 Qiuming Hu1Huazhong Ma2Jiawei Shen3†Zongming Zhuang1,4Jianqiang Li4Xinlan Huang4Xian Li5*†
Qiuming Hu1Huazhong Ma2Jiawei Shen3†Zongming Zhuang1,4Jianqiang Li4Xinlan Huang4Xian Li5*† Haoyu Li1,4*†
Haoyu Li1,4*†- 1Department of Ophthalmology, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
- 2Department of Ophthalmology, Pingguo People's Hospital, Baise, China
- 3Department of Stomatology, The Affiliated Hospital of Medical School, Ningbo University, Ningbo, China
- 4Department of Retina and Vitreous, Jingliang Eye Hospital, Guangxi Medical University, Nanning, China
- 5Division of Pharmacy and Optometry, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, United Kingdom
Background: Waardenburg Syndrome Type 1 (WS1) is a rare hereditary disease, which is usually caused by the mutations of PAX3 (paired box 3). Here, we reported a pedigree with WS1, which was caused by a novel mutation in PAX3.
Case Report: In this present report, a 10-year-old boy and his twin sister from a Han Chinese family presented with iris pigmentary abnormality, synophrys, and broad and high nasal root. Their father presented premature whitening of the hair, but no iris pigmentary abnormality. Their aunts presented the same clinical characteristics with the twins and premature graying of hair. However, none of the patients reported hearing loss. The clinical diagnosis of the four patients from this pedigree was WS1. The whole exome sequencing (WES) revealed a novel mutation (c.959-5T>G) in the PAX3 gene, which could be responsible for the observed pathogenic of WS1 in this pedigree. The genetic test confirmed the diagnosis of WS1 in the four patients from the studied pedigree.
Conclusion: This present study demonstrated that genetic test based on WES, an effective alternative to regular clinical examinations, helps diagnose WS1. The newly identified PAX3 gene mutation can expand the understanding of WS1.
Introduction
Waardenburg Syndrome (WS), also named auditory-pigmentary syndrome, is one of the most common causes of syndromic deafness, contributing to 2–5% of congenital deafness cases. This syndrome is mainly caused by monogenetic variants that are mostly inherited through autosomal dominance with incomplete penetrance (Read and Newton, 1997). In China, WS patients account for about 1% of the deaf population (Chen et al., 2020). WS can be classified into four types (WS1, OMIM# 193500; WS2, OMIM# 193510; WS3, OMIM# 148820, and WS4, OMIM# 277580) according to different clinical characteristics, and WS1 and WS2 are the most common types (Pingault et al., 2010). The first WS case was described as a syndrome of a disorder combining anomalies of the eyelids, eyebrows, and nasal root with congenital deafness, which is now known as WS1 (Waardenburg, 1951). WS2 is similar to WS1, but hearing loss is more common in the former, and widely spaced eyes occur more common in the latter (Arias, 1971). WS3, also known as Klein-Waardenburg syndrome, often exhibits all the clinical features of WS1 and upper limb deformity. WS4, also known as Waardenburg-Hirschsprung disease, often exhibits all the clinical characteristics of WS2 in addition to megacolon or gastrointestinal atresia (Ma et al., 2019). WS is usually caused by mutations of the following six genes (Pingault et al., 2010): PAX3 (paired box 3), EDN3 (endothelin 3), EDNRB (endothelin receptor type B), MITF (microphthalmia-associated transcription factor), SOX10 (SRY Box10), and SNAI2 (snail homolog 2). PAX3 gene mutations are the common genetic causes of WS1 and WS3 (Li et al., 2020).
WS is often described as an autosomal dominantly inherited disorder of neural crest (NC) cells. PAX3 protein contains two highly conserved DNA binding domains (Apuzzo and Gros, 2007): a pairing domain (PD, Amino acids at position 33–160) and a homologous domain (HD, Amino acids at position 219-276). Genes, including PAX3, play a vital role in the development and differentiation of melanocytes derived from embryonic NC cells. The corresponding protein function will be affected, if a mutation occurs in the functional region, which will cause WS (Liu et al., 2020). Until now, over 150 mutations of the PAX3 gene associated with WS have been reported (Li et al., 2020), and most of them are located at exons 2–6 (Pingault et al., 2010). In this report, a novel mutation (c.959-5T>G) of the PAX3 gene was detected in a Chinese family with WS1. The discovery of this study may provide valuable information for genetic counseling of WS1 families.
Case Presentation
In the studied family, a 10-year-old boy (the proband, III-1), his father (II-1), aunts (II-8 and II-10), and twin sister (III-2) presented with iris pigmentary abnormality, synophrys, and broad and high nasal root (Figure 1). This studied family members were Han Chinese and had no history of medication use, infectious disease, severe constipation, blockage of the intestine, or abnormalities or limits of mobility in the limbs. The iris pigmentary abnormality, synophrys, and broad and high nasal root of the proband were observed, the iris color in his right eye was off-white, and the color in his left eye was brown (Figure 2). Like the proband, patient III-2 was with the same clinical features, the iris color in her left eye was off-white, and the color in her right eye was brown (Figure 2). Based on iris pigmentary abnormality, synophrys, and broad and high nasal root, both II-8 and II-10 had premature whitening of hair. However, there was only premature whitening of the hair but no iris pigmentary abnormality in II-1. Notably, none of the patients in this pedigree complained of hearing loss, and they refused pure-tone audiometry. The clinical features are summarized in Supplementary Table 1. The patients III-1 and III-2 received a comprehensive ophthalmological examination, which included comprehensive medical optometry, anterior segment photography, optical coherence tomography, and ultra-widefield laser scanning imaging. Except for abnormal iris color and fundus depigmentation in the corresponding eye, no other abnormalities or refractive errors were found (Figure 3).
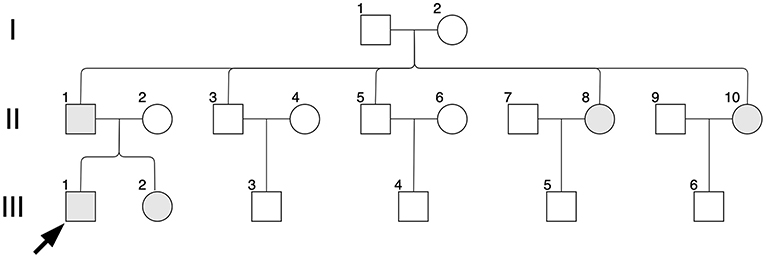
Figure 1. Pedigree diagram. Black arrow indicates the proband, II-1; shapes in gray indicate affected individuals, II-1, II-8, II-10, III-1 and III-2.
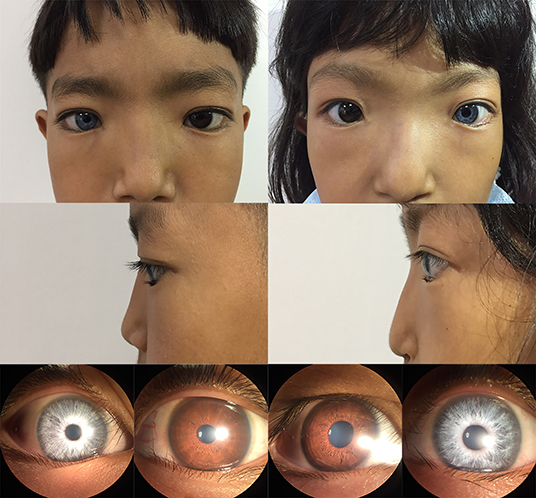
Figure 2. Photographic features of appearance and iris pigment in III-1 and III-2. Synophrys, and broad, and high nasal root without forehead white hair can be observed in III-1 (proband) and III-2. The iris color in the right eye of III-1 is off-white and the left eye is brown. The iris color in the right eye of III-2 is brown and the left eye is off-white.
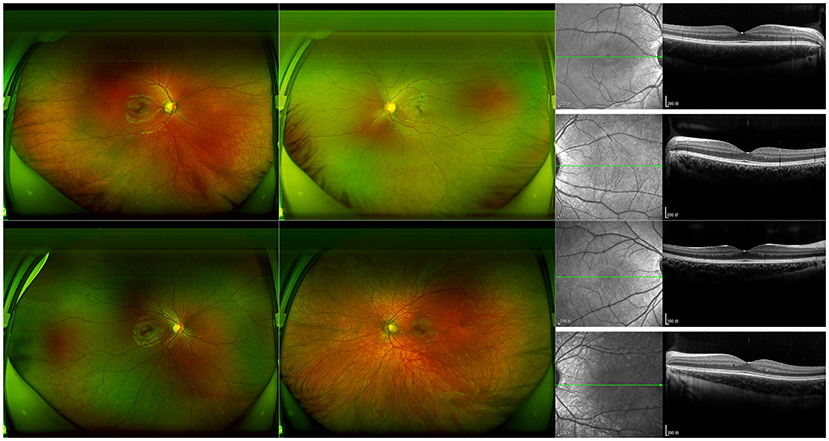
Figure 3. Photographic features of fundus in III-1 and III-2. Ultra-widefield laser scanning image and B-scan results of optical coherence tomography of III-1 (proband) and III-2 are shown. The hypopigmented fundus on the same side as the depigmentation of the iris can be observed.
In addition to clinical examinations, analysis of mutations via whole exome sequencing (WES) using peripheral blood was performed for II-1, II-2, II-8, II-10, III-1, and III-2, which was conducted by Aegicare Technology Co., Ltd. (Shenzhen, China). The DNA was extracted from the patients' blood sample, and 20,000 exons and the region about 20 bps upstream and downstream were sequenced. The reference genome GRCh37 was used for sequence alignment. The Aegicare's Weaver algorithm was used to detect copy number variations at the exon level. The sequencing depth was over 30 × average depth. Then the sequencing results were verified through Sanger sequencing. In this study, the most major mutation associated with the clinical features located at PAX3 (c.959-5T>G). The c.959-5T>G mutation was a novel mutation of NO. 959-5 nucleotide from thymine to guanine, which occurred in intron 6 of the PAX3 gene. Other mutations identified in this study included COL5A1 (c.698C>G, p.Ala233Gly), UBE3B (c.3086G>A, p.Arg1029His), WDR73 (c.935G>A, p.Arg312Gln) and ZNF469 (c.1663G>A, p.Asp555Asn and c.4910G>A, p.Arg1637Gln). The candidate variants are summarized in Table 1, and the results of Sanger sequencing are shown in Supplementary Figure 1. The identified mutation of the PAX3 gene was found in II-1, II-8, II-10, III-1, and III-2, while II-2 (the mother of the proband) had the wild-type PAX3. Therefore, the PAX3 mutation of the twins was from their father.
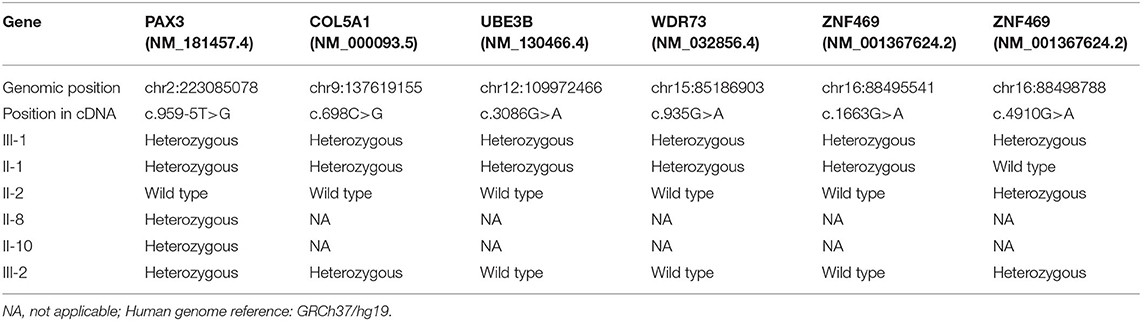
Table 1. Summary of the six candidate variants of five genes in the Waardenburg Syndrome Type 1 pedigree.
In addition to the previous publication, this PAX3 mutation was not recorded in gnomAD_exome, gnoAD_gnome, ExAc, or 1000 Genomes Project database. A series of prediction tools were used to evaluate the possible functional impacts of mutations in this study (Ng and Henikoff, 2003; Reva et al., 2007; Chun and Fay, 2009; Schwarz et al., 2010; Shihab et al., 2013; Choi and Chan, 2015). The results of the prediction are summarized in Supplementary Table 2. According to the prediction results of SpliceAI [DS_AL (acceptor loss) score = 0.5457] (Jaganathan et al., 2019), the detected mutation has a greater possibility of affecting the splicing. A DS_AL score >2 indicates a possibility of affecting the splicing; a score >5 indicates a possibility to cause a splicing-related disease. Splice variants frequently give rise to alternative splicing and affect protein coding. Consistent with the results, the mutations identified in Ehlers-Danlos-syndrome-related gene COL5A1 (Tuna et al., 2019; Angwin et al., 2020) and Kaufman-Oculocerebrofacial-syndrome-related gene UBE3B (Cheon et al., 2019; Ambrozkiewicz et al., 2020) also had a higher risk of disease. However, no results of the PAX3 mutation were available through the prediction tools. The American College of Medical Genetics and Genomics (ACMG) guidelines were used for the interpretation of variants (Richards et al., 2015).
Discussion
Diagnosis of WS is often established by clinical features. For WS1, the diagnosis requires two major criteria or one major plus two minor criteria (Farrer et al., 1992; Saleem, 2019). There are five major criteria: (1) congenital sensorineural hearing loss; (2) white forelock; (3) abnormal iris pigment; (4) dystopia canthorum; and (5) affected first-degree relative. The five minor criteria are (1) cutaneous hypopigmentation; (2) synophrys or medial eyebrow flare; (3) broad/high nasal root or low columella; (4) hypoplastic nasal alae; and (5) premature gray hair. In this studied family, all the patients met the criteria of WS1 diagnosis, although they presented different clinical characteristics. According to previous research, congenital sensorineural hearing loss is the most frequent clinical feature of WS1 patients (Oysu et al., 2000); however, none of the patients in this family presented this feature. In addition to hearing loss, dystopia canthorum is still considered the most reliable part for WS1, although it is a controversial diagnostic criterion (Sun et al., 2016; Minami et al., 2019). According to the Waardenburg consortium, dystopia canthorum should be evaluated by the W index. In this study, all the patients could be diagnosed with WS1 without considering this unagreeable indicator, the W index. Consistent with the previous research (Shields et al., 2013), the ocular features of III-1 and III-2 were described in this report that abnormal iris color and fundus depigmentation in the corresponding eye.
The sensorineural hearing loss is one of the typical phenotypes of WS1; however, none of the patients in this family complained of hearing loss. The rough hearing test also showed no abnormalities. Regretful, they refused the pure-tone audiometry. We deduced that the patients' hearing is generally normal, or the damage is too slight to detect.
The melanocyte, one of the cells affected by WS1, is derived from the NC. The NC can produce a series of cells and tissues, including melanocytes, neurons, the enteric nervous system, the facial skeleton, and other structures (Mica et al., 2013). Melanocytes exist in human skin, eyes, and cochlea. Melanocytes in the epidermis and iris contribute to skin and eye color variation, respectively (Lin and Fisher, 2007; Shields et al., 2013); melanocytes in the stria vascularis of the inner ear contribute to normal hearing (Tachibana, 2001). The biological activities of melanocytes are mediated by a series of genes, including MITF, PAX3, and other genes (Tachibana et al., 1996). MITF protein is essential for the survival and function of melanocytes, while transcription factors, such as PAX3, regulate MITF expression via extracellular signaling (Hou and Pavan, 2008). Additionally, PAX3 is broadly expressed in several other lineages of NC cells, and early expression of PAX3 is critical for developing melanocytes, craniofacial structure, and formation of the upper limbs (Wildhardt et al., 2013). Therefore, it is not difficult to understand that the abnormal expression of MITF or its regulatory genes can lead to WS characterized by depigmentation. In this study, the PAX3 gene mutation (c.959-5T>G) was identified through WES and Sanger sequencing, and it was considered the cause of WS1.
In this study, several mutations were discovered. According to ACMG guidelines and clinical manifestations, mutations with lower risk or inconsistent characteristics were excluded. Consistent with the clinical phenotypes and genetic traits, the proband's PAX3 conversion came from his father and was inherited autosomal dominantly. However, this novel mutation of PAX3 was rare, and due to the low frequency of population database and the mutation was co-segregated with the disease in multiple family members, according to the ACMG guidelines, it was classified as a variant of uncertain significance (VUS). Further study is needed to verify our results.
Comprehensive management of patients is required. As WS1 is a high-risk indicator for hearing loss, hearing screening and auditory diagnostic assessments are needed. In addition to the auditory system, wearing contact lenses or sunglasses to relieve photophobia and appropriate integumentary system protection and treatments are necessary. A previous study indicated that the pathogenic PAX3 alleles might increase the risk of severe neural tube defects in the patients' offspring, associated with folate-response (Hart and Miriyala, 2017). Thus, daily folic acid supplementation is recommended to all childbearing age women (Saleem, 2019).
In conclusion, this present study reports a novel mutation, c.959-5T>G of the PAX3 gene in a Han Chinese family with WS1. Discovering and reporting novel WS1-associated mutations facilitate the analysis of correlations between WS genotypes and phenotypes, helping further genetic consultation and diagnosis. Besides, our results showed that WES is a useful approach for congenital disease diagnosis and is of great benefit to disease screening, genetic diagnosis, and counseling.
Data Availability Statement
The datasets for this report are not publicly available due to concerns regarding participant anonymity. The datasets generated for this study can be available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethics Committee of Jingliang Eye Hospital Affiliated to Guangxi Medical University. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HM, JL, XH, and ZZ cared for the patients and performed medical examinations. QH took the lead in writing the manuscript. HM, JS, XL, and HL revised the manuscript. XL and HL designed the present research and guided the entire essay. All authors reviewed the manuscript and provided critical feedback and agreed on the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to express their gratitude to the family for their participation in this study. The authors also sincerely thank Aegicare (Shenzhen) Technology Co., Ltd. (Shenzhen, China) for technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.609040/full#supplementary-material
Supplementary Figure 1. The c.959-5T>G mutation in the PAX3 gene, which is identified in III-1 (proband), II-1, II-8, II-10 and III-2. The wild type is identified in II-2.
References
Ambrozkiewicz, M. C., Borisova, E., Schwark, M., Ripamonti, S., Schaub, T., Smorodchenko, A., et al. (2020). The murine ortholog of Kaufman oculocerebrofacial syndrome protein Ube3b regulates synapse number by ubiquitinating Ppp3cc. Mol. Psychiatry. doi: 10.1038/s41380-020-0714-8. [Epub ahead of print].
Angwin, C., Brady, A. F., Pope, F. M., Vandersteen, A., Baker, D., Cheema, H., et al. (2020). Arterial complications in classical Ehlers-Danlos syndrome: a case series. J. Med. Genet. 57, 769–776. doi: 10.1136/jmedgenet-2019-106689
Apuzzo, S., and Gros, P. (2007). Cooperative interactions between the two DNA binding domains of Pax3: helix 2 of the paired domain is in the proximity of the amino terminus of the homeodomain. Biochemistry 46, 2984–2993. doi: 10.1021/bi062107q
Arias, S. (1971). Genetic heterogeneity in the Waardenburg syndrome. Birth Defects Orig. Artic. Ser. 7, 87–101.
Chen, S., Jin, Y., Xie, L., Xie, W., Xu, K., Qiu, Y., et al. (2020). A novel spontaneous mutation of the SOX10 gene associated with Waardenburg Syndrome Type II. Neural. Plast. 2020:9260807. doi: 10.1155/2020/9260807
Cheon, S., Kaur, K., Nijem, N., Tuncay, I. O., Kumar, P., Dean, M., et al. (2019). The ubiquitin ligase UBE3B, disrupted in intellectual disability and absent speech, regulates metabolic pathways by targeting BCKDK. Proc. Natl. Acad. Sci. U.S.A. 116, 3662–3667. doi: 10.1073/pnas.1818751116
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. doi: 10.1093/bioinformatics/btv195
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi: 10.1101/gr.092619.109
Farrer, L. A., Grundfast, K. M., Amos, J., Arnos, K. S., Asher, J. H. Jr., et al. (1992). Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS consortium. Am. J. Hum. Genet. 50, 902–913.
Hart, J., and Miriyala, K. (2017). Neural tube defects in Waardenburg syndrome: a case report and review of the literature. Am. J. Med. Genet. A 173, 2472–2477. doi: 10.1002/ajmg.a.38325
Hou, L., and Pavan, W. J. (2008). Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: do all roads lead to Mitf? Cell Res. 18, 1163–1176. doi: 10.1038/cr.2008.303
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell 176, 535–548 e524. doi: 10.1016/j.cell.2018.12.015
Li, S., Guo, M., Ruan, B., Liu, Y., Cui, X., Han, W., et al. (2020). A Novel PAX3 Mutation in a Chinese family with Waardenburg Syndrome Type 1. Genet. Test. Mol. Biomarkers 24, 249–255. doi: 10.1089/gtmb.2019.0231
Lin, J. Y., and Fisher, D. E. (2007). Melanocyte biology and skin pigmentation. Nature 445, 843–850. doi: 10.1038/nature05660
Liu, Q., Cheng, J., Lu, Y., Zhou, J., Wang, L., Yang, C., et al. (2020). The clinical and genetic research of Waardenburg syndrome type I and II in Chinese families. Int. J. Pediatr. Otorhinolaryngol. 130:109806. doi: 10.1016/j.ijporl.2019.109806
Ma, J., Lin, K., Jiang, H. C., Yang, Y., Zhang, Y., Yang, G., et al. (2019). A novel mutation of the PAX3 gene in a Chinese family with Waardenburg Syndrome Type I. Mol. Genet. Genomic Med. 7:e00798. doi: 10.1002/mgg3.798
Mica, Y., Lee, G., Chambers, S. M., Tomishima, M. J., and Studer, L. (2013). Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell. Rep. 3, 1140–1152. doi: 10.1016/j.celrep.2013.03.025
Minami, S. B., Nara, K., Mutai, H., Morimoto, N., Sakamoto, H., Takiguchi, T., et al. (2019). A clinical and genetic study of 16 Japanese families with Waardenburg syndrome. Gene 704, 86–90. doi: 10.1016/j.gene.2019.04.023
Ng, P. C., and Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Oysu, C., Baserer, N., and Tinaz, M. (2000). Audiometric manifestations of Waardenburg's syndrome. Ear Nose Throat. J. 79, 704–709. doi: 10.1177/014556130007900909
Pingault, V., Ente, D., Dastot-Le Moal, F., Goossens, M., Marlin, S., and Bondurand, N. (2010). Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 31, 391–406. doi: 10.1002/humu.21211
Read, A. P., and Newton, V. E. (1997). Waardenburg syndrome. J. Med. Genet. 34, 656–665. doi: 10.1136/jmg.34.8.656
Reva, B., Antipin, Y., and Sander, C. (2007). Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 8:R232. doi: 10.1186/gb-2007-8-11-r232
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Saleem, M. D. (2019). Biology of human melanocyte development, Piebaldism, and Waardenburg syndrome. Pediatr. Dermatol. 36, 72–84. doi: 10.1111/pde.13713
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Shields, C. L., Nickerson, S. J., Al-Dahmash, S., and Shields, J. A. (2013). Waardenburg Syndrome: iris and choroidal hypopigmentation: findings on anterior and posterior segment imaging. JAMA Ophthalmol. 131, 1167–1173. doi: 10.1001/jamaophthalmol.2013.4190
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. doi: 10.1002/humu.22225
Sun, L., Li, X., Shi, J., Pang, X., Hu, Y., Wang, X., et al. (2016). Molecular etiology and genotype-phenotype correlation of Chinese Han deaf patients with type I and type II Waardenburg Syndrome. Sci. Rep. 6:35498. doi: 10.1038/srep35498
Tachibana, M. (2001). Cochlear melanocytes and MITF signaling. J. Investig. Dermatol. Symp. Proc. 6, 95–98. doi: 10.1046/j.0022-202x.2001.00017.x
Tachibana, M., Takeda, K., Nobukuni, Y., Urabe, K., Long, J. E., Meyers, K. A., et al. (1996). Ectopic expression of MITF, a gene for Waardenburg syndrome type 2, converts fibroblasts to cells with melanocyte characteristics. Nat. Genet. 14, 50–54. doi: 10.1038/ng0996-50
Tuna, F., Doganlar, Z. B., Özdemir, H., Demirbag Kabayel, D., and Doganlar, O. (2019). Ehlers-Danlos syndrome-related genes and serum strontium, zinc, and lithium levels in generalized joint hypermobility: a case-control study. Connect. Tissue Res. 62, 215–225. doi: 10.1080/03008207.2019.1675648
Waardenburg, P. J. (1951). A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am. J. Hum. Genet. 3, 195–253.
Keywords: Waardenburg Syndrome Type 1, Heterochromia iridis, PAX3 gene, whole exome sequencing, case report
Citation: Hu Q, Ma H, Shen J, Zhuang Z, Li J, Huang X, Li X and Li H (2021) Case Report: A Novel PAX3 Mutation Associated With Waardenburg Syndrome Type 1. Front. Genet. 12:609040. doi: 10.3389/fgene.2021.609040
Received: 22 September 2020; Accepted: 12 February 2021;
Published: 04 March 2021.
Edited by:
Sascha Meyer, Saarland University Hospital, GermanyReviewed by:
Chiara Di Resta, Vita-Salute San Raffaele University, ItalyMonkol Lek, Yale University, United States
Copyright © 2021 Hu, Ma, Shen, Zhuang, Li, Huang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xian Li, eGlhbi5saS0yQHBvc3RncmFkLm1hbmNoZXN0ZXIuYWMudWs=; Haoyu Li, bWVkLmRyLmxpaHlAZ21haWwuY29t
†ORCID: Jiawei Shen orcid.org/0000-0002-8780-2553
Xian Li orcid.org/0000-0001-8410-1586
Haoyu Li orcid.org/0000-0002-9826-181X