 Francisca Diaz-Gonzalez1,2
Francisca Diaz-Gonzalez1,2 Javier M. Sacedo-Gutiérrez2,3
Javier M. Sacedo-Gutiérrez2,3 Stephen R. F. Twigg4Eduardo Calpena4
Stephen R. F. Twigg4Eduardo Calpena4 Fernando E. Carceller-Benito2,3Manuel Parrón-Pajares2,5Fernando Santos-Simarro1,2,6
Fernando E. Carceller-Benito2,3Manuel Parrón-Pajares2,5Fernando Santos-Simarro1,2,6 Karen E. Heath1,2,6*
Karen E. Heath1,2,6*- 1Institute of Medical & Molecular Genetics (INGEMM), Hospital Universitario La Paz, Universidad Autónoma de Madrid, IdiPAZ, Madrid, Spain
- 2Skeletal Dysplasia Multidisciplinary Unit (UMDE) and ERN-BOND, Hospital Universitario La Paz, Madrid, Spain
- 3Department of Neurosurgery, Hospital Universitario la Paz, Universidad Autónoma de Madrid, IdiPAZ, Madrid, Spain
- 4Clinical Genetics Group, MRC Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom
- 5Department of Radiology, Hospital Universitario La Paz, Universidad Autónoma de Madrid, Madrid, Spain
- 6Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER, U753), Instituto Carlos III, Madrid, Spain
Introduction: Saethre-Chotzen syndrome, a craniosynostosis syndrome characterized by the premature closure of the coronal sutures, dysmorphic facial features and limb anomalies, is caused by haploinsufficiency of TWIST1. Although the majority of variants localize in the coding region of the gene, two variants in the 5′ UTR have been recently reported to generate novel upstream initiation codons.
Methods: Skeletal dysplasia Next-generation sequencing (NGS) panel was used for genetic analysis in a patient with bicoronal synostosis, facial dysmorphisms and limb anomalies. The variant pathogenicity was assessed by a luciferase reporter promoter assay.
Results: Here, we describe the identification of a third ATG-creating de novo variant, c.-18C>T, in the 5′ UTR of TWIST1 in the patient with a clinical diagnosis of Saethre-Chotzen syndrome. It was predicted to create an out-of-frame new upstream translation initiation codon resulting in a 40 amino acid larger functionally inactive protein. We performed luciferase reporter promoter assays to demonstrate that the variant does indeed reduce translation from the main open reading frame.
Conclusion: This is the third variant identified in this region and confirms the introduction of upstream ATGs in the 5′ UTR of TWIST1 as a pathogenic mechanism in Saethre-Chotzen syndrome. This case report shows the necessity for performing functional characterization of variants of unknown significance within national health services.
Introduction
Craniosynostosis syndromes encompass a large group of heterogeneous conditions characterized by the premature closure of one or more cranial sutures, which leads to abnormal skull shape and distinctive craniofacial deformities. Their combined estimated incidence is approximately one in 2100 live births (Wilkie et al., 2017). Six genes, FGFR2, FGFR3, TWIST1, EFBN1, TCF12 and ERF, account for approximately 84% of all genetically diagnosed cases (Paumard-Hernández et al., 2014; Wilkie et al., 2017; Hyder et al., 2021). Recently, a seventh gene has been added to this list, SMAD6 (Calpena et al., 2020).
Heterozygous loss-of-function variants in TWIST1 cause Saethre-Chotzen syndrome (SCS, MIM 101400), characterized by coronal suture synostosis, dysmorphic facial features, ptosis, hypertelorism and distal limb abnormalities such as brachydactyly or cutaneous syndactyly (El Ghouzzi et al., 1997; Howard et al., 1997).
TWIST1, located on chromosome 7p21.1 (Bourgeois et al., 1996), encodes a 202 amino acid class II basic helix-loop-helix (bHLH) transcription factor involved in calvarial development. TWIST1 plays an important role in the formation of the fronto-parietal boundary within the developing coronal suture (Merrill et al., 2006; Yen et al., 2010), and exerts an anti-osteogenic function preventing its premature closure (Yousfi et al., 2001; Bialek et al., 2004; Ting et al., 2009; Teng et al., 2018).
The major underlying pathogenic mechanism responsible for the craniofacial and limb anomalies observed in SCS patients is haploinsufficiency, usually caused by heterozygous variants affecting the coding region of the gene or structural variants, such as inversions or translocations encompassing TWIST1 regulatory elements, leading to functional loss of one gene copy (Hyder et al., 2021; Hirsch et al., 2022).
However, two pathogenic variants, in the 5′ untranslated region (5′ UTR) of TWIST1 have been recently reported in two patients with SCS (Zhou et al., 2018). Both were shown to create novel upstream initiation sites and a reduction in translation of TWIST1. Here, we report the clinical, genetics and functional characterization of a third variant in the 5′ UTR of TWIST1, in a patient with bicoronal craniosynostosis.
Patient and methods
Case report
The female proband, the second child of non-consanguineous healthy parents, was referred at 6 months of age from the neurosurgery department for molecular studies of craniosynostosis. She was born at term weighing 2500 g and craniosynostosis was noted. At 3 months old, cranial computed tomography (CT), performed prior to craniofacial surgery, revealed premature closure of both coronal sutures leading to an abnormal, brachycephalic head shape, large opening along the sagittal suture with a large anterior and posterior fontanel and several intra-sutural bones. An inter-parietal or Inca bone was also evident along with parietal and occipital foramina (Figure 1A). Physical examination showed frontal bossing, midface hypoplasia, mild pectus excavatum and slightly widened thumbs. No other skeletal anomalies were observed and neurological assessment was normal. No relevant familiar clinical history was reported. A clinical suspicion of Muenke syndrome (MIM 602849) or Saethre-Chotzen syndrome (MIM 101400) was suggested. The child underwent successful cranial surgery at 4 and 11 months old. The timeline of the case is described in supp table 1.
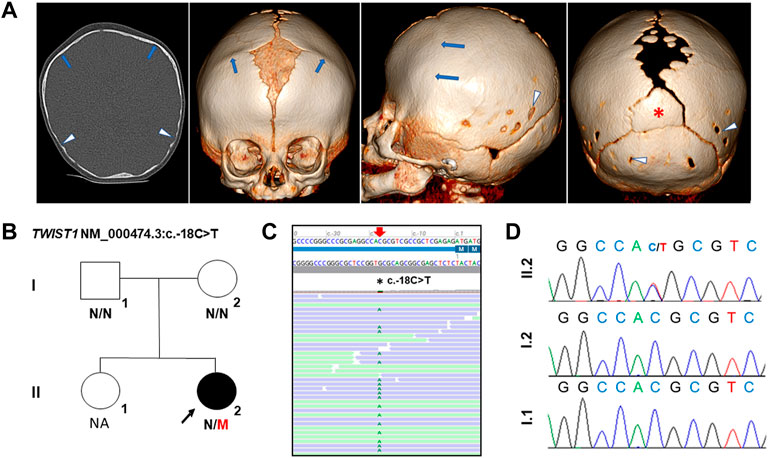
FIGURE 1. Clinical and genetic analysis of the proband with SCS. (A) Preoperative cranial CT images of the proband, age 3 months. From left to right: Frontal, lateral and axial images. Premature closure of both coronal sutures (blue arrows) is evident which lead to a brachycephalic cranial shape. Large anterior fontanel and superior part of metopic suture are present. Posterior part of sagittal suture and posterior fontanel are also enlarged, with several intrasutural bones. Note the presence of an inter-parietal or Inca bone (red asterisk). Posterior parietal bones and squamous part of occipital bone show thinning, parietal and occipital foramina (white arrow heads). (B) Pedigree of family. M, mutant; N, normal. (C) Alignment of NGS sequences of the proband highlighting the c.-18C>T variant. NGS panel: Average read depth 760X, 99.06% of bases >20X coverage and the variant reads 66:61 (reference:alternative). (D) Chromatograms showing the de novo TWIST1 5′ UTR variant, NM_000474.3:c.-18C>T in the proband. NA: Not available for genetic analysis.
Genetic analysis
The performed studies were reviewed and approved by the Hospital La Paz Ethical Committee (PI-5107). The participants provided their written informed consent to participate in this study.
Molecular screening of the proband was performed by a custom-designed NGS skeletal dysplasia gene panel (SKELETALSEQ.V11, n = 439 genes) using SeqCap EZ technology (Roche Nimblegen, Madison, WI, United States) and sequenced on a HiSeq platform (Illumina, San Diego, CA, United States). Analysis and variant filtering were performed as previously described (Sentchordi-Montané et al., 2021). The identified variant was subsequently validated by Sanger sequencing along with parental segregation.
In silico prediction analysis of translation initiation site
Three online prediction tools: ATGpr (https://atgpr.dbcls.jp/), DNA functional site miner (DNAFSMiner, https://dnafsminer.bic.nus.edu.sg/) and NetStart (https://www.cbs.dtu.dk/services/NetStart/) were used to evaluate the potential initiation activity of the identified variant.
Functional characterization of TWIST1 5′ UTR variants
Plasmid constructions
The psiCHECK-2 empty vector (EV), as well as the human TWIST1 5′ UTR wild type (psiCHECK2-TWIST1-5′UTR-WT) and TWIST1 c.-263C>A positive control mutant (psiCHECK2-TWIST1-c.-263C>A) were as previously described (Zhou et al., 2018; Calvo et al., 2018). The c.-18C>T mutant was generated by site-directed mutagenesis of the WT vector following the manufacturer’s indications (QuikChange II Site-Directed Mutagenesis Kit, Agilent technologies). Mutagenesis primers (mutated base underlined) were 5′- GCCCCGGGCCCGCGAGGCCATGCGTCGCCGCTCGAGAGATG - 3' (forward) and the other being the reverse complement. Incorporation of the variant into the construct was verified by Sanger sequencing.
Cell culture, transient transfection and luciferase reporter assays
HEK293 cells were maintained in 1X Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco, Life technologies, CA, United States) at 37°C and 5% CO2. Twenty-4 hours prior to transfection, cells were seeded in 12-well plates at 2 x 105 cells/well, and 2 µg of vector was transfected using FuGENE 6 (Promega, Madison, WI) at a 1:2 ratio of cDNA to FuGENE 6 following the manufacturer’s instructions.
HEK293 cells were transiently transfected either with the EV, the WT, the c.-18C>T or the c.-263C>A mutant as a positive control. Twenty-4 hours after transfection, cells were collected, lysed and the Renilla and Firefly luciferase activities were measured with the Dual-Luciferase® Reporter assay (Promega, Madison, WI, United States) according to the manufacturer’s protocol. Each construct was transfected in triplicate, measured in triplicate and a total of three biological replicates were performed. Data are presented as percentages relative to WT and statistical analysis was performed by Student’s t-test with values considered significantly different at p < 0.05.
Results
Genetic and molecular findings
A heterozygous variant located in the 5′ UTR of TWIST1 was identified in the proband: NM_000474.3:c.-18C>T p.? but absent from the parents (Figures 1B,C) (ClinVar SCV002589128). This variant is absent from large population databases (gnomAD, https://gnomad.broadinstitute.org) and has not been previously reported (HGMD Professional, Qiagen, CA, United States); ClinVar, www.ncbi.nlm.nih.gov/clinvar/LOVD, www.lovd.nl). The variant was classified as a variant of unknown significance (VUS) according to the recommendations of the American College of Medical Genetics and Genomics (Richards et al., 2015).
In silico analysis of the c.-18C>T variant
The variant creates an ATG upstream the main open reading frame (mORF), so we evaluated whether it could create an alternative translation initiation codon by firstly determining if it alters the Kozak consensus sequence (Kozak, 1986). A purine (G/A) at -3 from the ATG is considered functionally critical and a G at +4 is preferred. The sequence flanking the c.-18C>T variant indeed has a G at -3 but not a G at +4 (Figure 2B). Three online tools: ATGpr, DNAFSMiner and NetStart, predicted the variant to create an alternative out-of-frame initiation site upstream of the TWIST1 mORF (Figure 2B). Furthermore, if this aberrant transcript is translated then it would generate a mutant protein of 242 amino acids in size, entirely out of frame and using a stop codon 120bp downstream of the wildtype stop codon (ATPgr, https://atgpr.dbcls.jp/).
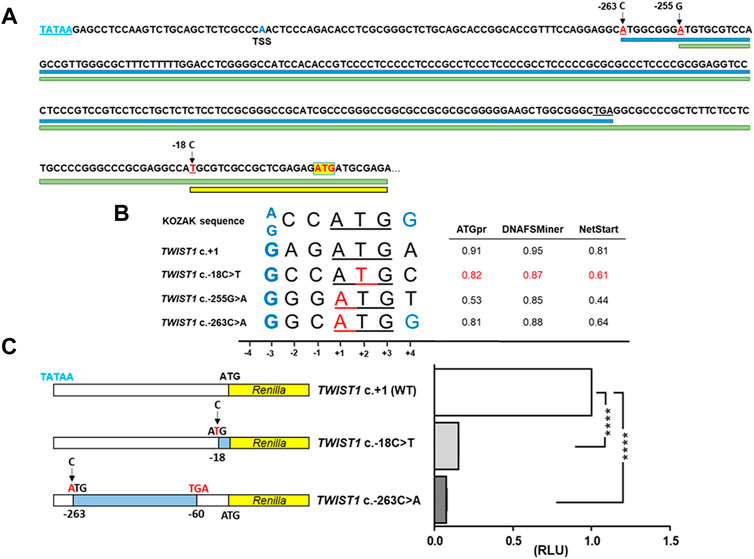
FIGURE 2. Effect on translation of the TWIST1 5′ UTR variant, c.-18C>T. (A) Genomic sequence showing the three upstream ATG-generating variants reported here and previously (mutated bases marked in red). Main wildtype TWIST1 start codon (ATG) is indicated by a yellow box. Blue, green and yellow lines indicate the ORFs generated from the ATG sites of the c.-263, c.-255 and c.-18 variants respectively. TATAA box (blue letters) and transcription start site (TSS) are indicated (B) Alignment of the Kozak consensus sequence with the ATGs of the variant c.-18C>T, the TWIST1 main start codon (+1) and the c.-255G>A and c.-263C>A variants. Mutated positions indicated in red. The important purine (G/A) at -3 from the ATG and a G at +4 are considered functionally critical (denoted in blue). In the right panel, evaluation of the translation initiation strength by ATGpr, DNAFSminer and NetStart, of the newly created upstream ATGs for the c.-18C>T variant (scores in red) compared to the WT sequence and the previously described positive controls. (C) Luciferase reporter DNA constructs psiCHECK2-TWIST1-5′UTR-WT, c.-18C>T and c.-263C>A positive control (left panel). The c.-263G>A variant creates an ORF of 68 codons that ends at a highly conserved stop codon at position c.-60 from the mORF (blue shading). The normalized luciferase reporter assay results of the WT, c.-18C>T mutant and the c.-263C>T positive control mutant are shown in the right panel. Each construct was transfected in triplicate, measured in triplicate in the luciferase assay and a total of three biological replicates were performed. RLU: relative luciferase units. The c.-18C>T mutant showed significantly reduced levels of Renilla activity (RLU: relative luciferase units) compared to WT (0.1537 ± 0.0010), similar to that observed with the c.-263C>A mutant (0.0759 ± 0.0041). RLU were significantly reduced (p < 0.0001) for both mutants compared to the WT.
Functional analysis of c.-18C>T variant on the translation of TWIST1 mORF
To test whether the TWIST1 c.-18C>T variant affects protein translation from the mORF, we performed a dual-luciferase reporter assay in HEK293 cells transiently transfected with a construct containing the 5′ UTR of TWIST1. We compared luciferase activities for WT, c.-18C>T or the previously reported variant, c.-263C>A, as a positive control (Figure 2C). The c.-18C>T mutant showed significantly reduced levels of Renilla activity compared to WT (0.1537 ± 0.0010), similar to that observed with the c.-263C>A mutant (0.0759 ± 0.0041 Figure 2C).
Discussion
SCS is one of the most common craniosynostosis syndromes. Although the vast majority of SCS causing variants are located within the coding region of TWIST1 (Zhou et al., 2018), two pathogenic variants, c.-263C>A and c.-255G>A, have been identified in the non-coding regulatory region in the 5′ UTR of TWIST1, located in exon 1. Both variants create an alternative upstream ATG that affects translation of the TWIST1 mORF (Zhou et al., 2018).
Here, we report a third pathogenic variant in the 5′ UTR of TWIST1 (c.-18C>T), very close to the main start site, which is predicted to create an out-of-frame upstream ATG codon. The de novo variant was identified in a proband with SCS, who presented with bilateral coronal synostosis with large fontanels, intrasutural bones, and extensive parietal and occipital foramina (Figure 1A).
The context and the position of the ATG play a key role in the initiation of translation (Kozak, 1981). In eukaryotes, the flanking sequence of the ATG initiation codons has a conserved pattern of nucleotides known as the Kozak consensus (A/GCCATGG) (Kozak, 1986). Thus, the efficiency of an alternative upstream ATG to serve as translation initiation codon is influenced by the strength of the Kozak consensus. The optimal context requires a purine (A/G) at position -3 and a guanine at +4 is also preferable (Kozak, 1986) (Figure 2B). All three variants have a guanine at -3 of the Kozak sequence, whilst only one of the three, c.-263C>A has a guanine at +4. In silico analysis suggested that the c.-18C>T variant could act as potential competitor with the main ATG start site similarly to the c.-263C>A control mutant (Figure 2B).
The presence of an upstream ATG codon might reduce or abolish translation from the downstream main initiation site (Lomedico and McAndrew, 1982) so we tested whether the variant could reduce TWIST1 translation by comparing the luciferase reporter activity in HEK293 cells transfected with either the c.-18C>T variant, WT or the previously reported variant c.-263C>A as a positive control (Zhou et al., 2018). We observed that Renilla luciferase activity was significantly reduced for the c-18C>T mutant compared to WT, similar to that observed for the positive control (>80% and >85%, respectively) (Figure 2C).
Previously, it was shown that the length of the upstream ORF and the distance between its stop codon and the wild type start initiation site are important for the repressive effect on translation of TWIST1 (Zhou et al., 2018). Translation of the majority of eukaryotic mRNAs occurs by binding of the 43S preinitiation complex to the 5′ cap, and then scanning along the leader sequence in search for the first AUG codon available. Upstream ATGs and ORFs can also influence ribosome scanning efficiency, and thus modulate the translation levels of the main ORF (Iacono et al., 2005).
If translated, the c.-18C>T variant is predicted to result in an aberrant protein of 242 amino acids, while the c.-255G>A variant produces an 85 amino acid longer TWIST1 protein through the generation of an in-frame ATG. In contrast, the c.-263C>A variant creates an out-of-frame ORF which terminates in a stop codon located 59 bases upstream of the mORF. An in vitro transcription-translation assay and subsequent visualization of the protein products would determine if an aberrant protein is produced. If so, this aberrant mutant protein may be degraded or lack the dimerization and DNA binding capability.
With the three variants now identified to date in TWIST1, it is important that the entire 5′ UTR is screened for variants in patients with SCS, especially in genetically underdiagnosed SCS cases negative for coding or copy-number/structural variants. Although this region in TWIST1 is exonic, it is a region that may be excluded from analysis for various reasons: it is not necessarily included in customized panel designs, bioinformatic analysis often excludes these regions and finally it is poorly covered in exomes. Pathogenic variants in the 5′ UTR of two other craniosynostosis genes also create upstream ORF-creating variants, EFBN1 (Twigg et al., 2013; Romanelli Tavares et al., 2019) and more recently, SMAD6 (Calpena et al., 2020). Thus, variants within the 5′ UTR of genes, and especially in haploinsufficient genes, should be carefully assessed in patients in whom the molecular defect has not been identified. Similar cases may be increasingly identified with the implementation of whole genome sequencing in healthcare but functional studies will be necessary to confirm the pathogenicity of these upstream ATG generating variants.
Conclusion
We have identified a third upstream ORF-generating variant in the 5′ UTR of TWIST1 in a child with SCS, which reduces the translation efficiency of the TWIST1 mORF. The child now has a definitive clinical diagnosis which enables both appropriate clinical monitorization and genetic counselling. Thus, this case report shows the necessity for the creation of platforms/services for the functional characterization of variants of unknown significance within national health systems.
Data availability statement
The dataset for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the dataset should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Hospital Universitario La Paz. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
JS-G, FC-B, FS-S, and MP-P performed clinical and radiological examinations of the patient. FD-G, ST, EC, and KH conducted the experimental design. FD-G and KH analyzed and interpreted the data. FD-G and KH wrote the paper. All authors revised and edited the manuscript.
Funding
This work was supported from grants (SAF 2017-84646-R, PID 2020-116263RB-I00) from the Ministerio de Ciencia y Innovación (to KH). FD-G was a recipient of a FPU Ph.D. studentship from the Ministerio de Educación. EC was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme, and ST by the VTCT Foundation.
Acknowledgments
The authors are very grateful to the family for their participation in this study. We are thankful to the DNA extraction, NGS, Sanger sequencing and bioinformatic services in the INGEMM.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1089417/full#supplementary-material
References
Bialek, P., Kern, B., Yang, X., Schrock, M., Sosic, D., Hong, N., et al. (2004). A twist code determines the onset of osteoblast differentiation. Dev. Cell 6 (3), 423–435. doi:10.1016/s1534-5807(04)00058-9
Bourgeois, P., Stoetzel, C., Bolcato-Bellemin, A. L., Mattei, M. G., and Perrin-Schmitt, F. (1996). The human H-twist gene is located at 7p21 and encodes a B-HLH protein that is 96% similar to its murine M-twist counterpart. Mamm. Genome 7 (12), 915–917. doi:10.1007/s003359900269
Calpena, E., Cuellar, A., Bala, K., Swagemakers, S., Koelling, N., McGowan, S. J., et al. (2020). SMAD6 variants in craniosynostosis: Genotype and phenotype evaluation. Genet. Med. 22 (9), 1498–1506. doi:10.1038/s41436-020-0817-2
Calvo, S. E., Pagliarini, D. J., and Mootha, V. K. (2009). Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. U. S. A. 106 (18), 7507–7512. doi:10.1073/pnas.0810916106
El Ghouzzi, V., Le Merrer, M., Perrin-Schmitt, F., Lajeunie, E., Benit, P., Renier, D., et al. (1997). Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat. Genet. 15 (1), 42–46. doi:10.1038/ng0197-42
Hirsch, N., Dahan, I., D'haene, E., Avni, M., Vergult, S., Vidal-García, M., et al. (2022). HDAC9 structural variants disrupting TWIST1 transcriptional regulation lead to craniofacial and limb malformations. Genome Res. 32 (7), 1242–1253. doi:10.1101/gr.276196.121
Howard, T. D., Paznekas, W. A., Green, E. D., Chiang, L. C., Ma, N., Ortiz de Luna, R. I., et al. (1997). Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 15 (1), 36–41. doi:10.1038/ng0197-36
Hyder, Z., Calpena, E., Pei, Y., Tooze, R. S., Brittain, H., Twigg, S. R. F., et al. (2021). Evaluating the performance of a clinical genome sequencing program for diagnosis of rare genetic disease, seen through the lens of craniosynostosis. Genet. Med. 23 (12), 2360–2368. doi:10.1038/s41436-021-01297-5
Iacono, M., Mignone, F., and Pesole, G. (2005). uAUG and uORFs in human and rodent 5' untranslated mRNAs. Gene 349, 97–105. doi:10.1016/j.gene.2004.11.041
Johnson, D., and Wilkie, A. (2011). Craniosynostosis. Eur. J. Hum. Genet. 19 (4), 369–376. doi:10.1038/ejhg.2010.235
Kozak, M. (1986). Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44 (2), 283–292. doi:10.1016/0092-8674(86)90762-2
Kozak, M. (1981). Possible role of flanking nucleotides in recognition of the AUG initiator codon by eukaryotic ribosomes. Nucleic Acids Res. 9 (20), 5233–5252. doi:10.1093/nar/9.20.5233
Lomedico, P. T., and McAndrew, S. J. (1982). Eukaryotic ribosomes can recognize preproinsulin initiation codons irrespective of their position relative to the 5' end of mRNA. Nature 299 (5880), 221–226. doi:10.1038/299221a0
Merrill, A. E., Bochukova, E. G., Brugger, S. M., Ishii, M., Pilz, D. T., Wall, S. A., et al. (2006). Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum. Mol. Genet. 15 (8), 1319–1328. doi:10.1093/hmg/ddl052
Paumard-Hernández, B., Berges-Soria, J., Barroso, E., Rivera-Pedroza, C., Pérez-Carrizosa, V., Benito-Sanz, S., et al. (2015). Expanding the mutation spectrum in 182 Spanish probands with craniosynostosis: Identification and characterization of novel TCF12 variants. Eur. J. Hum. Genet. 23 (7), 907–914. doi:10.1038/ejhg.2014.205
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Romanelli Tavares, V. L., Kague, E., Musso, C. M., Alegria, T., Freitas, R. S., Bertola, D. R., et al. (2019). Craniofrontonasal syndrome caused by introduction of a novel uATG in the 5’ UTR of EFNB1. Mol. Syndromol. 10 (1-2), 40–47. doi:10.1159/000490635
Sentchordi-Montané, L., Benito-Sanz, S., Aza-Carmona, M., Díaz-González, F., Modamio-Høybjør, S., de la Torre, C., et al. (2021). High prevalence of variants in skeletal dysplasia associated genes in individuals with short stature and minor skeletal anomalies. Eur. J. Endocrinol. 185 (5), 691–705. doi:10.1530/EJE-21-0557
Teng, C. S., Ting, M. C., Farmer, D. T., Brockop, M., Maxson, R. E., and Crump, J. G. (2018). Altered bone growth dynamics prefigure craniosynostosis in a zebrafish model of Saethre-Chotzen syndrome. eLife 7, e37024. doi:10.7554/eLife.37024
Ting, M. C., Wu, N. L., Roybal, P. G., Sun, J., Liu, L., Yen, Y., et al. (2009). EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development 136 (5), 855–864. doi:10.1242/dev.028605
Twigg, S. R., Babbs, C., van den Elzen, M. E., Goriely, A., Taylor, S., McGowan, S. J., et al. (2013). Cellular interference in craniofrontonasal syndrome: Males mosaic for mutations in the X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum. Mol. Genet. 22 (8), 1654–1662. doi:10.1093/hmg/ddt015
Wilkie, A., Johnson, D., and Wall, S. A. (2017). Clinical genetics of craniosynostosis. Curr. Opin. Pediatr. 29 (6), 622–628. doi:10.1097/MOP.0000000000000542
Yen, H. Y., Ting, M. C., and Maxson, R. E. (2010). Jagged1 functions downstream of Twist1 in the specification of the coronal suture and the formation of a boundary between osteogenic and non-osteogenic cells. Dev. Biol. 347 (2), 258–270. doi:10.1016/j.ydbio.2010.08.010
Yousfi, M., Lasmoles, F., Lomri, A., Delannoy, P., and Marie, P. J. (2001). Increased bone formation and decreased osteocalcin expression induced by reduced Twist dosage in Saethre-Chotzen syndrome. J. Clin. Invest. 107 (9), 1153–1161. doi:10.1172/JCI11846
Keywords: craniosynostosis, Saethre-Chotzen syndrome, Twist1, 5′ UTR, genetics
Citation: Diaz-Gonzalez F, Sacedo-Gutiérrez JM, Twigg SRF, Calpena E, Carceller-Benito FE, Parrón-Pajares M, Santos-Simarro F and Heath KE (2023) Case report: A third variant in the 5′ UTR of TWIST1 creates a novel upstream translation initiation site in a child with Saethre-Chotzen syndrome. Front. Genet. 13:1089417. doi: 10.3389/fgene.2022.1089417
Received: 04 November 2022; Accepted: 05 December 2022;
Published: 04 January 2023.
Edited by:
Jared C. Roach, Institute for Systems Biology (ISB), United StatesReviewed by:
Igor Fijalkowski, Ghent University, BelgiumGretchen MacCarrick, Johns Hopkins Medicine, United States
Ann Corsi, The Catholic University of America, United States
Copyright © 2023 Diaz-Gonzalez, Sacedo-Gutiérrez, Twigg, Calpena, Carceller-Benito, Parrón-Pajares, Santos-Simarro and Heath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen E. Heath, a2FyZW4uaGVhdGhAc2FsdWQubWFkcmlkLm9yZw==