 Anastasiya V. Snezhkina1*
Anastasiya V. Snezhkina1* Maria S. Fedorova1
Maria S. Fedorova1 Vladislav S. Pavlov1
Vladislav S. Pavlov1 Dmitry V. Kalinin2Alexander L. Golovyuk2
Dmitry V. Kalinin2Alexander L. Golovyuk2 Elena A. Pudova1
Elena A. Pudova1 Zulfiya G. Guvatova1
Zulfiya G. Guvatova1 Nataliya V. Melnikova1
Nataliya V. Melnikova1 Alexey A. Dmitriev1George S. Razmakhaev3
Alexey A. Dmitriev1George S. Razmakhaev3 Andrey A. Poloznikov3Galina S. Alekseeva3Andrey D. Kaprin3
Andrey A. Poloznikov3Galina S. Alekseeva3Andrey D. Kaprin3 George S. Krasnov1
George S. Krasnov1 Anna V. Kudryavtseva1*
Anna V. Kudryavtseva1*- 1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia
- 2Vishnevsky Institute of Surgery, Ministry of Health of the Russian Federation, Moscow, Russia
- 3National Medical Research Radiological Center, Ministry of Health of the Russian Federation, Moscow, Russia
Head and neck paragangliomas (HNPGLs) are rare neuroendocrine tumors that have a high degree of heritability and are predominantly associated with mutations in ten genes, such as SDHx, SDHAF2, VHL, RET, NF1, TMEM127, MAX, FH, MEN2, and SLC25A11. Elucidating the mutation prevalence is crucial for the development of genetic testing. In this study, we identified pathogenic/likely pathogenic variants in the main susceptibility genes in 102 Russian patients with HNPGLs (82 carotid and 23 vagal paragangliomas) using whole exome sequencing. Pathogenic/likely pathogenic variants were detected in 43% (44/102) of patients. We identified the following variant distribution of the tested genes: SDHA (1%), SDHB (10%), SDHC (5%), SDHD (24.5%), and RET (5%). SDHD variants were observed in the majority of the patients with bilateral/multiple paragangliomas. Thus, among Russian patients with HNPGLs the most frequently mutated gene was SDHD followed by SDHB, SDHC, RET, and SDHA.
Introduction
Head and neck (HN) paragangliomas (PGLs) are rare neuroendocrine tumors of four distinct localizations: carotid, vagal, laryngeal, and middle ear PGLs. Carotid paragangliomas (CPGLs) arise from the carotid glomus at the carotid artery bifurcation and are the most common form of HNPGLs (60%) (El-Naggar et al., 2017). Middle ear and vagal paragangliomas (MEPGLs and VPGLs) are less frequent than CPGLs and account for 29% and 13%, respectively; laryngeal PGLs are very rare (El-Naggar et al., 2017). All the HNPGLs are often characterized by slow growth and non-aggressive behavior, but exhibit metastatic potential. The overall metastatic rate for HNPGLs vary depending on the site of tumor localization: 2% for larynx and middle ear, 4–6% for carotid, and up to 16% for vagal PGLs (Williams, 2017). HNPGLs can also develop as bilateral or multiple tumors and pose significant treatment challenges.
HNPGLs can as familial or sporadic forms (Dahia, 2014). Familial HNPGLs together with pheochromocytomas (PCCs) account for about 40% and are associated with four types of hereditary paraganglioma syndromes (PGL1–5) caused by mutations in the following genes: SDHD (PGL1), SDHAF2 (PGL2), SDHC (PGL3), SDHB (PGL4), and SDHA (PGL5) (Burnichon et al., 2010; Boedeker et al., 2014). Patients with hereditary HNPGLs less frequently harbor germline mutations in TMEM127 (Bausch et al., 2017), RET (Kudryavtseva et al., 2019), MAX (Burnichon et al., 2012), FH (Castro-Vega et al., 2014), and SLC25A11 (Buffet et al., 2018). Germline mutations in VHL, NF1, and MEN2 have been detected in HNPGLs in association with other tumoral and clinical features (Boedeker et al., 2009).
In this study, we aimed to assess the frequency of variants in the main susceptibility genes for HNPGLs, such as SDHx, SDHAF2, VHL, RET, NF1, TMEM127, MAX, FH, MEN2, and SLC25A11, among a representative set of Russian patients with CPGLs and VPGLs.
Materials and Methods
Patients
In total, 102 Russian patients with HNPGLs, including 82 patients with CPGLs and 23 with VPGLs, were enrolled at the Vishnevsky Institute of Surgery, Ministry of Health of the Russian Federation. Informed consent was obtained from all patients. This study was approved by the Ethics Committee of Vishnevsky Institute of Surgery with ethics committee approval no. 004-2020, 03.07.2020, and was performed in accordance with the Declaration of Helsinki (1964) (World Medical Association, 2001). The clinicopathologic characteristics of the patients are presented in Table 1.
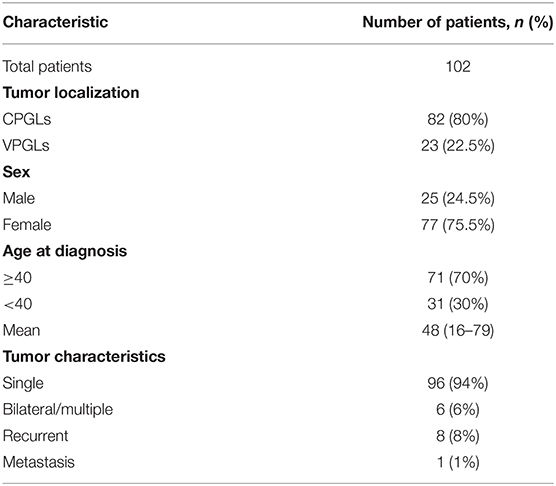
Table 1. Clinicopathologic characteristics of patients with HNPGLs.
Exome Library Preparation and Sequencing
DNA was extracted from formaldehyde fixed-paraffin embedded (FFPE) tumor tissues using High Pure FFPET DNA Isolation Kit (Roche, Basel, Switzerland) and quantified with a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA quality was assessed by quantitative PCR (qPCR) using QuantumDNA Kit (Evrogen, Moscow, Russia). Exome libraries were prepared from DNA using Rapid Capture Exome Kit (Illumina, San Diego, CA, USA) or TruSeq Exome Library Prep Kit (Illumina) according to the manufacturer's recommendations. High-throughput sequencing of the exome libraries was performed on a NextSeq 500 System (Illumina) in a paired-end mode of 76 × 2 bp. The average coverage for each sample was at least 300X. Sequencing data are available at NCBI Sequence Read Archive (SRA) BioProject PRJNA639937.
Mutation Analysis
Raw sequencing read quality was assessed using FastQC (v. 0.11.9). The reads were trimmed for quality (less than Q20), and adapter sequences were removed using Trimmomatic (v. 0.39) (Bolger et al., 2014). Alignment of reads to the reference human genome GRCh37.75/hg19 was performed with Burrows-Wheeler Aligner (v. 0.7.17) (Li and Durbin, 2010). To report alignment statistics and determine read duplicates, we applied SAMtools (v. 1.10) (Li et al., 2009) and Picard-tools (v. 2.23.4). Base quality score recalibration was carried out with GATK4 (v. 4.1.2) (McKenna et al., 2010) and dbSNP (common variants 2015-06-05) (Sherry et al., 2001). Variant calling was performed with GATK HaplotypeCaller (McKenna et al., 2010). We excluded false positives using StrandBiasBySample, StrandOddsRatio, and BaseQualityRankSumTest annotations, as well as mis-sequenced single-nucleotide variants in polyN motifs, such as GGGTG > GGGGG, CCCCG > CCCCC, and others. For functional annotation of variants, ANNOVAR (v. 20200316) (Wang et al., 2010) was used. Annotations included allele frequency data [gnomAD (Karczewski et al., 2020), Kaviar (Glusman et al., 2011), and ESP-6500 (http://evs.gs.washington.edu/EVS/)], information about reported genomic variations and its association with human pathologies [ClinVar (Landrum et al., 2018), dbSNP, and COSMIC (Tate et al., 2019)], score for the conservation of mutated sites [PhastCons (Siepel et al., 2005) and PhyloP (both PHAST v. 1.5) (Pollard et al., 2010)], localization of variants in protein domains [InterPro (v. 81.0) (Mitchell et al., 2019)], as well as pathogenicity prediction score [SIFT (v. 6.2.1) (Vaser et al., 2016), PolyPhen2 (v. 2.2.2) (Adzhubei et al., 2010), MutationTaster (v. 2013-03-20) (Schwarz et al., 2014), LRT (v. 0.2) (Chun and Fay, 2009), PROVEAN (Choi and Chan, 2015), MetaSVM and MetaLR (Dong et al., 2015), CADD (v. 1.6) (Kircher et al., 2014), and DANN (Quang et al., 2015)].
Sanger Sequencing
To validate the whole exome sequencing data, Sanger sequencing was performed in Evrogen. Primer sequences are available on request.
Results
A representative set of HNPGL samples were collected from 102 Russian patients diagnosed with CPGLs (n = 82) and VPGLs (n = 23), including 76 patients with single CPGL, 20 patients with single VPGL, and 6 patients with bilateral/multiple PGLs (three of them had both carotid and vagal paragangliomas) (Table 1). These tumor samples were analyzed for the presence of pathogenic/likely pathogenic variants in the main susceptibility genes for HNPGLs: SDHx, SDHAF2, VHL, RET, NF1, TMEM127, MAX, FH, MEN2, and SLC25A11. Variants were classified as pathogenic or likely pathogenic according to the annotations in the ClinVar database or by using the criteria of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) (Richards et al., 2015).
Pathogenic/likely pathogenic variants were revealed in 44 out of 102 (43%) patients with HNPGLs (Supplementary Table 1). The prevalence of variants was as follows: SDHA (1%, 1/102), SDHB (10%, 10/102), SDHC (5%, 5/102), SDHD (24.5%, 25/102), and RET (5%, 5/102). Almost all patients with bilateral/multiple paragangliomas (except a patient with no variants in any of the main susceptibility genes tested) demonstrated pathogenic variants in SDHD (Table 2). A pathogenic variant NM_003002: c.A305G, p.H102R (chr11: 111959726, rs104894302) was the most frequent SDHD mutation detected among the Russian patients. This variant has been found in nine patients with CPGLs and three with VPGLs, including two patients with bilateral/multiple paragangliomas (one patient among them had both tumors subjected to genetic testing). In addition, it is noteworthy that only one variant NM_020975: c.A2372T, p.Y791F (chr10: 43613908, rs77724903) was determined among all RET-mutated HNPGLs indicating its high frequency in this population.
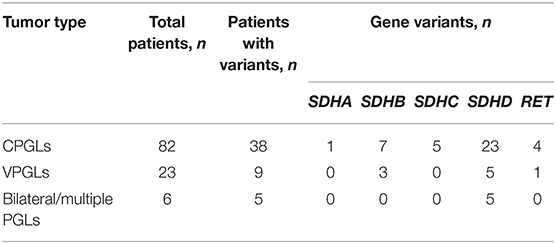
Table 2. Types of HNPGLs and number of patients with identified pathogenic/likely pathogenic variants.
Also, we analyzed the frequency of variants in the main susceptibility genes separately for CPGLs and VPGLs. In CPGLs, pathogenic/likely pathogenic variants were detected in 38 of 82 (46%) patients and were distributed as follows: SDHA (1%, 1/82), SDHB (8.5%, 7/82), SDHC (6%, 5/82), SDHD (28% 23/82), and RET (5%, 4/82) (Supplementary Table 1, Table 2). The majority of patients had pathogenic/likely pathogenic variants in one of these genes, however, in two patients, a likely pathogenic variant NM_020975: c.A2372T, p.Y791F (chr10: 43613908, rs77724903) in RET were corepresented with SDHA (Pat16) and SDHB (Pat142) variants. The same variant in RET was identified in two other patients (Pat35 and Pat155) with CPGLs. A likely pathogenic variant NM_003001: c.G149A, p.R50H (chr1: 161298257, rs769177037) in SDHC were also detected simultaneously in two patients (Pat102 and Pat152). Nine patients with CPGLs carried one pathogenic variant NM_003002: c.A305G, p.H102R (chr11: 111959726, rs104894302) in SDHD, including two patients (Pat1 and Pat5) with bilateral/multiple paragangliomas.
Pathogenic/likely pathogenic variants in SDHB, SDHD, and RET genes were found in 9 out of 23 (39%) patients with VPGLs (Supplementary Table 1, Table 2). The frequency of variants had the following distribution: SDHB (13%, 3/23), SDHD (22%, 5/23), and RET (4%, 1/23). All the patients with VPGLs carried pathogenic/likely pathogenic variants only in one of these genes. Notably, a likely pathogenic variant NM_020975: c.A2372T, p.Y791F (chr10: 43613908, rs77724903) in RET identified in four patients with CPGLs was also found in a patient with VPGL (Pat158). Moreover, three patients with VPGLs harbored the pathogenic variant NM_003002: c.A305G, p.H102R (chr11: 111959726, rs104894302) in the SDHD gene detected with high frequency in a set of CPGLs.
Next, we analyzed the age at diagnosis and sex ratio for SDHB, SDHC, SDHD, and RET variants within the cohort of patients with HNPGLs (Table 3). Variants in the SDHC and SDHD genes were diagnosed with approximately equal frequency in males and females taking into account 1:3 male to female ratio among the studied cohort. SDHB variants were found third-fold more frequent in females when RET variants were detected about two-fold more frequent in males. Variants in SDHB, SDHC, and SDHD were observed in all age groups and were more often detected in patients aged between 19–40 and 41–60 years. RET variants were identified in two groups of patients aged 19–40 and 41–60 years. Notably, frequency of variants in all the genes was the lowest in patients aged 61–80 years.
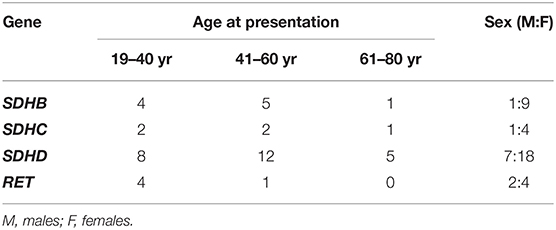
Table 3. Age and sex of patients with SDHB, SDHC, SDHD, and RET variants.
Discussion
In HNPGLs, the occurrence and frequency of mutations in the SDHx genes are extensively studied and are of importance for the diagnosis and management of the disease. The prevalence of mutations in other susceptibility genes has been poorly investigated. Notably, most studies have been focused on germline variants. In this work, we cannot establish germline and somatic mutation status for identified variants, since we used the archival collection of tumors for which blood or other normal tissues were not available. We obtained data on the overall frequency of pathogenic/likely pathogenic variants in the main susceptibility genes that allows to better understand molecular basis of the tumor development in Russian patients. A more similar study was performed for 24 Spanish patients with HNPGLs, who were subjected to genetic testing for germline and somatic mutations in the SDHx genes (Curras-Freixes et al., 2015). In contrast to our data, the majority of mutations were detected in SDHB (33%, 8/24) followed by SDHD (21%, 5/24) and SDHC (4%, 1/24). Two patients with SDHB mutations and one patient with SDHD mutation were characterized by metastatic tumors but no SDHA variants were detected.
In Russian patients, we revealed the likely pathogenic variant in RET in 5% of cases (5/102), including four patients with CPGLs and one patient with VPGL. Activation mutations in the proto-oncogene RET lead to the development of an autosomal dominant syndrome called multiple endocrine neoplasia type 2 (MEN2). Up to 50% of patients with MEN2 develop PCCs (Pedulla et al., 2014). HNPGLs have been rarely described in patients with MEN2 syndrome (Boedeker et al., 2009). However, several studies have reported RET mutations in HNPGLs without any association with MEN2 syndrome. A RET mutation was previously detected in a patient with multiple paragangliomas (Ding et al., 2019). Moreover, the likely pathogenic germline variant NM_020975: c.A2372T, p.Y791F (chr10: 43613908, rs77724903) in RET was identified in two out of four members of a family with multiple and malignant paragangliomas (Choi Jdo et al., 2014). All four members carried pathogenic SDHD mutations. In addition, in this family, the RET mutation was observed in the male adult with bilateral carotid body and jugulotympanic paragangliomas and his son with unilateral CPGLs. Here, we also detected this RET variant in all RET-mutated tumors. Moreover, this variant co-occurred with the pathogenic start-loss variant in SDHA and the pathogenic splice site variant in SDHB. Collectively, data from our study and previous studies suggest that this RET variant can occur both alone and together with SDHD, SDHA, and SDHB pathogenic variants. However, according to the ClinVar database, this RET mutation was annotated as pathogenic variant associated with the MEN2 syndrome and its pathogenicity has not been proved by any functional studies. Thus, the role of the RET variant in the development of HNPGLs is controversial taking into account that we detected this variant in patients, who were not diagnosed with the MEN2 syndrome.
No pathogenic/likely pathogenic variants in SDHAF2, VHL, MAX, MEN2, NF1, FH, TMEM127, and SLC25A11 were identified among the Russian patients. Variants in all the genes were previously reported only as germline at a very low frequency in HNPGLs (Boedeker et al., 2009; Burnichon et al., 2012; Castro-Vega et al., 2014; Bausch et al., 2017; Buffet et al., 2018; Guha et al., 2019).
In the study, we also analyzed male to female ratio and age groups for identified variants in the SDHx and RET genes. Currently, clinic-genetic correlations in association with mutations in these genes in HNPGLs have been poorly investigated. Moreover, reported data are related to germline mutations and can be compared only conditionally with our results. Thus, Mario Hermsen with colleagues studied 23 males and 51 females and with HNPGLs and found 8:6 and 5:9 ratios for SDHB and SDHD mutations, respectively (Hermsen et al., 2010). Taking into account the initial male to female ratio approximately 1:2, we can see that SDHD mutations were detected about equally in males and females that is concordance with our data. SDHB mutations were more frequently revealed in males compared with females, while we obtained opposite results. In addition, it was observed that SDHB and SDHD mutations were diagnosed predominantly in patients aged <50 years. We showed that patients aged <61 years carried variants in these genes more frequently. Also, age-related penetrance for SDHB and SDHD mutations was shown to be increased by 50 years in HNPGLs (Neumann et al., 2004). In our study, we also observed higher number of patients aged 41–60 years with SDHB and SDHD variants.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA639937.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Vishnevsky Institute of Surgery. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AS and AVK: conceptualization and writing—review and editing. GK, VP, AD, AS, and NM: formal analysis. MF, EP, and ZG: investigation. GR, DK, and AG: resources. AP, GA, and ADK: writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Russian Science Foundation, Grant Number 19-15-00419.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Vishnevsky Institute of Surgery for sample collection and National Medical Research Center of Radiology for assistance in data analysis and discussing. The authors thank the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, EIMB RAS for providing the computing power and techniques for the data analysis. This work was performed using the equipment of EIMB RAS “Genome” center (http://www.eimb.ru/rus/ckp/ccu_genome_c.php).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.614908/full#supplementary-material
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Bausch, B., Schiavi, F., Ni, Y., Welander, J., Patocs, A., Ngeow, J., et al. (2017). Clinical Characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 3, 1204–1212. doi: 10.1001/jamaoncol.2017.0223
Boedeker, C. C., Erlic, Z., Richard, S., Kontny, U., Gimenez-Roqueplo, A. P., Cascon, A., et al. (2009). Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J. Clin. Endocrinol. Metab. 94, 1938–1944. doi: 10.1210/jc.2009-0354
Boedeker, C. C., Hensen, E. F., Neumann, H. P., Maier, W., Van Nederveen, F. H., Suarez, C., et al. (2014). Genetics of hereditary head and neck paragangliomas. Head Neck 36, 907–916. doi: 10.1002/hed.23436
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buffet, A., Morin, A., Castro-Vega, L. J., Habarou, F., Lussey-Lepoutre, C., Letouze, E., et al. (2018). Germline mutations in the mitochondrial 2-oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res. 78, 1914–1922. doi: 10.1158/0008-5472.CAN-17-2463
Burnichon, N., Briere, J. J., Libe, R., Vescovo, L., Riviere, J., Tissier, F., et al. (2010). SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 19, 3011–3020. doi: 10.1093/hmg/ddq206
Burnichon, N., Cascon, A., Schiavi, F., Morales, N. P., Comino-Mendez, I., Abermil, N., et al. (2012). MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin. Cancer Res. 18, 2828–2837. doi: 10.1158/1078-0432.CCR-12-0160
Castro-Vega, L. J., Buffet, A., De Cubas, A. A., Cascon, A., Menara, M., Khalifa, E., et al. (2014). Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 23, 2440–2446. doi: 10.1093/hmg/ddt639
Choi Jdo, W., Tucker, K. M., Lee, T. T., and Chong, G. C. (2014). Hereditary paraganglioma-pheochromocytoma syndromes associated with SDHD and RET mutations. Head Neck 36, E99–E102. doi: 10.1002/hed.23598
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. doi: 10.1093/bioinformatics/btv195
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi: 10.1101/gr.092619.109
Curras-Freixes, M., Inglada-Perez, L., Mancikova, V., Montero-Conde, C., Leton, R., Comino-Mendez, I., et al. (2015). Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J. Med. Genet. 52, 647–656. doi: 10.1136/jmedgenet-2015-103218
Dahia, P. L. (2014). Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat. Rev. Cancer 14, 108–119. doi: 10.1038/nrc3648
Ding, Y., Feng, Y., Wells, M., Huang, Z., and Chen, X. (2019). SDHx gene detection and clinical Phenotypic analysis of multiple paraganglioma in the head and neck. Laryngoscope 129, E67–E71. doi: 10.1002/lary.27509
Dong, C., Wei, P., Jian, X., Gibbs, R., Boerwinkle, E., Wang, K., et al. (2015). Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137. doi: 10.1093/hmg/ddu733
El-Naggar, A. K., Chan, J. K. C., Grandis, J. R., Takata, T., and Slootweg, P. J. (2017). WHO Classification of Head and Neck Tumours. Lyon: International Agency for Research on Cancer.
Glusman, G., Caballero, J., Mauldin, D. E., Hood, L., and Roach, J. C. (2011). Kaviar: an accessible system for testing SNV novelty. Bioinformatics 27, 3216–3217. doi: 10.1093/bioinformatics/btr540
Guha, A., Musil, Z., Vicha, A., Zelinka, T., Pacak, K., Astl, J., et al. (2019). A systematic review on the genetic analysis of paragangliomas: primarily focused on head and neck paragangliomas. Neoplasma 66, 671–680. doi: 10.4149/neo_2018_181208N933
Hermsen, M. A., Sevilla, M. A., Llorente, J. L., Weiss, M. M., Grimbergen, A., Allonca, E., et al. (2010). Relevance of germline mutation screening in both familial and sporadic head and neck paraganglioma for early diagnosis and clinical management. Cell Oncol. 32, 275–283. doi: 10.3233/CLO-2009-0498
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kircher, M., Witten, D. M., Jain, P., O'roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Kudryavtseva, A. V., Lukyanova, E. N., Kalinin, D. V., Zaretsky, A. R., Pokrovsky, A. V., Golovyuk, A. L., et al. (2019). Mutational load in carotid body tumor. BMC Med. Genomics 12:39. doi: 10.1186/s12920-019-0483-x
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. doi: 10.1093/nar/gkx1153
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2019). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360. doi: 10.1093/nar/gky1100
Neumann, H. P., Pawlu, C., Peczkowska, M., Bausch, B., Mcwhinney, S. R., Muresan, M., et al. (2004). Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292, 943–951. doi: 10.1001/jama.292.8.943
Pedulla, G., Crocetti, D., Paliotta, A., Tarallo, M. R., De Gori, A., Cavallaro, G., et al. (2014). Surgical treatment of pheochromocytoma in MEN 2. Ann. Ital. Chir. 85, 443–447.
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R., and Siepel, A. (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. doi: 10.1101/gr.097857.109
Quang, D., Chen, Y., and Xie, X. (2015). DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761–763. doi: 10.1093/bioinformatics/btu703
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi: 10.1093/nar/29.1.308
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. doi: 10.1101/gr.3715005
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947. doi: 10.1093/nar/gky1015
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M., and Ng, P. C. (2016). SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9. doi: 10.1038/nprot.2015.123
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Williams, M. D. (2017). Paragangliomas of the head and neck: an overview from diagnosis to genetics. Head Neck Pathol. 11, 278–287. doi: 10.1007/s12105-017-0803-4
Keywords: head and neck paragangliomas, carotid and vagal paragangliomas, susceptibility genes, SDHx, mutation frequency, pathogenic mutations
Citation: Snezhkina AV, Fedorova MS, Pavlov VS, Kalinin DV, Golovyuk AL, Pudova EA, Guvatova ZG, Melnikova NV, Dmitriev AA, Razmakhaev GS, Poloznikov AA, Alekseeva GS, Kaprin AD, Krasnov GS and Kudryavtseva AV (2020) Mutation Frequency in Main Susceptibility Genes Among Patients With Head and Neck Paragangliomas. Front. Genet. 11:614908. doi: 10.3389/fgene.2020.614908
Received: 07 October 2020; Accepted: 26 November 2020;
Published: 18 December 2020.
Edited by:
Yuriy L. Orlov, I.M. Sechenov First Moscow State Medical University, RussiaReviewed by:
Lyudmila Gulyaeva, Novosibirsk State University, RussiaAleksandr Khudiakov, Almazov National Medical Research Centre, Russia
Copyright © 2020 Snezhkina, Fedorova, Pavlov, Kalinin, Golovyuk, Pudova, Guvatova, Melnikova, Dmitriev, Razmakhaev, Poloznikov, Alekseeva, Kaprin, Krasnov and Kudryavtseva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anastasiya V. Snezhkina, bGVmdGdlckByYW1ibGVyLnJ1; Anna V. Kudryavtseva, cmhpemFtb2ViYUBtYWlsLnJ1