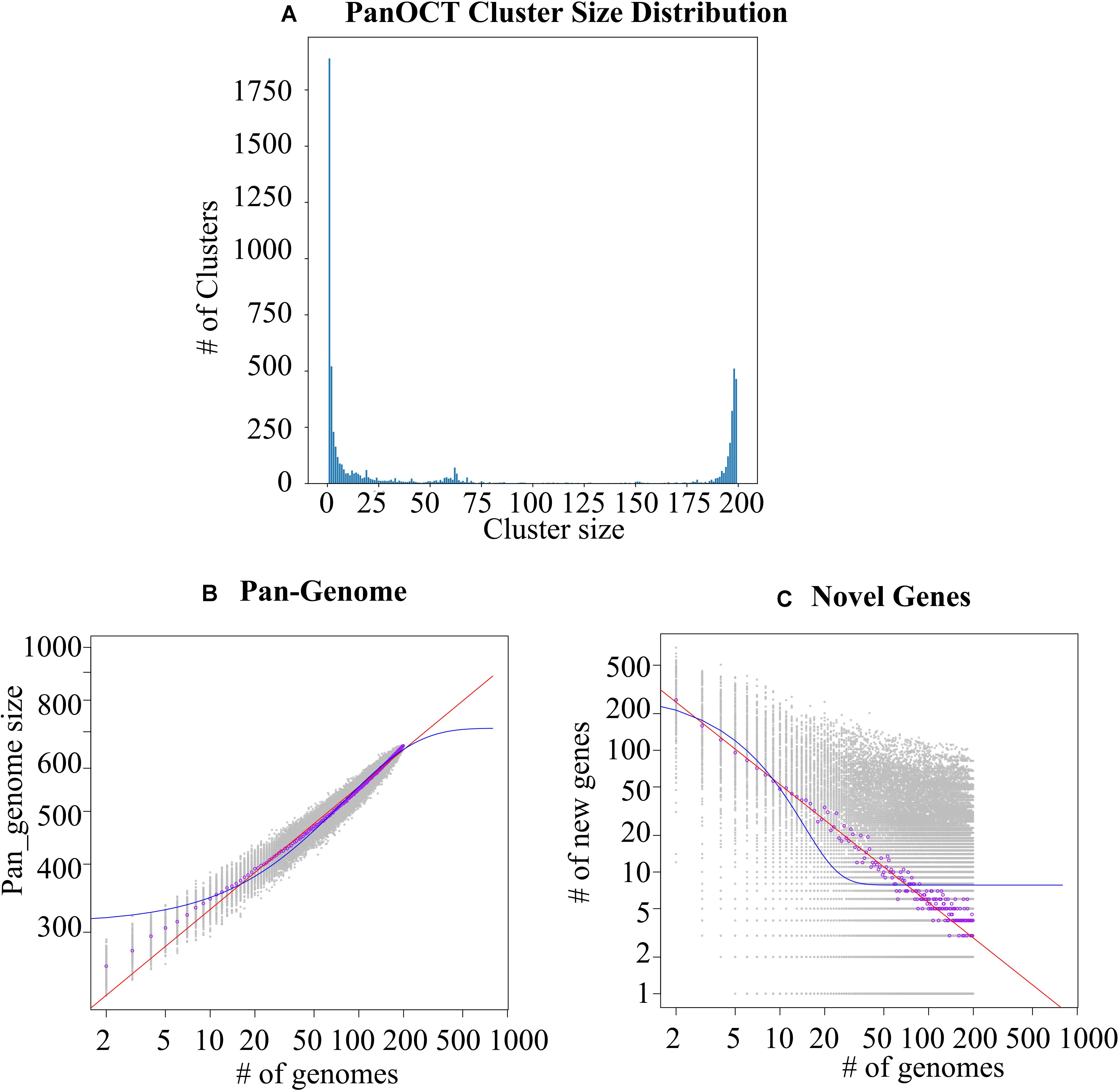
Figure 1. Analysis of the Staphylococcus epidermidis pan-genome. (A) The distribution of protein cluster sizes generated from the comparison of 198 S. epidermidis genomes using PanOCT. (B) The pan-genome size (left) and the number of novel genes discovered with the addition of each new genome (C, right) were estimated for all 198 genomes using a pan-genome model based on the original Tettelin et al. (2005) model.
The function of the genes within the variable genome was investigated by assigning all gene clusters to clusters of orthologous groups (COGs; Tatusov et al., 2003), and the results showed that novel genes were most likely to be assigned to categories (Figure 2 and Supplementary Tables S2, S3) such as mobilome, ribosomal structure and biogenesis, carbohydrate transport and metabolism, and nucleotide transport and metabolism, based on the result of Fisher’s exact test.
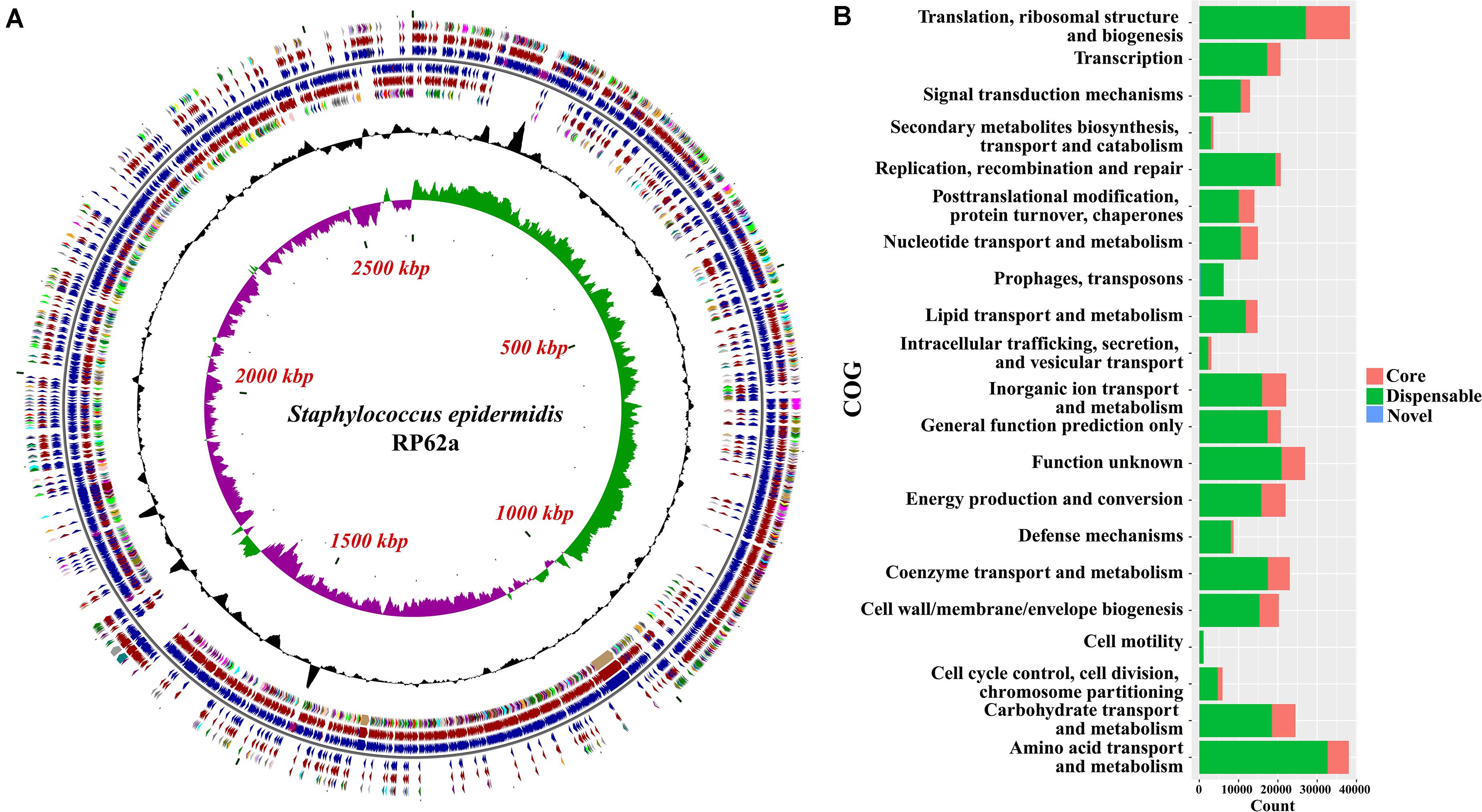
Figure 2. Functional analysis of the pan-genome of Staphylococcus epidermidis. (A) Distribution of core/accessory/novel genes in the type strain RP62a. Starting from the outermost ring the feature rings depict: (1) clusters of orthologous group (COG) functional categories for forward strand coding sequences; (2) Core (brown)/accessory (blue) genes for forward strand coding sequences; (3) Forward strand sequence features; (4) Reverse strand sequence features; (5) Core (brown)/accessory (blue) genes for reverse strand coding sequences; (6) COG functional categories for reverse strand coding sequences. (7) GC content; (8) GC skew. The colors of different COG functional categories were following the definition of Grant et al. (2012). (B) Numbers of core, accessory and novel genes for each clusters of orthologous group (COG) category. COGs significantly enriched (adjusted P-value < 0.05, Fisher exact test) in core, accessory or novel genes are marked with red asterisk.
Phylogenetic Relationship of S. epidermidis Isolates
To estimate the genetic relationships among S. epidermidis strains, we compared all 198 genomes by using a single nucleotide polymorphism-based phylogeny. SNPs were identified from the combined set of genome sequences by using kSNP. Nucleotide positions present in at least 80% of all genomes were used to build a Maximum-Likelihood phylogenetic tree with RAxML following the tutorial. Strikingly, the 198 S. epidermidis isolates formed two distinct groups (Figure 3), called Cluster A (solid line) and B (dotted line). Most of Sequence Type (ST) 2 nosocomial isolates were near-identical at the nucleotide level for all core genes (Supplementary Table S4). All of ST 2 strains in this study presented in Cluster A and had an extremely short evolutionary distance from each other (scaled branch lengths < 0.01), with 99.7% identical in sequence, indicating that these strains were probably derived from a recent common ancestor. By contrast, Cluster B represented a lineage with reduced virulence and all of ST 5 commensal strains presented in Cluster B and clustered together. The rest of Cluster B had a much longer evolutionary distance from ST 5 strains. This clade might have a more complex history of evolution and produce a variety of sub-groups.
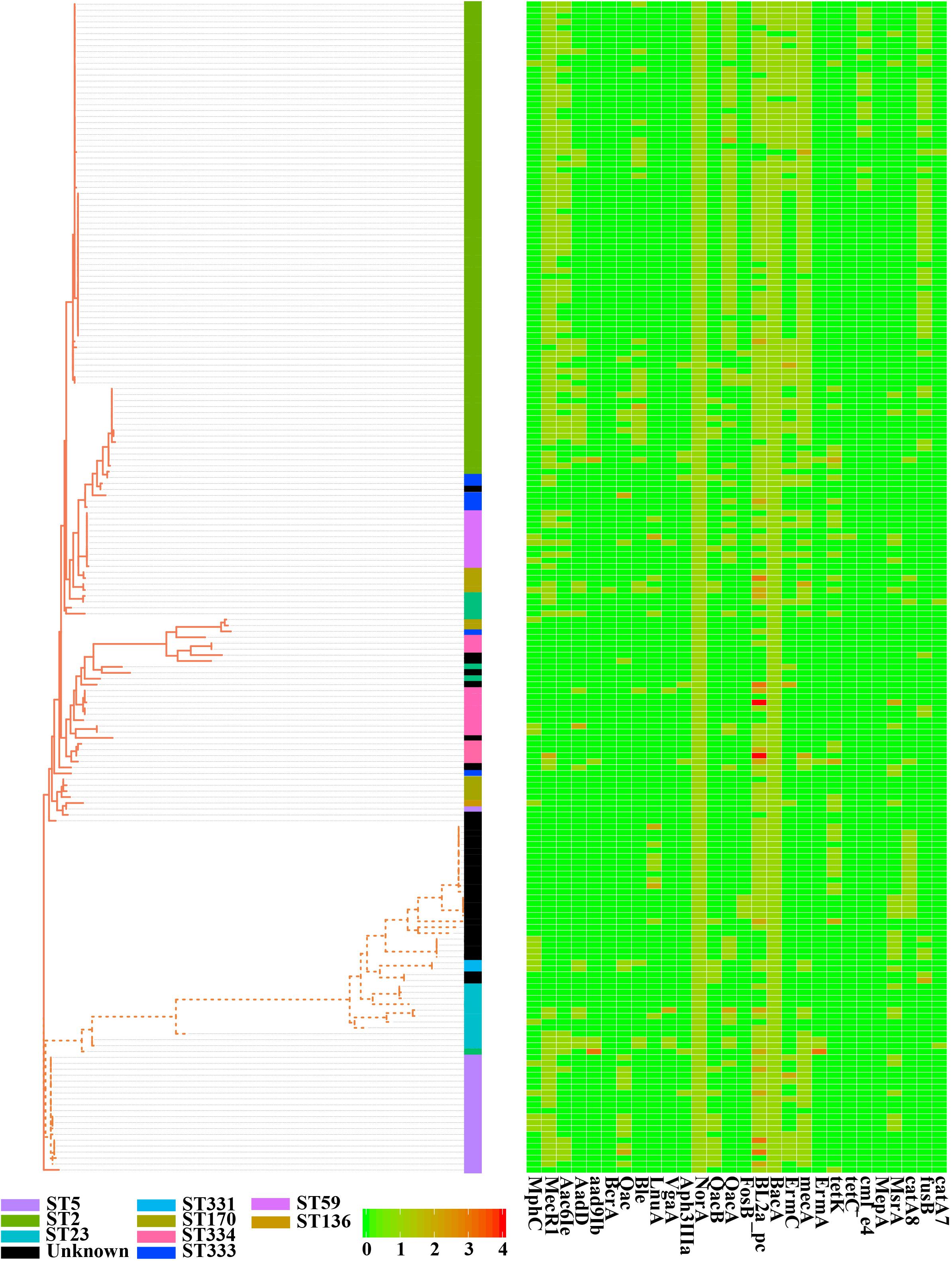
Figure 3. Phylogenetic single-nucleotide polymorphism (SNP) tree of Staphylococcus epidermidis strains. A whole-genome core SNP maximum likelihood tree was constructed for 198 genomes with kSNP and RAxML. Heatmap on the right indicates copies of 28 genes involved in antibiotic resistance. Legends on the bottom stand for copy number of resistant genes. The scale bar, which is an indicator of genetic distance, indicates 0.01.
Antimicrobial Resistance Across S. epidermidis
Antimicrobial resistance is very common among S. epidermidis isolates and often limits treatment options (Kleinschmidt et al., 2015). Given the clinical importance of AMR in S. epidermidis, we performed a genome-wide analysis of all known AMR genes within our genomic dataset. According to the analysis of ARDB, ResFinder and CARD databases, we found 28 different genes involved in 31 antibiotics (Figure 3). Nearly all isolates carried at least one antibiotic resistance gene. Among the genes involved in AMR, our data showed that there were two genes, norA (AAW53745.1) and bacA (AAW53717.1), conserved in all strains. Based on the enrichment analysis of strains from different niches, we found that strains from different sources (skin, blood, environment, and plant) had significantly different antibiotic resistance profiles: isolates from blood (9 antibiotic resistance genes) and skin (8 antibiotic resistance genes) had significantly enriched antibiotics (Supplementary Table S5), while isolates from environment had no significantly enriched antibiotics. First-line antibiotic therapy for catheter-related bloodstream infections was vancomycin. None of these isolates were resistant to the antibiotic at the genetic level, regardless of isolation source.
SCCmec and ACME in S. epidermidis
Staphylococcal chromosome cassette mec, or staphylococcal cassette chromosome mec, is a mobile genetic element that carries the central determinant for broad-spectrum beta-lactam resistance encoded by the mecA gene (AAW53314.1), a mobile genetic element of Staphylococcus bacterial species (IWG-SCC, 2009; Mcmanus et al., 2015). According to the completeness of genome in this study (only 7 complete genome sequences), we only analyzed the genes from well-defined SCCmec genomic islands (Kos et al., 2012). There were 58.6% (116/198) of S. epidermidis strains, in which complete mec gene complexes, mecA (AAW53314.1), and mecR1 (AAW53313.1) genes were detected (Supplementary Table S1). However, only 39.4% (78/198) of strains had ccr gene complex from type IV cassette, in which both ccrA and ccrB were present but not in RP62a. Similar to the previous results (Conlan et al., 2012; Du et al., 2013), nearly all of the ST 2 nosocomial isolates (94.6%, 70/74) had at least one copy of mecA from type IX cassette and mecR1 from Type VIII or IV-G cassette. On the other hand, a high prevalence (98%, 195/198) of ACME was found in S. epidermidis strains in this study, of which 22.7% (45/198) was type I and 75.8% (150/198) was type II.
Biofilm Formation of S. epidermidis
Biofilm formation is the major of virulence factor of S. epidermidis strains, which contributes to the persistence of clinical infection. Here, we analyze some well-known factors involved in biofilm formation such as adhesive molecules, including polysaccharide intercellular adhesin (icaABCD), proteinaceous factors (bhp (AAW53225.1), and aap (AAW53239.1), teichoic acids, extracellular DNA (Supplementary Table S6). The polysaccharide intercellular adhesion (icaABCD) genes that encode biofilm-associated genes for poly-N-acetylglucosamine synthesis were found in 60% of the commensal isolates, in agreement with previous studies (Du et al., 2013). Especially, any of the ica genes was not found in some ST 2 strains (Figure 4). Gene aap was enriched in the blood (adjusted P-value < 0.01) compared to the remaining isolates and therefore might be a potential biomarker for S. epidermidis infection. We analyzed the enrichment of all genes involved in virulence factors and found the icaABCD was significantly enriched despite the sources or sequence types.
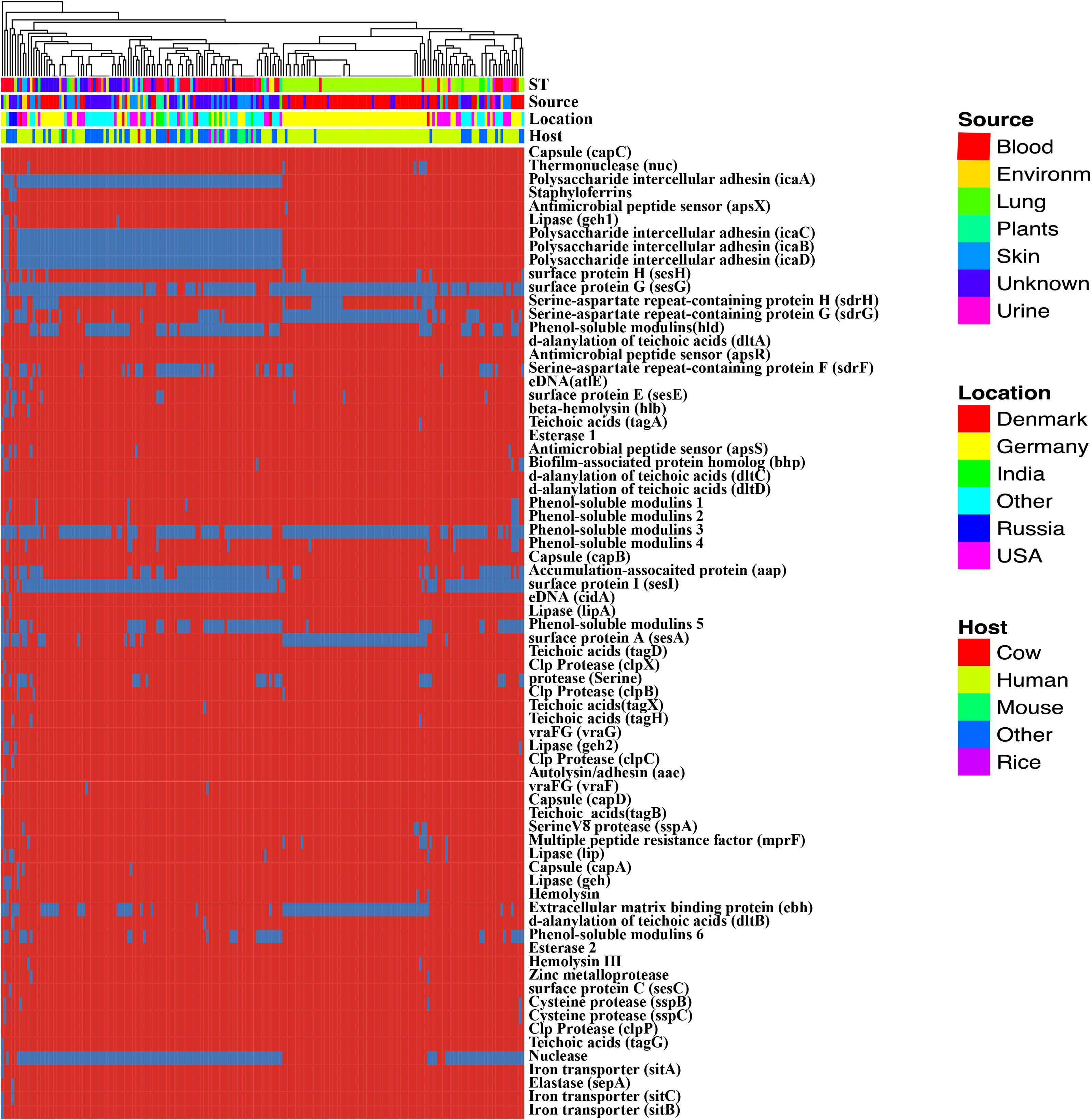
Figure 4. Heatmap of virulence factors among the Staphylococcus epidermidis strains. The dendrogram was generated using complete linkage clustering of copies of genes involved in virulence factors. The red color stands for genes that exist in the genomes and the blue color for missing ones. Legends on the right stand for colors of different host, isolates and geographic information.
Human-Bacterium and Bacterium-Bacterium Interactions in S. epidermidis
Staphylococcus epidermidis is the major colonization microorganisms in the human skin with complex human-bacterium and bacterium-bacterium interactions. We analyzed the genes (Figure 5, Table 1 and Supplementary Table S5) involved in resistance against AMPs that can inhibit the growth of most skin microorganism including S. epidermidis. We also identified 25 genes related to biofilm formation (7/17) and cell toxicity (8/17), which were significantly enriched in the blood and skin, were reported to assist the strain to survive on the skin surface (Joo and Otto, 2015). We also analyzed genes involved in bacterium-bacterium interactions. We found that the genes involved in short-chain fatty acids biosynthesis and extracellular proteases (e.g., Esp) had no difference among the isolates.
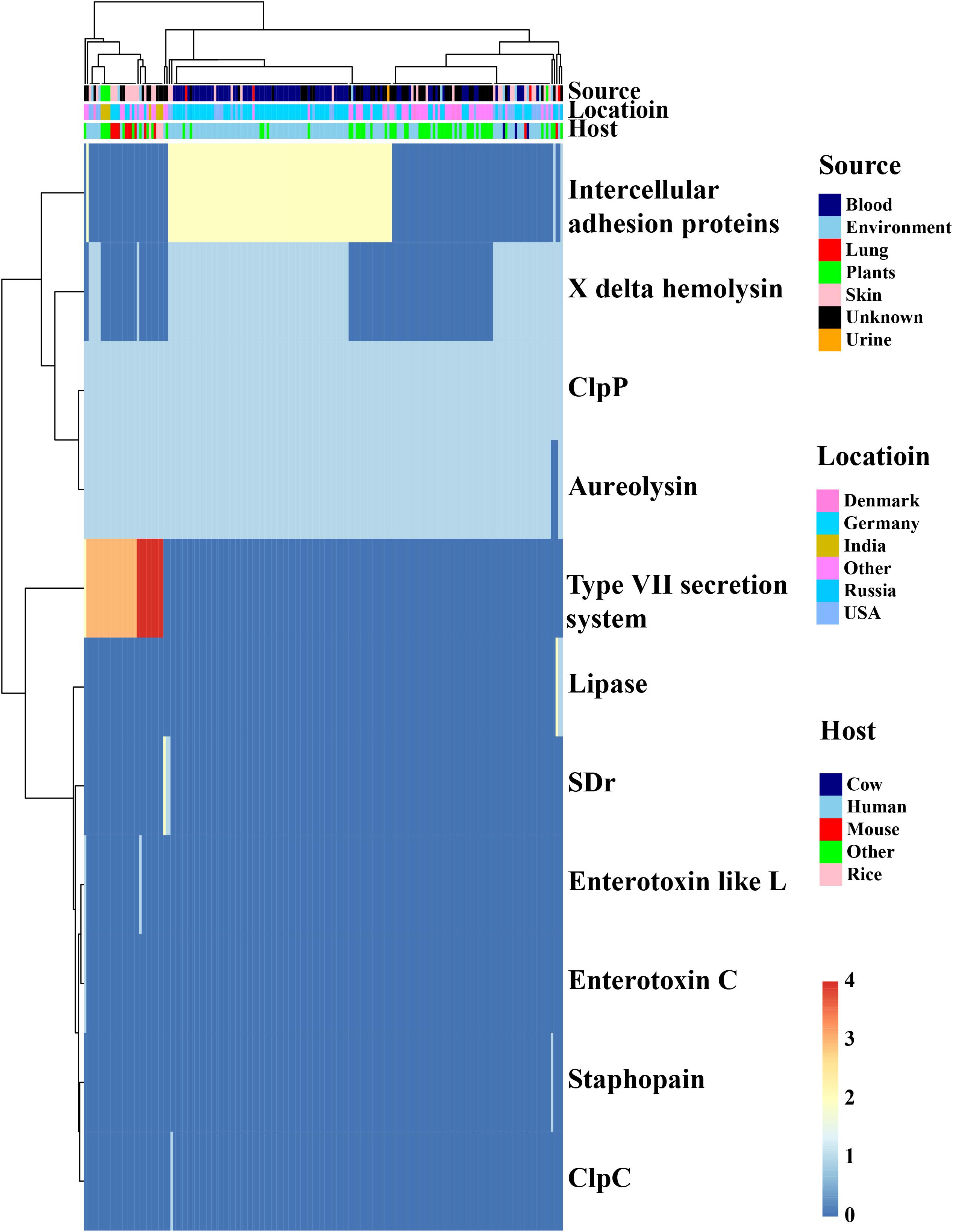
Figure 5. In silico analysis of virulence factors of the Staphylococcus epidermidis strains. The types of virulence factors were following the VFDBs database. Legends on the right stand for colors of different host, isolates, and geographic information. Different colors stand for copy number of each virulence factors.
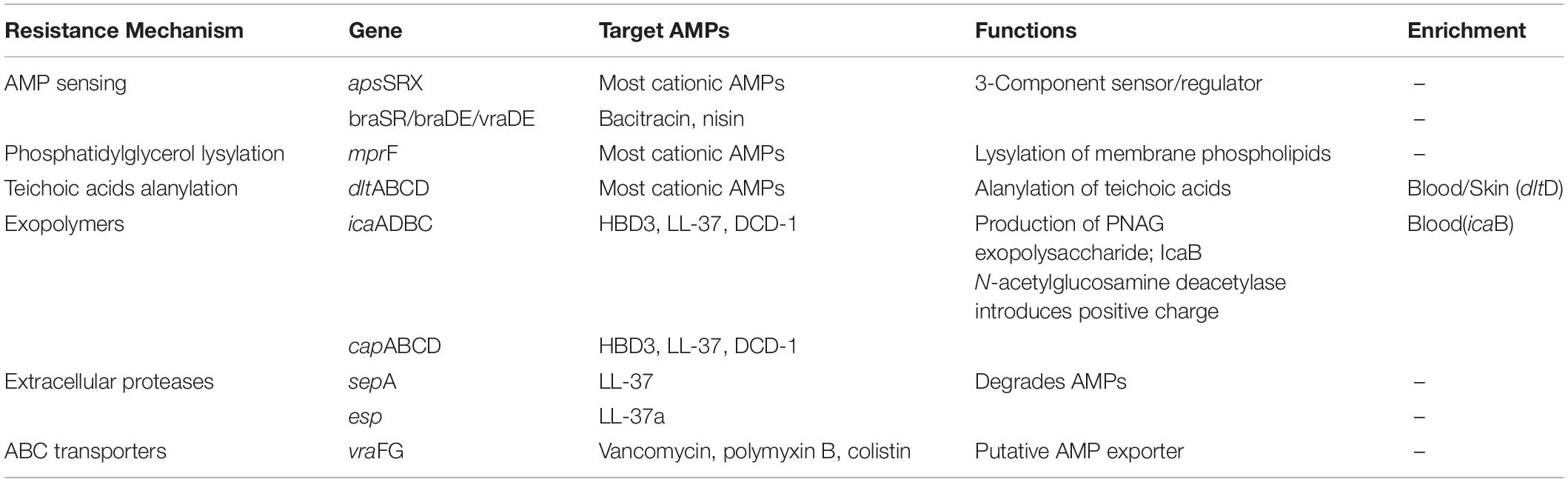
Table 1. Staphylococcus epidermidis resistance mechanisms that target antimicrobial peptides (AMPs).
Discussion
Staphylococcus epidermidis is a coagulase-negative and Gram-positive staphylococcus that is part of the normal mucosa and skin microflora in humans and other mammals (Oh et al., 2016). It is the second leading cause of nosocomial infections (Ziebuhr et al., 2006). Although S. epidermidis is a saprophyte and an opportunistic pathogen with plenty of antibiotic resistance and virulence factors (Namvar et al., 2014), this natural skin colonizer plays a critical role in balancing the epithelial microflora (Otto, 2009, 2010). As an innocuous commensal microorganism, S. epidermidis has long been seen as an avirulent species. With the accumulation of genomic sequences, we can now further explore the genetic mechanisms of environmental adaptability of S. epidermidis, the evolution process during the outbreak, and the molecular biomarkers for clinical diagnosis (Otto, 2009; Didelot et al., 2012).
In our current pan-genome analysis, S. epidermidis had a relatively compact genome with a size of about 2.5 Mb, and yet almost 20% of this genome was in flux, exchanging with a large pool of various genes. These findings were similar to what had been reported by Conlan et al. (2012). The significant number of genes involved in mobilome makes horizontal gene transfer easier among Staphylococcus stains and lead to the increase of the “open” pan-genome (Palmer et al., 2010). Besides, mobile genetic elements, such as SCCmec, ACME, and plasmids, make the genome structure more unpredictable (Qin et al., 2016). High-resolution phylogenetic tree constructed from genome-wide SNPs revealed important details not seen by traditional multi-locus sequence typing (MLST) or single gene marker (16S rDNA). From the phylogenetic tree, we found the ST 2 isolates had an extremely short evolutionary distance from each other.
The genetic markers mecA and icaA, which are used to predict the AMR and biofilm phenotypes, have been shown to be enriched in hospital isolates than in non-hospital isolates; however, these markers have much less power to distinguish infection isolates from commensally available isolates that contaminate clinical specimens (Tolo et al., 2016). According to our enrichment analysis, we found it was possible to distinguish the strains of blood from those of skin, with 17 potential biomarkers related to biofilm formation and cell toxicity, demonstrating the potential for identifying risk genotypes. Whole genome sequencing has been proved to be a more powerful routine diagnostic tool than the traditional MLST or RT-PCR because it can rapidly identify the infection source and antibiotic resistance in an affordable manner (Rasko et al., 2011; Pankhurst et al., 2016). As more genetic data of S. epidermidis have been available and a new machine learning algorithm has been developed (Tolo et al., 2016), WGS may help to predict the infection isolation sources and antibiotic resistance in a quicker and more accurate manner. On the other hand, S. epidermidis has very complicated relationship with human and other bacteria. The prevalence of S. epidermidis gene content and other genetic features exhibited strain specificity, suggesting functional specialization to the niche. AMPs play an important role in providing immunity to bacterial colonization on human epithelia (Liu et al., 2020; Zhou et al., 2020). Recent research has shown that Staphylococci have multiple systems to combat AMP activity, including AMP sensor that can regulate the expressions of genes involved in AMP resistance depending on the presence of AMPs (Joo and Otto, 2015). We analyzed the distribution of gene involved in AMP resistance and found the enrichment was significant in blood and skin and variable among different strains, which may be the consequence of coevolution of human’s immune system. On the other side, S. epidermidis strains also can inhibit the growth of other bacterium to be dominant species on the skin surface. Serine protease Esp, which is secreted by S. epidermidis, has been found to be able to inhibit the biofilm formation of S. aureus and destroy pre-existing S. aureus biofilms (Iwase et al., 2010). Other mechanisms are also involved in fighting against pathogens and maintaining homeostasis (Otto et al., 2001; Wang et al., 2014). S. epidermidis was also found to be a reservoir of antibiotic resistance, with its virulence determinants shared with other more pathogenic species such as S. aureus, as demonstrated in previous studies (Conlan et al., 2012). In particular, SCCmec, ACME elements conferring β-lactam resistance, and other genes are transferred frequently between Staphylococcus strains, enabling rapid evolution and adaptation against antibiotic selection pressure and provide additional competitive advantage. For instance, type III of SCCmec carries a phenol soluble modulin psm-mec, which may affect the virulence of S. aureus (Qin et al., 2016). This provides strong support for pathogen carriage and increased infection risks elsewhere in the body, such as of methicillin-resistant S. aureus in the nares, as well as for the contextual microbiome affecting infection risk via HGT of pathogenicity reservoirs.
Conclusion
Our current study provides information on the molecular characteristics of S. epidermidis strains isolated from different environments from all over the world. From a genomic perspective, the pan-genome analysis of the S. epidermidis reveals a high level of diversity among the generic and strain-specific genes and provides novel insights into the adaptation and evolution of S. epidermidis isolates as opportunistic, multidrug-resistant nosocomial pathogens. S. epidermidis strains from habitats are not equivalent and pathogenic sub-population acquired genetic elements and related phenotypes that promote infection. We identified 17 potential biomarkers related to biofilm formation and cell toxicity, which may help to distinct blood strains from skin stains. A better understanding of the mechanisms of gene transfer will help to control the epidemic of pan-drug-resistant S. epidermidis strains.
Data Availability Statement
The original contributions generated in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding authors.
Author Contributions
FS, HX, and HC designed the project. FS conceived the experiment and analyzed the genome data. RT, YY, HL, GS, YL, BH, XX, XC, and GZ interpreted the results and drafted the manuscript. All authors reviewed the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (Grant: 2018YFC2000505), the Fundamental Research Funds for the Central Universities (No: 3332019121), the Applied Research Program of Capital Clinical Features (Grant: Z18110001718172), and CAMS Innovation Fund for Medical Sciences (Grant: 2018-I2M-1-002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.566080/full#supplementary-material
Supplementary Table 1 | Basic information of all strains analyzed used in this study.
Supplementary Table 2 | Result of clusters of orthologous group (COG) enrichment analysis across all strains.
Supplementary Table 3 | Result of KEGG enrichment analysis across all strains.
Supplementary Table 4 | Cluster of orthologous proteins produced by PanOCT.
Supplementary Table 5 | Enrichment analysis about antibiotic resistances from different sources.
Supplementary Table 6 | Enrichment analysis about genes related in biofilm formation.
Abbreviations
SCCmec, staphylococcal chromosome cassette mec; AMPs, antimicrobial peptides; SNPs, single-nucleotide polymorphisms; COGs, clusters of orthologous groups; ST, sequence type; AMR, antimicrobial resistance.
Footnotes
- ^ https://sourceforge.net/projects/ksnp/
- ^ https://sourceforge.net/projects/panoct/
- ^ http://kobas.cbi.pku.edu.cn/
References
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Barbier, F., Lebeaux, D., Hernandez, D., Delannoy, A. S., Caro, V., Francois, P., et al. (2011). High prevalence of the arginine catabolic mobile element in carriage isolates of methicillin-resistant Staphylococcus epidermidis. J. Antimicrob. Chemother. 66, 29–36. doi: 10.1093/jac/dkq410
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., et al. (2013). GenBank. Nucleic Acids Res. 41, D36–D42. doi: 10.1093/nar/gks1195
Bloemendaal, A. L., Brouwer, E. C., and Fluit, A. C. (2010). Methicillin resistance transfer from Staphylocccus epidermidis to methicillin-susceptible Staphylococcus aureus in a patient during antibiotic therapy. PLoS One 5:e11841. doi: 10.1371/journal.pone.0011841
Chen, L., Xiong, Z., Sun, L., Yang, J., and Jin, Q. (2012). VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40, D641–D645. doi: 10.1093/nar/gkr989
Conlan, S., Mijares, L. A., Program, N. C. S., Becker, J., Blakesley, R. W., Bouffard, G. G., et al. (2012). Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome Biol. 13:R64. doi: 10.1186/gb-2012-13-7-r64
Didelot, X., Bowden, R., Wilson, D. J., Peto, T. E., and Crook, D. W. (2012). Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13, 601–612. doi: 10.1038/nrg3226
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739. doi: 10.1016/s0140-6736(06)68231-7
Du, X., Zhu, Y., Song, Y., Li, T., Luo, T., Sun, G., et al. (2013). Molecular analysis of Staphylococcus epidermidis strains isolated from community and hospital environments in China. PLoS One 8:e62742. doi: 10.1371/journal.pone.0062742
Ehsan Vafadarnejad, G. R., Laura, K., Panagiota, A., Vallery, A. N., Dirk, S., Melanie, R., et al. (2020). Time-resolved single-cell transcriptomics uncovers dynamics of cardiac neutrophil diversity in murine myocardial infarction. bioRxiv [Preprint]. doi: 10.1101/738005
Foster, T. J. (2017). Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 41, 430–449. doi: 10.1093/femsre/fux007
Fouts, D. E., Brinkac, L., Beck, E., Inman, J., and Sutton, G. (2012). PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res. 40:e172. doi: 10.1093/nar/gks757
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Gill, S. R., Fouts, D. E., Archer, G. L., Mongodin, E. F., Deboy, R. T., Ravel, J., et al. (2005). Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187, 2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005
Grant, J. R., Arantes, A. S., and Stothard, P. (2012). Comparing thousands of circular genomes using the CGView comparison tool. BMC Genom. 13:202. doi: 10.1186/1471-2164-13-202
Iwase, T., Uehara, Y., Shinji, H., Tajima, A., Seo, H., Takada, K., et al. (2010). Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465, 346–349. doi: 10.1038/nature09074
IWG-SCC (2009). Classification of staphylococcal cassette chromosome mec (SCCmec): guidelines for reporting novel SCCmec elements. Antimicrob. Agents Chemother. 53, 4961–4967. doi: 10.1128/aac.00579-09
Jack, R. W., Tagg, J. R., and Ray, B. (1995). Bacteriocins of gram-positive bacteria. Microbiol. Rev. 59, 171–200. doi: 10.1128/mmbr.59.2.171-200.1995
Jetten, A. M., and Vogels, G. D. (1972). Mode of action of a Staphylococcus epidermidis bacteriocin. Antimicrob. Agents Chemother. 2, 456–463. doi: 10.1128/aac.2.6.456
Joo, H. S., and Otto, M. (2015). Mechanisms of resistance to antimicrobial peptides in Staphylococci. Biochim. Biophys. Acta 1848, 3055–3061. doi: 10.1016/j.bbamem.2015.02.009
Kleinschmidt, S., Huygens, F., Faoagali, J., Rathnayake, I. U., and Hafner, L. M. (2015). Staphylococcus epidermidis as a cause of bacteremia. Future Microbiol. 10, 1859–1879. doi: 10.2217/fmb.15.98
Kos, V. N., Desjardins, C. A., Griggs, A., Cerqueira, G., Van Tonder, A., Holden, M. T., et al. (2012). Comparative genomics of vancomycin-resistant Staphylococcus aureus strains and their positions within the clade most commonly associated with Methicillin-resistant S. aureus hospital-acquired infection in the United States. mBio 3:e00112. doi: 10.1128/mBio.00112-12
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/jcm.06094-11
Lee, J. Y. H., Monk, I. R., Goncalves Da Silva, A., Seemann, T., Chua, K. Y. L., Kearns, A., et al. (2018). Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat. Microbiol. 3, 1175–1185. doi: 10.1038/s41564-018-0230-7
Liu, B., and Pop, M. (2009). ARDB–antibiotic resistance genes database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Liu, Q., Liu, Q., Meng, H., Lv, H., Liu, Y., Liu, J., et al. (2020). Staphylococcus epidermidis contributes to healthy maturation of the nasal microbiome by stimulating antimicrobial peptide production. Cell Host Microb. 27, 68–78.e65. doi: 10.1016/j.chom.2019.11.003
Mcmanus, B. A., Coleman, D. C., Deasy, E. C., Brennan, G. I., Brain, O. C., Monecke, S., et al. (2015). Comparative genotypes, Staphylococcal cassette chromosome mec (SCCmec) Genes and antimicrobial resistance amongst Staphylococcus epidermidis and Staphylococcus haemolyticus isolates from infections in humans and companion animals. PLoS One 10:e0138079. doi: 10.1371/journal.pone.0138079
Meric, G., Mageiros, L., Pensar, J., Laabei, M., Yahara, K., Pascoe, B., et al. (2018). Disease-associated genotypes of the commensal skin bacterium Staphylococcus epidermidis. Nat. Commun. 9:5034. doi: 10.1038/s41467-018-07368-7
Meric, G., Miragaia, M., De Been, M., Yahara, K., Pascoe, B., Mageiros, L., et al. (2015). Ecological overlap and horizontal gene transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 7, 1313–1328. doi: 10.1093/gbe/evv066
Miragaia, M., Thomas, J. C., Couto, I., Enright, M. C., and De Lencastre, H. (2007). Inferring a population structure for Staphylococcus epidermidis from multilocus sequence typing data. J. Bacteriol. 189, 2540–2552. doi: 10.1128/jb.01484-06
Namvar, A. E., Bastarahang, S., Abbasi, N., Ghehi, G. S., Farhadbakhtiarian, S., Arezi, P., et al. (2014). Clinical characteristics of Staphylococcus epidermidis: a systematic review. GMS Hyg. Infect. Control 9:Doc23.
Oh, J., Byrd, A. L., Park, M., Program, N. C. S., Kong, H. H., and Segre, J. A. (2016). Temporal stability of the human skin microbiome. Cell 165, 854–866. doi: 10.1016/j.cell.2016.04.008
Otto, M. (2009). Staphylococcus epidermidis – the “accidental” pathogen. Nat. Rev. Microbiol. 7, 555–567. doi: 10.1038/nrmicro2182
Otto, M. (2010). Staphylococcus colonization of the skin and antimicrobial peptides. Expert. Rev. Dermatol. 5, 183–195. doi: 10.1586/edm.10.6
Otto, M., Echner, H., Voelter, W., and Gotz, F. (2001). Pheromone cross-inhibition between Staphylococcus aureus and Staphylococcus epidermidis. Infect. Immun. 69, 1957–1960. doi: 10.1128/iai.69.3.1957-1960.2001
Palmer, K. L., Kos, V. N., and Gilmore, M. S. (2010). Horizontal gene transfer and the genomics of Enterococcal antibiotic resistance. Curr. Opin. Microbiol. 13, 632–639. doi: 10.1016/j.mib.2010.08.004
Pankhurst, L. J., Del Ojo Elias, C., Votintseva, A. A., Walker, T. M., Cole, K., Davies, J., et al. (2016). Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: a prospective study. Lancet Respir. Med. 4, 49–58. doi: 10.1016/s2213-2600(15)00466-x
Qin, L., Mccausland, J. W., Cheung, G. Y., and Otto, M. (2016). PSM-Mec-A virulence determinant that connects transcriptional regulation, virulence, and antibiotic resistance in Staphylococci. Front. Microbiol. 7:1293. doi: 10.3389/fmicb.2016.01293
Rasko, D. A., Webster, D. R., Sahl, J. W., Bashir, A., Boisen, N., Scheutz, F., et al. (2011). Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365, 709–717. doi: 10.1056/NEJMoa1106920
Schommer, N. N., and Gallo, R. L. (2013). Structure and function of the human skin microbiome. Trends Microbiol. 21, 660–668. doi: 10.1016/j.tim.2013.10.001
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinform. 4:41. doi: 10.1186/1471-2164-13-041
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tolo, I., Thomas, J. C., Fischer, R. S., Brown, E. L., Gray, B. M., and Robinson, D. A. (2016). Do Staphylococcus epidermidis genetic clusters predict isolation sources? J. Clin. Microbiol. 54, 1711–1719. doi: 10.1128/jcm.03345-15
Ugolotti, E., Larghero, P., Vanni, I., Bandettini, R., Tripodi, G., Melioli, G., et al. (2016). Whole-genome sequencing as standard practice for the analysis of clonality in outbreaks of meticillin-resistant Staphylococcus aureus in a paediatric setting. J. Hosp. Infect. 93, 375–381. doi: 10.1016/j.jhin.2016.04.003
Wang, Y., Kuo, S., Shu, M., Yu, J., Huang, S., Dai, A., et al. (2014). Staphylococcus epidermidis in the human skin microbiome mediates fermentation to inhibit the growth of Propionibacterium acnes: implications of probiotics in acne vulgaris. Appl. Microbiol. Biotechnol. 98, 411–424. doi: 10.1007/s00253-013-5394-8
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Yao, Y., Sturdevant, D. E., Villaruz, A., Xu, L., Gao, Q., and Otto, M. (2005). Factors characterizing Staphylococcus epidermidis invasiveness determined by comparative genomics. Infect. Immun. 73, 1856–1860. doi: 10.1128/iai.73.3.1856-1860.2005
Yu, G., Lam, T. T., Zhu, H., and Guan, Y. (2018). Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol. Biol. Evol. 35, 3041–3043. doi: 10.1093/molbev/msy194
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, Y. Q., Ren, S. X., Li, H. L., Wang, Y. X., Fu, G., Yang, J., et al. (2003). Genome-based analysis of virulence genes in a non-biofilm-forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol. 49, 1577–1593. doi: 10.1046/j.1365-2958.2003.03671.x
Zhou, W., Spoto, M., Hardy, R., Guan, C., Fleming, E., Larson, P. J., et al. (2020). Host-specific evolutionary and transmission dynamics shape the functional diversification of Staphylococcus epidermidis in Human Skin. Cell 180, 454–470.e18. doi: 10.1016/j.cell.2020.01.006
Keywords: Staphylococcus epidermidis, antimicrobial resistance, pan-genome, mobile genetic element, comparative genome analysis
Citation: Su F, Tian R, Yang Y, Li H, Sun G, Li Y, Han B, Xu X, Chen X, Zhao G, Cui H and Xu H (2020) Comparative Genome Analysis Reveals the Molecular Basis of Niche Adaptation of Staphylococcus epidermidis Strains. Front. Genet. 11:566080. doi: 10.3389/fgene.2020.566080
Received: 22 July 2020; Accepted: 21 October 2020;
Published: 09 November 2020.
Edited by:
Rongling Wu, Pennsylvania State University (PSU), United StatesReviewed by:
Prabhu B. Patil, Institute of Microbial Technology (CSIR), IndiaYong Li, Henan Agricultural University, China
Copyright © 2020 Su, Tian, Yang, Li, Sun, Li, Han, Xu, Chen, Zhao, Cui and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyuan Cui, Y3VpaG9uZ3l1YW4zOTIxQGJqaG1vaC5jbg==; Y3VpaG9uZ3l1YW4xOTgzQDEyNi5jb20=; Hongtao Xu, eHVob25ndGFvMjkxMUBiamhtb2guY24=
†These authors have contributed equally to this work