 Nannan Qian1†
Nannan Qian1† Taohua Wei2†
Taohua Wei2† Wenming Yang2,3*Jiuxiang Wang2Shijie Zhang2
Wenming Yang2,3*Jiuxiang Wang2Shijie Zhang2 Shan Jin2Wei Dong1,2Wenjie Hao1
Shan Jin2Wei Dong1,2Wenjie Hao1 Yue Yang1Ru Huang4
Yue Yang1Ru Huang4- 1Graduate School, Anhui University of Traditional Chinese Medicine, Hefei, China
- 2The First Affiliated Hospital of Anhui University of Traditional Chinese Medicine, Hefei, China
- 3Key Laboratory of Xin’an Medicine Ministry of Education, Hefei, China
- 4V-Medical Laboratory Co., Ltd, Hangzhou, China
Autosomal recessive cerebellar ataxia type 1 (ARCA-1), also known as autosomal recessive spinocerebellar ataxia type 8 (SCAR8), is caused by spectrin repeat containing nuclear envelope protein 1 (SYNE1) gene mutation. Nesprin-1, encoded by SYNE1, is widely expressed in various tissues, especially in the striated muscle and cerebellum. The destruction of Nesprin-1 is related to neuronal and neuromuscular lesions. It has been reported that SYNE1 gene variation is associated with Emery-Dreifuss muscular dystrophy type 4, arthrogryposis multiplex congenita, SCAR8, and dilated cardiomyopathy. The clinical manifestations of SCAR8 are mainly characterized by relatively pure cerebellar ataxia and may be accompanied by upper and/or lower motor neuron dysfunction. Some affected people may also display cerebellar cognitive affective syndrome. It is conventionally held that the age at the onset of SCAR8 is between 6 and 42 years (the median age is 17 years). Here, we report a pedigree with SCAR8 where the onset age in the proband is 48 years. This case report extends the genetic profile and clinical features of SCAR8. A new pathogenic site (c.7578del; p.S2526Sfs*8) located in SYNE1, which is the genetic cause of the patient, was identified via whole exome sequencing (WES).
Introduction
Hereditary ataxia is a group of highly heterogeneous genetic degenerative diseases characterized by chronic progressive ataxia. Its phenotype includs ataxic gait and uncoordinated eye movement, language and hand movement, and often involves pathological changes mainly affecting the spinal cord, cerebellum, and brainstem (Jayadev and Bird, 2013). Hereditary ataxia includes autosomal dominant ataxia, autosomal recessive ataxia, X-linked ataxia, and ataxia associated with mitochondrial diseases (Braga Neto et al., 2016). SCAR8 is a type of autosomal recessive ataxia, which is clinically characterized by mild cerebellar ataxia and may be accompanied by upper and/or lower motor neuron dysfunction. Some people may also experience cerebellar cognitive affective syndrome (Laforce et al., 2010). It is reported that the incidence of autosomal recessive ataxia is 0–7.2/100,000 (Ruano et al., 2014). Friedreich ataxia and ataxia-telangiectasia are the most common autosomal recessive ataxia, while other types such as SCAR8 are less common (Palau and Espinós, 2006). SYNE1, the gene responsible for SCAR8 first reported in French-Canadian patients in Quebec, Canada (Gros-Louis et al., 2007), was considered to be limited to a specific section of the French-Canadian population in a long time (Synofzik et al., 2019). With the development of high-throughput sequencing technology, genetic variations in SYNE1 have been reported worldwide (Gama et al., 2016; Algahtani et al., 2017; Peng et al., 2018; Swan et al., 2018; Arias et al., 2019; Duan et al., 2021). According to a multicenter study of SYNE1 mutations (Mademan et al., 2016), the age of onset is between 6 and 42 years (the median age is 17 years). Not all patients show the classical SYNE1 phenotype of pure cerebellar ataxia, but all exhibit ataxia and motor neuron disease, which involves upper and/or lower motor neuron dysfunction. Here, we report a Chinese SCAR8 patient carrying a new homozygous mutation (c.7578del; p.S2526Sfs*8) in SYNE1, which has just been observed for the first time. The patient showed late-onset at the age of 48, and the clinical symptoms were characterized by slowly progressive cerebellar ataxia accompanied by motor neuron dysfunction.
Methods
This study was approved by the Ethics Committee of The First Affiliated Hospital of Anhui University of Chinese Medicine (batch number: 2021AH-60). The patient and her relatives were informed and provided written consent for the study and the publication of this report.
Case Description
At the age of 48 years, a female patient began to experience unsteady gait and trouble walking up stairs after prolonged standing. The patient did not pay attention to the symptoms at the time and did not seek medical treatment. At the age of 53 years, the patient found herself a symptom of slurred speech. After 1 year she went to the local hospital where on admission CT showed cerebellar atrophy. The cause was not clear, so the patient was discharged after symptomatic treatment consisting of oral butylphthalide soft capsules and pyridostigmine bromide tablets. Despite adhering to outpatient follow-up, the patient’s symptoms did not improve. The patient continued having difficulty with the stairs, dizziness after prolonged standing, and occasional falls, so she was admitted to our hospital for seeking treatment. On admission, the patient demonstrated an ataxic gait, poor distance discrimination, abnormal finger-to-nose test, dysphagia, slurred speech, and negative nystagmus. The physical exam of the patient showed that she had a decreased muscle tone in the lower extremities with weak tendon reflex, unstable heel-knee-shin test, and dysdiadochokinesia. Moreover, during the Romberg’s test, the patient was unstable in the stage of closing eyes and opening eyes. Her evaluation on the Scale for the Assessment and Rating of Ataxia (SARA) demonstrated 15 points out of a total of 40. In addition, she got 19/30 on the Mini Mental State Exam (MMSE) and 14/30 on the Montreal Cognitive Assessment (MoCA), respectively (the patient was illiterate). The patient self-reported no family history of neurological disease, and the brain MRI showed severe cerebellar atrophy (Figure 1A). Her father had succumbed to cerebral infarction at the age of 70 and her mother had died due to lung cancer at the age of 65. The patient stated that her parents had a non-consanguineous marriage, and neither of her parents showed symptoms of cerebellar ataxia. The patient has a 58-year-old brother and a 51-year-old sister. Neither of them showed symptoms of cerebellar ataxia, as examined by the chief physician. The patient’s 33-years-old daughter and two granddaughters (7 and 5 years old, respectively) showed no symptoms of cerebellar ataxia, as ascertained after examination by the chief physician. The patient was examined for short tandem repeat expansion in the responsible genes for SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA10, SCA12, SCA17, and DRPLA, which are the most common subtypes of spinocerebellar ataxia (SCA) in China. The results showed no abnormalities (Supplementary Figure S1).
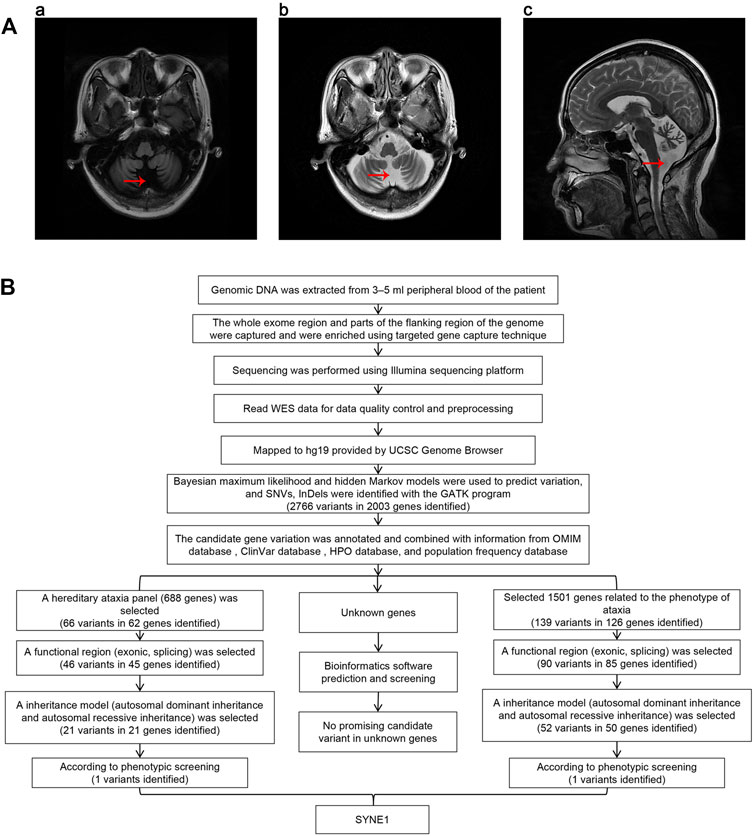
FIGURE 1. (A) Brain MRI of proband. The red arrows in the charts of “a,” “b,” and “c” show that the sulcus in both cerebellar hemispheres is significantly increased and widened, indicating cerebellar atrophy. (B) The workflow of analysis of whole exome sequencing data.
Whole-Exome Sequencing
Genomic DNA was extracted from 3–5 ml peripheral blood of the patient treated with the anticoagulant EDTA. All exonic regions and parts of their flanking intronic regions were captured and enriched by using Agilent SureSelect Human All Exon V6 Kit. The point mutations in exons, some flanking regions of selected genes, and insertion and deletion variations within 20 bp (small variation) were analyzed by using the Illumina sequencing platform (San Diego, California, United States). The average sequencing depth of this test was 210.56×, and the coverage rate of the target fragment reaching 20× was 97.74%. The Q30 score was greater than 90%. Data quality control and preprocessing were performed, followed by strict quality control of the sequencing data with removal of joint contamination and low-quality sequencing data to ensure the authenticity and reliability of the data. The high-quality sequencing data was compared with the human genome reference sequence (GRCh37/hg19) provided by the UCSC Genome Browser database (http://genome.ucsc.edu/) using the Burrows-Wheeler transform (BWT) algorithm. The coverage and the sequencing quality of the target region were evaluated at the same time. Bayesian maximum likelihood and hidden Markov models were used to predict variation, and single nucleotide variants (SNVs) and InDels (insertions and deletions) were identified using HaplotypeCalleralgorithm in GATK (http://www.broadinstitute.org/gsa/wiki/index.php/Home_Page). The candidate gene variants were annotated and combined with information from the Online Mendelian Inheritance in Man (OMIM) database (https://omim.org/), ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), Human Phenotype Ontology (HPO) database (https://hpo.jax.org/app/), and population frequency database (http://www.allelefrequencies.net/). Next, we chose two methods to screen candidate genes. One way is to screen candidates in a selected gene panel related to hereditary ataxia including 688 known genes (Supplementary Table S1). First, we identified 66 variants in 62 genes within this panel. Then functional regions (exonic, splicing) were selected for a following screening, and candidates were narrowed down to 46 variants in 45 genes. In third step, we selected variants according to their inheritance patterns, such as autosomal dominant inheritance, autosomal recessive inheritance and X-linked inheritance, and 21 variants in 21 genes were kept after this round screening. Finally, according to the patient’s clinical phenotypes, SYNE1 mutation was screened out among 21 variants. The other way is to screen 1,501 known genes related to the phenotype of ataxia in the databases (Supplementary Table S2). We could identify 139 variants in 126 genes after the first round screening. Then a functional regions (exonic, splicing) were selected for a following screening, and 90 variants in 85 genes were kept. In third round, we screened variants according to their inheritance patterns, candidates were narrowed down to 52 variants in 50 genes. Lastly, SYNE1 mutation was picked out among 52 variants according to the patient’s clinical phenotypes. In addition, we performed bioinformatics analysis and prediction for unknown genes as well. We selected variants with absent or frequency <1% in all the population databases, including dbSNP, gnomAD, ExAC, g1000 and ESP6500, Exac_all, Exac_eas. We used bioinformatics prediction software (SIFT, Polyphen-2, Mutation Taster, LR pred, Ljb23_metasvm, REVEL, dbscSNVada and dbscSNV_rf) to predict the pathogenicity of variation. However, we did not find any promising candidate variant (Figure 1B). The mutation separation were verified among family members by Sanger sequencing.
Results
The NM_033071:c.7578del (p.S2526Sfs*8) variant in SYNE1 was spotted by two screening methods. The result of Sanger sequencing confirmed the patient is a homozygous carrier of NM_033071:c.7578del (p.S2526Sfs*8) variant. In order to further clarify the genetic status within the family, the available family members (the patient’s brother, sister, daughter, and one of granddaughters) were tested for SYNE1 gene. The result showed that her brother, sister, and daughter carried one copy of the NM_033071:c.7578del variant in SYNE1, while her 5-year-old granddaughter (IV-2) did not have this variant (Figures 2A,B). Furthermore, no pathogenic variant was detected in responsible genes for SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA10, SCA12, SCA17 and DRPLA by filter screening using whole exome sequencing data in the patient (data not shown).
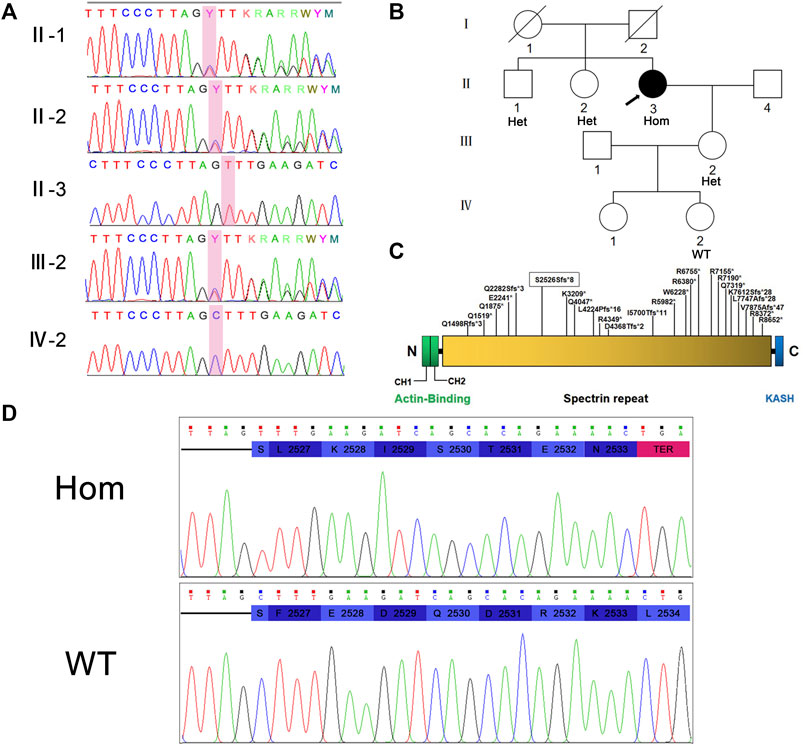
FIGURE 2. (A) Sanger sequencing peaks of family members. (B) Pedigree of family. II-3 is the proband. (C) Nesprin-1 and the location of previously reported truncated variants are illustrated. The variations found in this study are marked with boxes. (D) Translation pattern diagram. The mutation c.7578del (p.S2526Sfs*8) occurred in exon 51 of SYNE1 gene, leaving the serine at position 2,526 unchanged. The subsequent codon frameshift mutation resulted in the suspension of translation of 7 amino acids and the deletion of protein fragments (including KASH domain).
Hence, the mutation in exon 51 of SYNE1 was considered to be the genetic cause for the patient. This mutation did not affect the coding of the serine at position 2,526 per se, but a frameshift mutation occurred and changed the reading of subsequent codons. This resulted in transcription and translation errors which, in turn, affected protein function (PVS1) (null variant, including nonsense, frameshift, canonical+/−1 or 2 splice sites, initiation codon, single or multi-exon deletion in a gene where loss of function (LOF) is a known mechanism of disease). This mutation is not included in many population databases including 1,000 Genomes database, ExAC database, gnomAD database, Esp6500 database, HGMD database, and ClinVar database. The mutation does not belong to polymorphic loci, and the frequency of occurrence in the population is very low (PM2) (absent from controls, or at extremely low frequency if recessive in Exome Sequencing Project, 1,000 Genomes or ExAC). Based on the above evidence, the variant was classified as a likely pathogenic (ACMG criteria PVS1 and PM2) (Richards et al., 2015). The pathogenicity of SYNE1 gene mutation was predicted by using the online bioinformatics software LR pred, Mutation Taster, and Ljb23_metasvm, and the results revealed it as a disease-causing mutation.
Discussion
SYNE1, also known as nuclear envelope spectrin 1 (Nesprin-1), which is located at 6q25.2, is one of the largest genes in the human genome. It has 146 exons, encoding 27,652 base pairs of messenger RNA, translated into a protein of 8,797 amino acids, Nesprin-1. Nesprin-1 is widely expressed in various tissues, especially in striated muscles and the cerebellum (King et al., 2014). Nesprin-1 is located on the outer nuclear membrane and binds to F-actin through the N-terminal. Through the association between its C-terminal (Klarsicht-ANC-Syne-homology) KASH domain and the SUN domain of the perinuclear space SUN1/2, the linker of nucleoskeleton and cytoskeleton (LINC) complex is formed. The LINC complex connects the nuclear membrane to the cytoskeleton elements, which include actin filaments and actin microtubule networks (Nikolova-Krstevski et al., 2011). If the SYNE1 gene is mutated, the structure of Nesprin-1 protein will be changed, and in turn the stability, size, and shape of nucleus will also be affected. Thus the defected composition of the nuclear layer and the nuclear membranes will result in various degenerative diseases which affect the development of striated muscles, peripheral nerves, bones and fat, as well as premature aging syndrome (Sur et al., 2014). Drosophila larvae with the deletion of KASH domain exhibits motor disturbance of myasthenia, which proves the importance of KASH in structural dyskinesia phenotype. Further experiments showed that abnormal neuromuscular transmission and postsynaptic imbalance of glutamate receptors were observed at the neuromuscular junctions in Drosophila larvae with the deletion of KASH, and the percentage of receptors containing glutamate receptor type A (GluRIIA) decreased. The dyskinesia phenotype of KASH deletion was prevented by GluRIIA overexpression, which indicated that the motor disturbance associated with KASH domain deletion was caused by the decrease in GluRIIA expression (Morel et al., 2014).
It has been confirmed that SYNE1 gene variation is associated with Emery-Dreifuss muscular dystrophy type 4 (EDMD4) (OMIM#612998), myogenic-type arthrogryposis multiplex congenita-3 (AMC3) (OMIM#618484), and SCAR8 (OMIM#610743) (Gama et al., 2016; Indelicato et al., 2019; Noreau et al., 2013). In addition, it has been reported that SYNE1 gene mutation may be associated with dilated cardiomyopathy (DCM) (Zhao et al., 2021). Nesprin-1 binds emerin and lamins A/C to form a network in muscles, connecting nucleoskeleton to inner nuclear membrane (INM), outer nuclear membrane (ONM), membrane organelle, sarcomere, and actin to form cytoskeleton (Warren et al., 2005). In addition, the functional and structural abnormalities of nesprin/lamin/emerin interaction may play a role in the muscle-specific pathogenesis of Emery-Dreifuss muscular dystrophy type 4 (Zhang et al., 2007). The C-terminal heterozygous missense variant of SYNE1 gene has been identified to be associated with sporadic dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy type 4, and a patient with C-terminal truncation mutation of SYNE1 gene reportedly developed cardiomyopathy (Indelicato et al., 2019). Three novel C-terminal missense mutations were identified in 218 patients with sporadic dilated cardiomyopathy. The expression of these mutants disrupted the Nesprin-1/lamin/SUN interaction and led to nuclear morphological defects (Zhou et al., 2017). In another report, a nonsense homozygous mutation at the C-terminal in a patient with AMC was identified, resulting in truncation of the KASH domain. Interestingly, mRNA analysis showed that the transcript of the mutant was expressed at the same level of wild type (Baumann et al., 2017). We found that the truncation caused by previously reported SYNE1 mutations near the N-terminal of Nesprin-1 resulted in late-onset, while those near the C-terminal of Nesprin-1 resulted in early-onset (Figure 2C, Supplementary Figure S2, Supplementary Table S1). Nesprin-1 has multiple internal promoters which can produce shorter isoforms with a common C-terminal region, but truncated N-terminal, such as Nesprin-1-giant, Nesprin-1α2, Nesprin-1α1, Nesprin-1β2, Nesprin-1γ, p53 KASHNesp1, and p53 KASHNesp1 (Rajgor et al., 2012; Baumann et al., 2017). Some studies have shown that the dysfunction of a KASH-LESS Nesprin1 giant (KLNes1g) is the basis for pathogenesis of SCAR8, and it has been confirmed that KLNes1g is related to the function of vesicles and dendritic membrane (Razafsky and Hodzic, 2015). There are different distribution patterns of Nesprin-1 isoforms in INM and ONM. Based on the points above, we hypothesized when the truncation is close to the N-terminal of Nesprin-1, the influence of truncation mutation to the Nesprin-1 isoforms is less or it only affects the function of Nesprin-1giant and KLNes1g. Furthermore, the structural homologue Nesprin-2-giant has a certain compensation effect on the function of Nesprin-1giant (Razafsky and Hodzic, 2015); hence, the onset caused by their defect is later. On the contrary, when the truncation was close to the C-terminal of Nesprin-1, the mutation not only affected the function of Nesprin-1giant and KLNes1g, but also the function of all other Nesprin-1 isoforms with early-onset. Therefore, the hypothesis needs to be further studied. The short α isoforms of Nesprin, Nesprin-1α2 and Nesprin-2α2, are almost exclusively expressed in the heart and skeletal muscle (Apel et al., 2000). EDMD showed only cardiac and skeletal muscle involvement. All known Nesprin mutations associated with EDMD or DCM are located in the α isoform sequence (Duong et al., 2014). The tissue-specific distribution of α isoforms and the location of pathogenic SYNE1 mutations may indicate that α isoforms have some specific functions that are not available in the full-length “giant” form. The loss of this function may be the cause of EDMD and DCM. Furthermore, it can be inferred that the tissue-specific distribution of α isoform and the location of pathogenic SYNE1 mutation may be the reasons for the pleiotropy of SYNE1 gene mutation. However, further research is needed to address this issue in the future. The frameshift mutation c.7578del (p.S2526Sfs*8) of the SYNE1 gene we reported results in an early interruption in the process of Nesprin-1 translation. However, it may also lead to mRNA degradation through the nonsense-mediated mRNA decay (NMD) pathway (Kölbel et al., 2019), resulting in the inability of protein translation. Both mechanisms need to be verified via biological experiments in the future.
The typical manifestation of SCAR8 includes cerebellar syndromes, such as cerebellar ataxia, dysarthria, dyslexia, saccade, etc. Other features include upper motor neuron dysfunction (spasm, hyperreflexia, Babinski sign) and/or lower motor neuron dysfunction (muscular atrophy, impaired reflex, muscle bundle tremor). Some people may display cerebellar cognitive affective syndrome, that is, obvious attention deficit. Executive function, language working memory, and visual space/visual construction skills may also be affected (Beaudin et al., 1993; Algahtani et al., 2017). In this work, we report that the SYNE1 gene with a mutation of c.7578del (p.S2526Sfs*8) in exon 51 has an early-translation interruption in the Nesprin-1 translation process, resulting in a deletion of protein fragment including the KASH domain (Figure 2D). It was previously reported that the onset age of SCAR8 ranges from 6 to 42 years, and the median age is 17 years. The proband in this family displayed a late-onset slowly progressive cerebellar ataxia with dysfunction of lower motor neurons, had a 7-years history of progression, cerebellar atrophy, which were caused by carrying the homozygous SYNE1 mutation of NM_033071:c.7578del (p.S2526Sfs*8). It is interesting to note that there was no consanguineous marriage between the parents of the proband. But they came from the same geographical region, which indicates that they may originate from a common ancestor. However, uniparental disomy (UPD) cannot be ruled out as a possible mechanism. As the whole exome sequencing data of the proband is available, this could be a new hypothesis to investigate in the future. The proband’s brother and sister are heterozygous carriers and neither of them nor the deceased parents developed the same disease. Therefore, carrying the homozygous SYNE1 mutation is the main genetic cause of SCAR8 for this patient.
At present, the effective drug treatment in degenerative cerebellar diseases is very limited, and the treatment is confined to only specific type of diseases and symptoms (Ilg et al., 2014). Physiotherapy is the main treatment of ataxia currently (Synofzik and Ilg, 2014). The characteristics of ataxia gait include difficulties with inter- and intra-limb coordination, a slow walking speed, irregular steps and reduced postural stability (Ioffe et al., 2007). Studies have shown that a series of physiotherapy interventions have a positive effect on ataxia (Cassidy et al., 2018). The difficulty of inter- and intra-limb coordination is one of the main manifestations of cerebellar ataxia. Gait coordination is related to the function of paleocerbellum and neocerebellum. Gait coordination can be improved through exercises to improve postural stability, reduction of degrees of freedom (such as positioning, splinting), and providing external support (such as orthotics, walking aids) (Delatycki et al., 2005; Ilg et al., 2010; Serrao et al., 2017). In one study, investigators assessed the long-term effects of a 4-weeks intensive coordination training and subsequent 1-year continuation of training at home. The results showed that there was a trend towards lower SARA scores in patients with 4 weeks of training and 1 year of training compared to pre training. And patients walked longer distances unaided (Ilg et al., 2010). Patients with ataxia may experience reduced ability of adapting to environmental changes. The ability of adapting to environmental changes is mainly controlled by the neocerebellum. The ability of adapting to environmental changes can be improved through adaptive gait training. A study showed that 10 patients with degenerative cerebellar ataxia were trained in gait adaptation. Visual step targets and obstacles are projected on the surface of the treadmill running belt. After 10 times of training within 5 weeks (1 h per time), the success rate of patients avoiding obstacles has increased significantly (Fonteyn et al., 2014a). The motor learning ability of patients with cerebellar injury will also be impaired. Motor learning abilities were related to the neocerebellum. Stepwise prompts to learn and high levels of repetition exercises should be used (Kelly and Shanley, 2016). When patients experience a decrease in consolidation motor skills, they need to practice the necessary skills with high intensity and pay attention to walking exercises consciously (Im et al., 2017). These training sessions primarily improved neocerebellum function. Patients with reduced posture control and muscle tone can be given special strength and balance training as well as compensatory strategies to increase stability (Kelly and Shanley, 2016). These training sessions primarily improved paleocerebellum function. Physiotherapy usually combines balance, gait, coordination, strength, endurance and posture (Fonteyn et al., 2014b). Often, the combination of multiple training methods can improve ataxia (Miyai et al., 2012; Schroeder, 2013). Recently, several commercial video training games have been used to train patients with ataxia. Commercial video training games can provide a novel and beneficial treatment tool (Ilg et al., 2012; Synofzik et al., 2013; Schatton et al., 2017). In addition, exercises like Tai Chi and Chinese acupuncture treatment may have potential benefits for patients with ataxia (Winser et al., 2018; Wang et al., 2021).
Since the occurrence of SYNE1-related SCAR8 cases in Canada (Gros-Louis et al., 2007), up to September 2021, a total of 229 SYNE1 gene mutations have been reported according to the professional edition of HGMD database (Stenson et al., 2020). The frameshift mutation c.7578del (p.S2526Sfs*8) we reported is not mentioned in this database, and hence, is a new type of mutation. The symptoms of the patient reported here are similar to those of typical cerebellar ataxia previously reported, and are additionally accompanied by lower motor neuron dysfunction. More importantly, the onset age of this patient exceeds the onset age range previously reported, and our report expands the clinical-pathological features and gene profile in SCAR8.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of The First Affiliated Hospital of Anhui University of Chinese Medicine (batch number: 2021AH-60). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
YW designed the study. QN and WT interpreted the data and wrote the manuscript. ZS, JS, DW, HW, and YY conducted follow-up studies, and collected and evaluated clinical and genetic evidence. YW, WJ and HR modified the manuscript. The final draft was read and approved by all the authors.
Funding
This work is supported by the National Natural Science Foundation of China (No. 81973825), the evidence-based capacity Building Project of traditional Chinese Medicine of the State Administration of traditional Chinese Medicine: evidence-based capacity improvement and platform Construction of traditional Chinese Medicine Encephalopathy (No. 2019XZZX-NB001), and the Collaborative Innovation Project of Anhui Universities (No. GXXT-2020-81973825).
Conflict of Interest
RH was employed by V-Medical Laboratory Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank V-Medical Laboratory (Guangzhou) Co., Ltd for assistance with the experiments involved in this work. We also sincerely thank the patient and the family for their help in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.795188/full#supplementary-material
References
Algahtani, H., Marzouk, Y., Algahtani, R., Salman, S., and Shirah, B. (2017). Autosomal Recessive Cerebellar Ataxia Type 1 Mimicking Multiple Sclerosis: A Report of Two Siblings with a Novel Mutation in SYNE1 Gene in a Saudi Family. J. Neurol. Sci. 372, 97–100. doi:10.1016/j.jns.2016.11.036
Apel, E. D., Lewis, R. M., Grady, R. M., and Sanes, J. R. (2000). Syne-1, a Dystrophin- and Klarsicht-Related Protein Associated with Synaptic Nuclei at the Neuromuscular junction. J. Biol. Chem. 275 (41), 31986–31995. doi:10.1074/jbc.M004775200
Arias, M., Mir, P., Fernández-Matarrubia, M., Arpa, J., García-Ramos, R., Blanco-Arias, P., et al. (2019). Heredoataxia cerebelosa recesiva ARCA1/SCAR8: primeras familias detectadas en España. Neurología S0213-4853 (19), 30050–30057. doi:10.1016/j.nrl.2019.01.004
Baumann, M., Steichen-Gersdorf, E., Krabichler, B., Petersen, B.-S., Weber, U., Schmidt, W. M., et al. (2017). Homozygous SYNE1 Mutation Causes Congenital Onset of Muscular Weakness with Distal Arthrogryposis: A Genotype-Phenotype Correlation. Eur. J. Hum. Genet. 25, 262–266. doi:10.1038/ejhg.2016.144
Beaudin, M., Gamache, P., Gros-Louis, F., and Dupré, N. (1993). SYNE1 Deficiency (Seattle. Seattle (WA): University of Washington.
Braga Neto, P., Pedroso, J. L., Kuo, S.-H., Marcondes Junior, C. F., Teive, H. A. G., and Barsottini, O. G. P. (2016). Current Concepts in the Treatment of Hereditary Ataxias. Arq. Neuro-psiquiatr. 74, 244–252. doi:10.1590/0004-282X20160038
Cassidy, E., Naylor, S., and Reynolds, F. (2018). The Meanings of Physiotherapy and Exercise for People Living with Progressive Cerebellar Ataxia: An Interpretative Phenomenological Analysis. Disabil. Rehabil. 40, 894–904. doi:10.1080/09638288.2016.1277400
Delatycki, M. B., Holian, A., Corben, L., Rawicki, H. B., Blackburn, C., Hoare, B., et al. (2005). Surgery for Equinovarus Deformity in Friedreich's Ataxia Improves Mobility and Independence. Clin. Orthop. Relat. Res. 430, 138–141. doi:10.1097/01.blo.0000150339.74041.0e
Duan, X., Hao, Y., Cao, Z., Zhou, C., Zhang, J., Wang, R., et al. (2021). Autosomal Recessive Cerebellar Ataxia Type 1: Phenotypic and Genetic Correlation in a Cohort of Chinese Patients with SYNE1 Variants. Cerebellum 20, 74–82. doi:10.1007/s12311-020-01186-8
Duong, N. T., Morris, G. E., Lam, L. T., Zhang, Q., Sewry, C. A., Shanahan, C. M., et al. (2014). Nesprins: Tissue-specific Expression of Epsilon and Other Short Isoforms. PLOS ONE 9, e94380. doi:10.1371/journal.pone.0094380
Fonteyn, E. M. R., Heeren, A., Engels, J.-J. C., Boer, J. J. D., van de Warrenburg, B. P. C., and Weerdesteyn, V. (2014a). Gait Adaptability Training Improves Obstacle Avoidance and Dynamic Stability in Patients with Cerebellar Degeneration. Gait & Posture 40, 247–251. doi:10.1016/j.gaitpost.2014.04.190
Fonteyn, E. M. R., Keus, S. H. J., Verstappen, C. C. P., Schöls, L., de Groot, I. J. M., and van de Warrenburg, B. P. C. (2014b). The Effectiveness of Allied Health Care in Patients with Ataxia: A Systematic Review. J. Neurol. 261, 251–258. doi:10.1007/s00415-013-6910-6
Gama, M. T. D., Houle, G., Noreau, A., Dionne-Laporte, A., Dion, P. A., Rouleau, G. A., et al. (2016). SYNE1 Mutations Cause Autosomal-Recessive Ataxia with Retained Reflexes in Brazilian Patients. Mov Disord. 31, 1754–1756. doi:10.1002/mds.26810
Gros-Louis, F., Dupré, N., Dion, P., Fox, M. A., Laurent, S., Verreault, S., et al. (2007). Mutations in SYNE1 lead to a Newly Discovered Form of Autosomal Recessive Cerebellar Ataxia. Nat. Genet. 39, 80–85. doi:10.1038/ng1927
Ilg, W., Bastian, A. J., Boesch, S., Burciu, R. G., Celnik, P., Claaßen, J., et al. (2014). Consensus Paper: Management of Degenerative Cerebellar Disorders. Cerebellum 13, 248–268. doi:10.1007/s12311-013-0531-6
Ilg, W., Brötz, D., Burkard, S., Giese, M. A., Schöls, L., and Synofzik, M. (2010). Long-term Effects of Coordinative Training in Degenerative Cerebellar Disease. Mov. Disord. 25, 2239–2246. doi:10.1002/mds.23222
Ilg, W., Schatton, C., Schicks, J., Giese, M. A., Schöls, L., and Synofzik, M. (2012). Video Game-Based Coordinative Training Improves Ataxia in Children with Degenerative Ataxia. Neurology 79, 2056–2060. doi:10.1212/WNL.0b013e3182749e67
Im, S.-J., Kim, Y.-H., Kim, K.-H., Han, J.-W., Yoon, S.-J., and Park, J.-H. (2017). The Effect of a Task-specific Locomotor Training Strategy on Gait Stability in Patients with Cerebellar Disease: A Feasibility Study. Disabil. Rehabil. 39, 1002–1008. doi:10.1080/09638288.2016.1177124
Indelicato, E., Nachbauer, W., Fauth, C., Krabichler, B., Schossig, A., Eigentler, A., et al. (2019). SYNE1-ataxia: Novel Genotypic and Phenotypic Findings. Parkinsonism Relat. Disord. 62, 210–214. doi:10.1016/j.parkreldis.2018.12.007
Ioffe, M. E., Chernikova, L. A., and Ustinova, K. I. (2007). Role of Cerebellum in Learning Postural Tasks. Mcer 6, 87–94. doi:10.1080/14734220701216440
Jayadev, S., and Bird, T. D. (2013). Hereditary Ataxias: Overview. Genet. Med. 15, 673–683. doi:10.1038/gim.2013.28
Kelly, G., and Shanley, J. (2016). Rehabilitation of Ataxic Gait Following Cerebellar Lesions: Applying Theory to Practice. Physiother. Theor. Pract. 32, 430–437. doi:10.1080/09593985.2016.1202364
King, S. J., Nowak, K., Suryavanshi, N., Holt, I., Shanahan, C. M., and Ridley, A. J. (2014). Nesprin-1 and Nesprin-2 Regulate Endothelial Cell Shape and Migration. Cytoskeleton 71, 423–434. doi:10.1002/cm.21182
Kölbel, H., Abicht, A., Schwartz, O., Katona, I., Paulus, W., Neuen-Jacob, E., et al. (2019). Characteristic Clinical and Ultrastructural Findings in Nesprinopathies. Eur. J. Paediatric Neurol. 23, 254–261. doi:10.1016/j.ejpn.2018.12.011
Laforce, R., Buteau, J. P., Bouchard, J.-P., Rouleau, G. A., Bouchard, R. W., and Dupré, N. (2010). Cognitive Impairment in ARCA-1, a Newly Discovered Pure Cerebellar Ataxia Syndrome. Cerebellum 9, 443–453. doi:10.1007/s12311-010-0184-7
Mademan, I., Harmuth, F., Giordano, I., Timmann, D., Magri, S., Deconinck, T., et al. (2016). MultisystemicSYNE1ataxia: Confirming the High Frequency and Extending the Mutational and Phenotypic Spectrum. Brain 139, e46. doi:10.1093/brain/aww115
Miyai, I., Ito, M., Hattori, N., Mihara, M., Hatakenaka, M., Yagura, H., et al. (2012). Cerebellar Ataxia Rehabilitation Trial in Degenerative Cerebellar Diseases. Neurorehabil. Neural Repair 26, 515–522. doi:10.1177/1545968311425918
Morel, V., Lepicard, S., N. Rey, A., Parmentier, M.-L., and Schaeffer, L. (2014). Drosophila Nesprin-1 Controls Glutamate Receptor Density at Neuromuscular Junctions. Cell. Mol. Life Sci. 71, 3363–3379. doi:10.1007/s00018-014-1566-7
Nikolova-Krstevski, V., Leimena, C., Xiao, X.-H., Kesteven, S., Tan, J. C., Yeo, L. S., et al. (2011). Nesprin-1 and Actin Contribute to Nuclear and Cytoskeletal Defects in Lamin A/C-deficient Cardiomyopathy. J. Mol. Cell Cardiol. 50, 479–486. doi:10.1016/j.yjmcc.2010.12.001
Noreau, A., Bourassa, C. V., Szuto, A., Levert, A., Dobrzeniecka, S., Gauthier, J., et al. (2013). SYNE1Mutations in Autosomal Recessive Cerebellar Ataxia. JAMA Neurol. 70, 1296–1231. doi:10.1001/jamaneurol.2013.3268
Palau, F., and Espinós, C. (2006). Autosomal Recessive Cerebellar Ataxias. Orphanet J. Rare Dis. 1, 47. doi:10.1186/1750-1172-1-47
Peng, Y., Ye, W., Chen, Z., Peng, H., Wang, P., Hou, X., et al. (2018). Identifying SYNE1 Ataxia with Novel Mutations in a Chinese Population. Front. Neurol. 9, 1111. doi:10.3389/fneur.2018.01111
Rajgor, D., Mellad, J. A., Autore, F., Zhang, Q., and Shanahan, C. M. (2012). Multiple Novel Nesprin-1 and Nesprin-2 Variants Act as Versatile Tissue-specific Intracellular Scaffolds. PLOS ONE 7, e40098. doi:10.1371/journal.pone.0040098
Razafsky, D., and Hodzic, D. (2015). A Variant of Nesprin1 Giant Devoid of KASH Domain Underlies the Molecular Etiology of Autosomal Recessive Cerebellar Ataxia Type I. Neurobiol. Dis. 78, 57–67. doi:10.1016/j.nbd.2015.03.027
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Ruano, L., Melo, C., Silva, M. C., and Coutinho, P. (2014). The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 42, 174–183. doi:10.1159/000358801
Schatton, C., Synofzik, M., Fleszar, Z., Giese, M. A., Schöls, L., and Ilg, W. (2017). Individualized Exergame Training Improves Postural Control in Advanced Degenerative Spinocerebellar Ataxia: A Rater-Blinded, Intra-individually Controlled Trial. Parkinsonism Relat. Disord. 39, 80–84. doi:10.1016/j.parkreldis.2017.03.016
Schroeder, T. (2013). Rehabilitation of a U.S. Army Soldier Diagnosed with Cerebellar Atrophy with an Ataxic Gait. Mil. Med. 178, e879–e883. doi:10.7205/MILMED-D-12-00404
Serrao, M., Casali, C., Ranavolo, A., Mari, S., Conte, C., Chini, G., et al. (2017). Use of Dynamic Movement Orthoses to Improve Gait Stability and Trunk Control in Ataxic Patients. Eur. J. Phys. Rehabil. Med. 53, 735–743. doi:10.23736/S1973-9087.17.04480-X
Stenson, P. D., Mort, M., Ball, E. V., Chapman, M., Evans, K., Azevedo, L., et al. (2020). The Human Gene Mutation Database (HGMD): Optimizing its Use in a Clinical Diagnostic or Research Setting. Hum. Genet. 139, 1197–1207. doi:10.1007/s00439-020-02199-3
Sur, I., Neumann, S., and Noegel, A. A. (2014). Nesprin-1 Role in DNA Damage Response. Nucleus 5, 173–191. doi:10.4161/nucl.29023
Swan, L., Cardinal, J., and Coman, D. (2018). SYNE1-related Autosomal Recessive Cerebellar Ataxia, Congenital Cerebellar Hypoplasia, and Cognitive Impairment. Clin. Pract. 8, 91–93. doi:10.4081/cp.2018.1071
Synofzik, M., and Ilg, W. (2014). Motor Training in Degenerative Spinocerebellar Disease: Ataxia-specific Improvements by Intensive Physiotherapy and Exergames. Biomed. Res. Int. 2014, 1–11. doi:10.1155/2014/583507
Synofzik, M., Puccio, H., Mochel, F., and Schöls, L. (2019). Autosomal Recessive Cerebellar Ataxias: Paving the Way toward Targeted Molecular Therapies. Neuron 101, 560–583. doi:10.1016/j.neuron.2019.01.049
Synofzik, M., Schatton, C., Giese, M., Wolf, J., Schöls, L., and Ilg, W. (2013). Videogame-based Coordinative Training Can Improve Advanced, Multisystemic Early-Onset Ataxia. J. Neurol. 260, 2656–2658. doi:10.1007/s00415-013-7087-8
Wang, Y., Wan, J., Yang, T., Zhang, Z., Ren, D., and Zou, Y. (2021). Acupuncture for Spinocerebellar Ataxia Type 1: A Case Report. Acupunct. Med. 9645284211055758, 096452842110557. doi:10.1177/09645284211055758
Warren, D. T., Zhang, Q., Weissberg, P. L., and Shanahan, C. M. (2005). Nesprins: Intracellular Scaffolds that Maintain Cell Architecture and Coordinate Cell Function? Expert Rev. Mol. Med. 7, 1–15. doi:10.1017/S1462399405009294
Winser, S. J., Kannan, P., Pang, M., Smith, C., and Tsang, W. W. (2018). Potential Benefits and Safety of T'ai Chi for Balance and Functional Independence in People with Cerebellar Ataxia. J. Altern. Complement. Med. 24, 1221–1223. doi:10.1089/acm.2017.0396
Zhang, Q., Bethmann, C., Worth, N. F., Davies, J. D., Wasner, C., Feuer, A., et al. (2007). Nesprin-1 and -2 Are Involved in the Pathogenesis of Emery-Dreifuss Muscular Dystrophy and Are Critical for Nuclear Envelope Integrity. Hum. Mol. Genet. 16, 2816–2833. doi:10.1093/hmg/ddm238
Zhao, T., Ma, Y., Zhang, Z., Xian, J., Geng, X., Wang, F., et al. (2021). Young and Early‐onset Dilated Cardiomyopathy with Malignant Ventricular Arrhythmia and Sudden Cardiac Death Induced by the Heterozygous LDB3, MYH6, and SYNE1 Missense Mutations. Ann. Noninvasive Electrocardiol. 26, e12840. doi:10.1111/anec.12840
Keywords: case report, autosomal recessive cerebellar ataxia, ARCA-1, SYNE1 ataxia, SYNE1 gene, SCAR8
Citation: Qian N, Wei T, Yang W, Wang J, Zhang S, Jin S, Dong W, Hao W, Yang Y and Huang R (2022) Case Report: Late-Onset Autosomal Recessive Cerebellar Ataxia Associated With SYNE1 Mutation in a Chinese Family. Front. Genet. 13:795188. doi: 10.3389/fgene.2022.795188
Received: 14 October 2021; Accepted: 24 January 2022;
Published: 23 February 2022.
Edited by:
Babak Behnam, National Sanitation Foundation International, United StatesReviewed by:
Corrado Italo Angelini, University of Padua, ItalyHelio AG Teive, Federal University of Paraná, Brazil
Copyright © 2022 Qian, Wei, Yang, Wang, Zhang, Jin, Dong, Hao, Yang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenming Yang, eWFuZ3dtODgxMEAxMjYuY29t
†These authors have contributed equally to this work.