 Darshini A. Desai1†
Darshini A. Desai1† Vinay J. Rao
Vinay J. Rao Anil G. Jegga
Anil G. Jegga Perundurai S. Dhandapany
Perundurai S. Dhandapany Sakthivel Sadayappan
Sakthivel Sadayappan- 1Department of Internal Medicine, Division of Cardiovascular Health and Disease, University of Cincinnati, Cincinnati, OH, United States
- 2Cardiovascular Biology and Disease Theme, Institute for Stem Cell Science and Regenerative Medicine, Bangalore, India
- 3Division of Biomedical Informatics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
- 4Department of Pediatrics, College of Medicine, University of Cincinnati, Cincinnati, OH, United States
- 5The Knight Cardiovascular Institute, Oregon Health and Science University, Portland, OR, United States
Myosin binding protein-C (MyBP-C) is a sarcomeric protein which regulates the force of contraction in striated muscles. Mutations in the MYBPC family of genes, including slow skeletal (MYBPC1), fast skeletal (MYBPC2) and cardiac (MYBPC3), can result in cardiac and skeletal myopathies. Nonetheless, their evolutionary pattern, pathogenicity and impact on MyBP-C protein structure remain to be elucidated. Therefore, the present study aimed to systematically assess the evolutionarily conserved and epigenetic patterns of MYBPC family mutations. Leveraging a machine learning (ML) approach, the Genome Aggregation Database (gnomAD) provided variants in MYBPC1, MYBPC2, and MYBPC3 genes. This was followed by an analysis with Ensembl’s variant effect predictor (VEP), resulting in the identification of 8,618, 3,871, and 3,071 variants in MYBPC1, MYBPC2, and MYBPC3, respectively. Missense variants comprised 61%–66% of total variants in which the third nucleotide positions in the codons were highly altered. Arginine was the most mutated amino acid, important because most disease-causing mutations in MyBP-C proteins are arginine in origin. Domains C5 and C6 of MyBP-C were found to be hotspots for most mutations in the MyBP-C family of proteins. A high percentage of truncated mutations in cMyBP-C cause cardiomyopathies. Arginine and glutamate were the top hits in fMyBP-C and cMyBP-C, respectively, and tryptophan and tyrosine were the most common among the three paralogs changing to premature stop codons and causing protein truncations at the carboxyl terminus. A heterogeneous epigenetic pattern was identified among the three MYBP-C paralogs. Overall, it was shown that databases using computational approaches can facilitate diagnosis and drug discovery to treat muscle disorders caused by MYBPC mutations.
Introduction
Complex diseases stemming from genetic mutations have become a worldwide concern affecting the quality of life. Detecting such genetic diseases depends, in part, on information from online databases (Gudmundsson et al., 2021). Thus, combining such readily accessible data with advanced molecular technologies has helped in identifying various diseases caused by changes in DNA sequences. Indeed, understanding such genetic defects and detecting them at early stages have steadily progressed (Mendez et al., 2021; Yu et al., 2021). No less important is an understanding of conserved elements of the human genome in the context of disease etiology, as well as disease prevention and treatment (Mooney et al., 2010). Researchers now use next-generation sequencing (NGS) methods to determine the order of nucleotides in entire genomes or targeted regions of DNA or RNA, leading to the isolation of genetic mutations likely to develop diseases (Gagan and Van Allen, 2015). Such high-throughput technologies also make it easier to predict the nature of the complex diseases. NGS platforms carry out sequencing of the whole human genome, or a number of small fragments of DNA, at the same time, followed by mapping individual reads to the human reference genome. We can also choose a specific site of interest. Any size of gene can be sequenced to detect the presence of mutations. So far, NGS has successfully identified many disease-causing variants, leading to a better understanding of pathogenic effects and clinical consequences (Schmitt et al., 2012). Moreover, deep machine learning methods can be developed to predict genotype-phenotype outcomes (Al-Numair et al., 2016; de Marvao et al., 2021; Wang et al., 2021).
We herein focused on a group of myosin binding protein-C (MYBPC) paralogs which, together, constitute an immunoglobulin super-family of intracellular muscle proteins (Sadayappan and de Tombe, 2012). MYBPC has three paralogs encoded by unique genes, including slow skeletal (MYBPC1), fast skeletal (MYBPC2), and cardiac (MYBPC3) (Lin et al., 2013). Slow skeletal MyBP-C (sMyBP-C), fast skeletal MyBP-C (fMyBP-C) and cardiac MyBP-C (cMyBP-C) proteins are highly conserved with over 90% homology. They play unique muscle-specific structural and regulatory roles in actomyosin interactions and contractility in striated muscles, including both cardiac and skeletal muscles (Lin et al., 2013). MyBP-C protein is found in the cross-bridge-bearing zone (C region) of A bands in sarcomere of striated muscles (Offer et al., 1973). They provide thick filament stability by interacting with titin and the rod portion of sarcomeric myosin (light meromyosin) through MyBP-C’s C-terminal region (Flashman et al., 2004; Jiang et al., 2015). Ablating MYBPC2 (Song et al., 2021) and MYBPC3 (Harris et al., 2002) gene expression results in contractile dysfunction, suggesting the key role played by MyBP-C in striated muscles. It is well known that genetic variants in MYBPC genes cause various life-threatening cardiovascular and congenital muscular diseases. For example, mutations in MYBPC3 are linked to hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) (Bonne et al., 1995; Watkins et al., 1995; Barefield and Sadayappan, 2010; Harris et al., 2011). More than 45% of HCM cases can be attributed to mutations in the MYBPC3 gene (Spirito et al., 1997). Strikingly, 70% of genetic variants in MYBPC3 are nonsense mutations, including indels, frameshift, and splice-site mutations, leading to cMyBP-C truncations at the carboxyl terminus (Richard et al., 2010; Harris et al., 2011). It is, however, unclear why MYBPC3 variants predominantly result in protein truncations and whether any evolutionary reasons behind such preferential variants. In contrast, few variants have been reported in the skeletal paralogs (Desai et al., 2020). However, some recent studies suggest that mutations in MYBPC1 are linked to a congenital disease called distal arthrogryposis (Bamshad et al., 2009; Stavusis et al., 2019; Desai et al., 2020) and myogenic tremor (Geist Hauserman et al., 2021). Specifically, infants born with MYBPC1 variants developed with multiple joint contractures congenitally limiting muscular movement and affecting the quality of life (Bamshad et al., 2009; Markus et al., 2012). On the other hand, MYBPC2 has also been linked to skeletal muscular disorders like arthrogryposis (Bayram et al., 2016). Thus, it is well worth systematically determining the genetic variability among these three genes, such as frequency, a hot spot, differences in codon usage, and degree of pathogenicity.
To date, around 2,000 variants have been reported in the MYBPC3 gene (Helms et al., 2020), but the conserved pattern and biochemical characteristics of these variants have not been systematically reviewed. Therefore, in the present study, we analyzed variants of all three MYBPC gene isoforms for their effects, using the Variant Effect Predictor (VEP) to understand MyBP-C biology and evolutionary pattern (McLaren et al., 2016). The Genome Aggregation Database (GnomAD) (Gudmundsson et al., 2021) was used to collect the up-to-date MYBPC variants. We then performed data mining, queried the database of variants reported in these three gene isoforms, and carried out a comprehensive bioinformatics review of the evolutionary pattern of conserved variants in the MYBPC gene family. Variants and the similarities among them were compared among the three MYBPC paralogs, along with heterogeneous, gene-specific epigenetic patterns.
Materials and Methods
Accessing Variant Database and Data Extraction
Variant data for MYBPC1, MYBPC2, and MYBPC3 genes were directly downloaded from the Genome Aggregation Database (gnomAD) (Karczewski et al., 2020). This database is open source, and it aggregates and harmonizes exome and genome sequencing data from multiple large-scale sequencing projects. We also used Ensembl’s Variant Effect Predictor (VEP) (McLaren et al., 2016) to obtain annotations for all gnomAD variants from these three genes with an rsID. The collection of variants was genome-wide, including both coding and noncoding regions. We understand that homopolymeric regions pertaining to mitochondrial DNA (mtDNA) variants have been filtered out of gnomAD data, and we carried out our analyses accordingly.
Variant Identification and Analyses
Analysis was carried out using in-house scripts. The data were first processed by removing any duplicate variant entries. The longest isoforms of MYBPC1 (transcript ID ENST00000361466), MYBPC2 (transcript ID ENST00000357701), and MYBPC3 (transcript ID ENST0000545968) were identified from the resulting files. Variants impacting other genes could then be removed (AC117505.1 residing within MYBPC1; AC020909.1 and SPIB, both downstream of MYBPC2, and FAM71E1 upstream of MYBPC2; MADD downstream of MYBPC3 and SPI1 upstream of MYBPC3) (Supplementary Figure S1). Based on the resultant data, we wanted to discriminate among the variations observed across the three genes. First, we identified the frequency of each variant by category, including synonymous, missense, truncation, frameshift, and non-frameshift, indels and others, such as splice donors and acceptors, loss of start and stop codon, and protein-altering variants. Next, we identified the mutated nucleotides in each variant category, as well as codon position of nucleotides mutated in missense and synonymous variants. We also studied protein variants, classifying the amino acid (aa) variations in protein domains, the information of which was obtained through UniProt. Based on nucleotide data obtained earlier, we also investigated the proneness of certain exons to mutations. Last, with the help of variant consequence predictors, such as SIFT and Polyphen, we identified the distribution of pathogenic variants among the three genes (Supplementary Figure S2). Scripts will be provided upon request to the corresponding author.
Results
Variable Distribution of Genetic Variants in Myosin Binding Protein-C Paralogs
The MyBP-C protein family is a group of thick filament accessory proteins regulating striated muscle structure and function. cMyBP-C differs from sMyBP-C and fMyBP-C proteins by containing a unique C0 domain and 28 aa loops at the C5 domain (Figure 1). sMyBP-C is highly homologous to fMyBP-C, but its expression and functions differ (Dhoot et al., 1985; Weber et al., 1993). To determine the conserved pattern in MyBP-C structural biology, we analyzed 8,617, 3,870, and 3,070 variants in MYBPC1, MYBPC2, and MYBPC3 genes, respectively. Variants were collected from GnomAD, and annotations were calculated by VEP. Among coding variants across the three paralogs, VEP analysis revealed missense variants to be the most predominant (61%–66%), followed by synonymous variants (∼30%), frameshift and truncation variants (∼3%), and then in-frame indels and splice-site variants (2%–3%) (Figure 2). While MYBPC3 had the highest number of coding variants (Supplementary Figure S2), MYBPC1 had the highest number of variants with intronic variants, making up 90% of variants in MYBPC1 as compared to 75% in MYBPC2 and 62% in MYBPC3 (data not shown). Next, we analyzed the domain-wise frequency in all MyBP-C proteins (Figure 3). Interestingly, the C5 domain proved to be the most prone to mutations among all three proteins, despite not being the longest domain in sMyBP-C and fMyBP-C (Figures 3A–C).
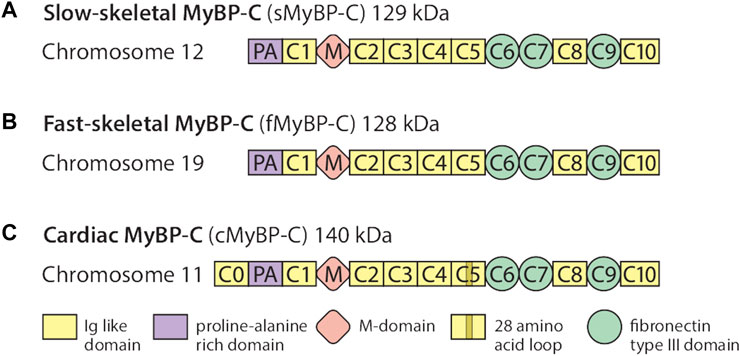
FIGURE 1. The MyBP-C family consists of one cardiac and two skeletal paralogs. sMyBP-C is a 129 kDa protein encoded by the MYBPC1 gene contained in chromosome 12 (A), and fMyBP-C is a skeletal muscle-specific protein encoded by the MYBPC2 gene contained in chromosome 19 (B). The two skeletal paralogs share similar domains, from C1 to C10, which contain three fibronectin type III domains (C6, C7, and C9) and seven immunoglobulin-like domains with one Proline-Alanine (PA)-rich domain and phosphorylation (M) domains. The 140 kDa cMyBP-C is encoded by the MYBPC3 gene (C) and also shares structural features similar to those of the skeletal paralogs, except it has one additional domain in its N′-region (C0). It also has a 28-amino acid loop in its C5 domain.
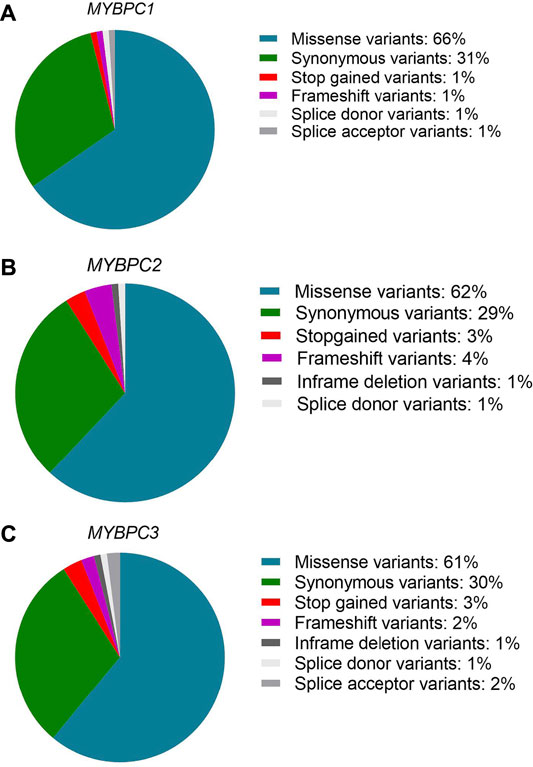
FIGURE 2. Missense variants predominate across paralogs. Variant ensemble predictor (VEP) analysis shows that the MYBPC family shares a similar pattern of variant distribution, predominantly comprised of single nucleotide variants (61%–66% missense variants, 30% synonymous variants). The remainder includes stop-gain, splicing variants, non-frameshift variants and frameshift variants in MYBPC1 (A), MYBPC2 (B), and MYBPC3 (C) genes.
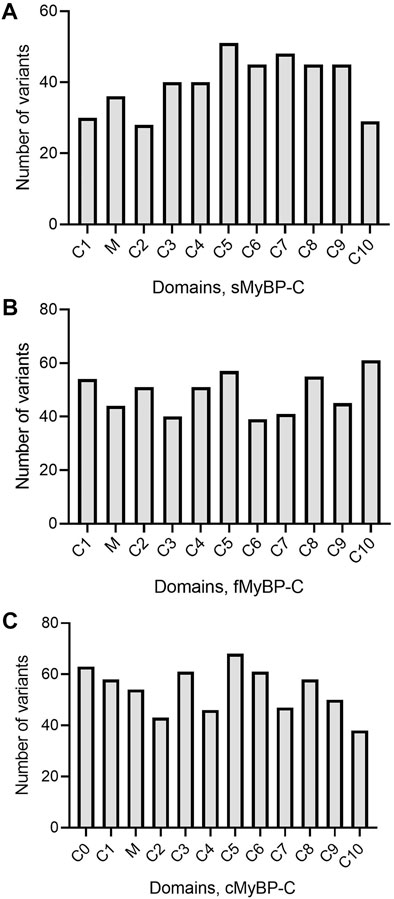
FIGURE 3. Variant frequency is characterized by heterogeneity in MyBP-C paralogs. Heterogeneous distribution of variant frequency observed in the domains of the MYBPC family. (A–C) sMyBP-C has the lowest number of variants across domains, compared to other paralogs, and C5 domain was the most mutated domain with the highest number of variants in the MYBPC family.
Paralog-Specific Alterations in Amino Acids
Next, variants were categorized into missense, frameshift, and truncation mutants based on variations in their amino acids. Our analyses revealed that Glu > Lys (E > K) and Ala > Thr (A > T) were the most frequent missense amino acid substitutions across the MyBP-C proteins and that Ile > Val (I > V) was the most frequent in sMyBP-C (Figures 4A–C). Interestingly, among the ten most frequent amino acid substitutions among the three paralogs, there lacks a mutation of the commonly post-translationally modifiable residues (Lys, Ser, Thr, Tyr), except for Arg, however we do commonly observe modifiable amino acids occurring in these proteins as a result of mutations. This feature could be explored as a potential therapeutic target since post-translational modifications are known to frequently activate or de-activate proteins.
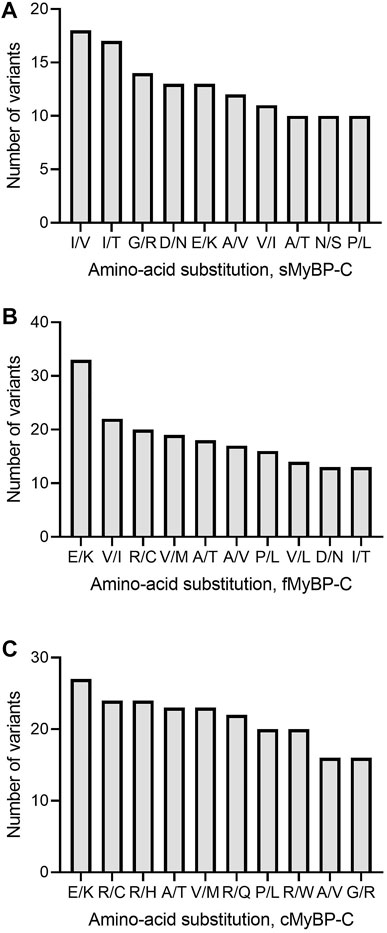
FIGURE 4. Top ten most frequent amino acid substitutions in the MyBP-C paralogs. E > K and A > T are the most common amino acid substitutions across paralogs. I/V, V/I and R/C are most frequent amino acid substitutions in sMyBP-C (A), fMyBP-C (B) and cMyBP-C (C), respectively.
Among missense variants, Cys, Phe, His, and Trp were the least mutated amino acids, while, again, Arg and Val were the most frequently mutated amino acids across all MYBPC genes (Figures 5A–C). sMyBP-C was shown to have other frequently mutated amino acids, including Ile, Gly, Val, Asp, Ala, and Glu, while fMyBP-C had frequent mutations in Pro, Glu and Val, and cMyBP-C showed no affinity towards mutations in any amino acids other than Val and Arg. This could be attributed to an excess number of codons coding for Arg. However, this pattern dramatically changed in frameshift variants with most mutations impacting Thr (sMyBP-C), Ile (fMyBP-C), and Pro (cMyBP-C), respectively (Figures 6A–C). Arg was largely unaffected by frameshift mutations.
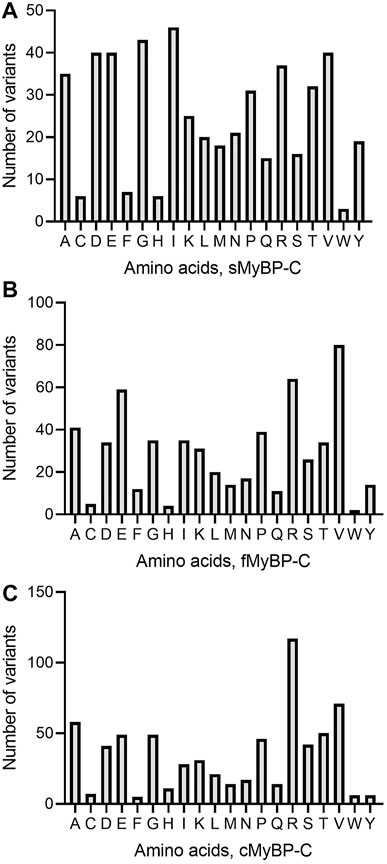
FIGURE 5. Prevalence of amino acids in missense variants across paralogs. Arg (R) was the most mutated amino acid in missense variants in the MyBP-C protein family with Val (V) being the second most mutated in sMyBP-C (A), fMyBP-C (B) and cMyBP-C (C). Iso (I) was another highly mutated amino acid in missense variants in the cMyBP-C protein.
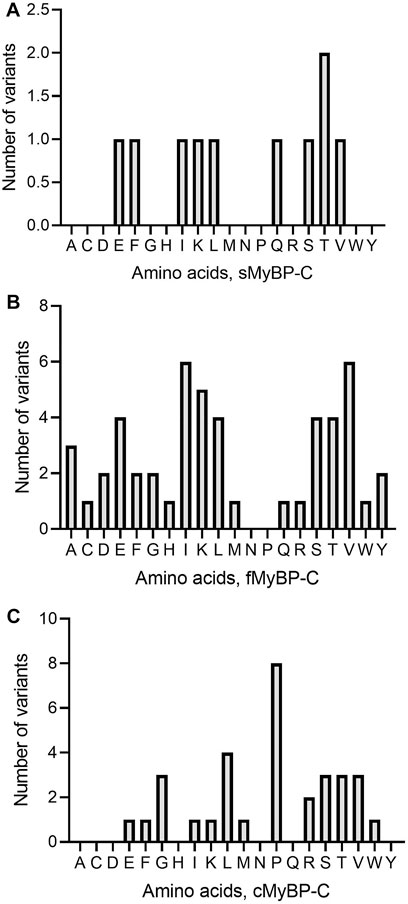
FIGURE 6. Prevalence of amino acids in frameshift variants in the MyBP-C gene family. Thr (T), Iso (I) and Pro (P) were the most mutated amino acids in frameshift variants in sMyBP-C (A), fMyBP-C (B) and cMyBP-C (C) proteins, respectively.
Another important category of mutation includes truncation variants since they might not include regulatory or functional domains in the translated protein. In the MyBP-C family, not surprisingly, we mostly observe Trp and Tyr mutations leading to the introduction of a premature stop codon (Figures 7A–C). In sMyBP-C, however, Arg variants leading to truncated variants are as common as Trp and Tyr (Figure 7A). Glu mutations leading to truncation could be observed in both fMyBP-C and cMyBP-C (Figure 7).
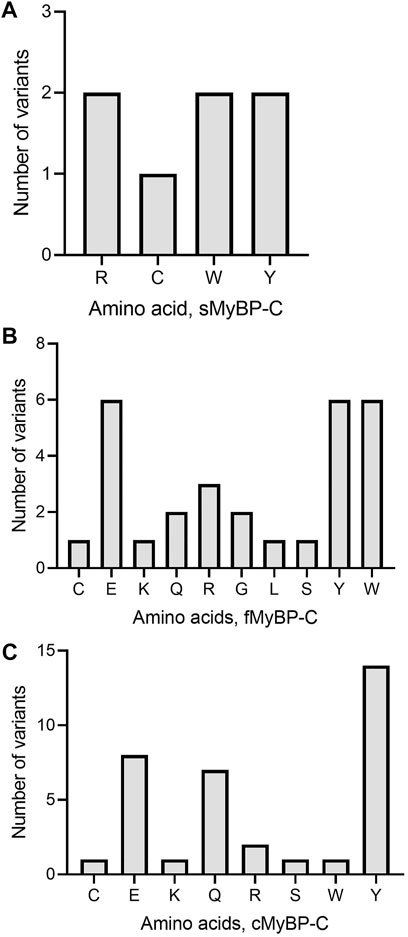
FIGURE 7. Prevalence of amino acids targeted to introduce premature termination in the MyBP-C gene family. Trp (W) and Arg (R) were the top amino acids introducing a prematurely terminated stop codon in sMyBP-C protein (A), and Glu (E) was the most targeted amino acid in both fMyBP-C (B) and cMyBP-C (C) protein. Lastly, Tyr (Y) was the most prevalent amino acid leading to premature termination and causing C′-terminal truncation in all three paralogs.
Variant Distribution Across Exons and Domains in the Paralogs
Next, we applied filters to the VEP files in order to categorize variants as “likely pathogenic” and analyzed which domains and exons were the most susceptible to mutation. Again, all three paralogs showed very heterogeneous distribution in the frequency of pathogenic variants (Figures 8D–F). However, while mutations in the C10 domain caused pathogenic variants in fMyBP-C, the C10 domain of cMyBP-C was the least mutated. Instead, C6 was the most mutated domain. Heterogeneous distribution characterized sMyBP-C in which domains comprising the N terminal of the protein were found to be the least mutated. Next, we investigated which exons contained the most pathogenic variants. Here, although MYBPC1 showed very mixed distribution, exons 21 and 29 contained most pathogenic variants. Exons 8, 10, 26, and 27 contained the most pathogenic variants for MYBPC2, whereas exon 25 was clearly the most pathogenic variant-containing exon for MYBPC3, followed by exons 2 and 29. (Figures 8A–C). Distal Arthrogryposis has been attributed to pathogenic variants in MYBPC1, and MYBPC3 is known to cause a series of cardiomyopathies, including HCM, DCM and congenital heart defects. Mutations in MYBPC2 were not very well annotated in terms specific diseases, but a few variants were linked to cognitive dysfunction, according to the VEP files.
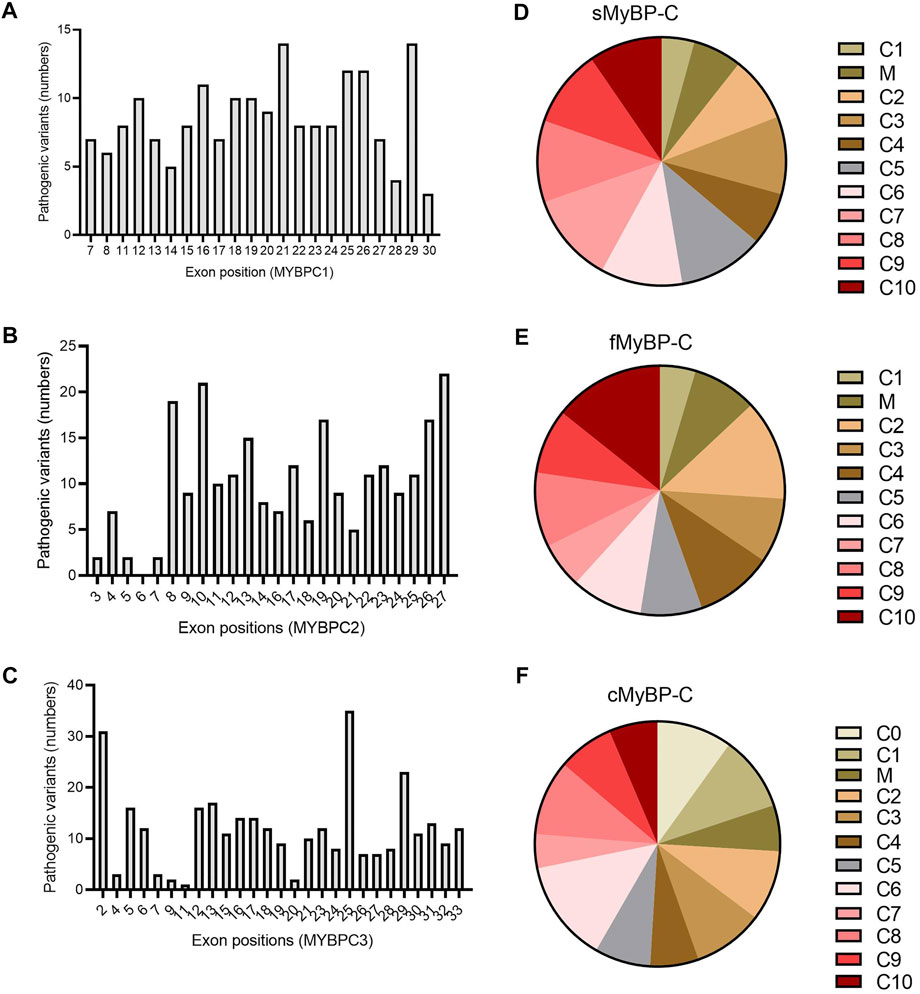
FIGURE 8. Prevalence of exons in the genes and domains of proteins susceptible to pathogenic variants in MYBPC gene paralogs. In MYBPC1 (A), exons 21 and 29 had the highest number of hits with pathogenic variants. However, in MYBPC2 (B), exons 8, 10, 19, 26, and 27 were the prevalent targets with pathogenic variants. In MYBPC3 (C), exon 2 and exon 25 had the highest number of variants. In terms of domain-wise frequency of pathogenic variants, the C6 domain was the most highly mutated domain leading to pathogenic variants across paralogs, followed by C5 and C7 in sMyBP-C (D), C2 and C10 in fMyBP-C (E) and C0, C1, and C6 in cMyBP-C (F) proteins.
Discussion
MYBPC paralogs play a major role in striated muscle contraction. Increasing evidence suggests that genetic alterations in MYBPC paralogs are directly linked to myopathies. However, no systematic analyses have been carried out to determine nucleotide pattern, codon, and amino acid changes in existing genetic mutations among these three proteins. We wanted to understand the patterns of genetic variants arising from evolutionarily conserved amino acids in MyBP-C structural biology. To this end, we analyzed around 3,000 variants in each paralog obtained from GnomAD and calculated the annotations using the variant effect predictor (VEP). The collected data were analyzed by comparing all three paralogs. Mapping variant frequency in the domains of all MyBP-C proteins revealed a heterogeneous distribution, indicating that all domains in the MyBP-C protein are equally susceptible to mutation. Very few studies have reported on the conserved sequences among the three MYBPC paralogs (Okagaki et al., 1993; Weber et al., 1993; Shaffer and Gillis, 2010; Lin et al., 2013). However, in the present study, the conserved pattern of MYBPC mutations also showed a very heterogeneous distribution in all three paralogs. Missense variants predominated with Ile as the most mutated amino acid in sMyBP-C, Val in fMBP-C and Arg in cMyBP-C.
(Shaffer and Gillis, 2010) reported a high level of conserved sequences. The M-domain, otherwise known as the MyBP-C motif, contains a unique set of 100 amino acids at the N terminus between domains C1 and C2. This region is essential for actomyosin interactions. The M-domain binds myosin S2, as well as actin, to regulate cross-bridge formation during contraction and relaxation. Upon phosphorylation by kinases like PKA, the bond between M-domain and S2, or actin, is broken, allowing cross-bridge formation (Gruen et al., 1999; Korte et al., 2003; Stelzer et al., 2007; Shaffer et al., 2009). Many regions of the M domain are highly conserved, including 293–300 and 331–353 in humans, which may, or may not, carry functional importance. However, some regions of MyBP-C are not well conserved and are unique to the cardiac paralog. The cardiac cMyBP-C isoforms contain an additional ∼100 amino acid domain at the extreme N terminus called the C0 domain which is absent in the slow and fast skeletal paralogs (Figure 1). In the same evolutionary study by Shaffer and Gillis, the phylogenetic analysis of MyBP-C sequences revealed MyBP-C paralogs to be monophyletic, while the fast and slow skeletal MyBP-C protein paralogs clustered in a group that deviated from that of cMyBP-C. This indicated that cMyBP-C is the ancestral form of MyBP-C. They also predicted that gene duplication events caused changes in the sequence of cMyBP-C, resulting in the differentiation of slow skeletal from the cardiac paralog. Differences in the sequence of cMyBP-C enable it to carry out its specialized cardiac muscle function (Shaffer and Gillis, 2010).
Previous alignment studies noted a significant degree of conserved sequences across all three paralogs (Shaffer and Gillis, 2010). A high number of amino acids were found to be conserved in MyBP-C, depending on the species. Altogether, eight residues in mammalian cMyBP-C switch from nonpolar amino acids to charged amino acids in the other two isoforms (Shaffer and Gillis, 2010). For example, in human cMyBP-C, as well as all other mammalian cardiac isoforms, Gly-354 (or its equivalent) can be observed, while an Arg residue can be found at the equivalent site in nonmammalian cardiac isoforms, as well as all fast and slow skeletal isoforms. The functional importance of these amino acids is unknown (Shaffer and Gillis, 2010). This could explain why Arg was the most mutated amino acid in cMyBP-C and the other paralogs.
Life-threatening diseases have been attributed to mutations in the MYBPC paralog proteins. For example, mutations in the MYBPC3 gene are also linked to HCM, DCM and sudden cardiac death. Missense mutations cause stable proteins to incorporate into the sarcomere and lead to various functional defects. However, frameshift mutations result in a prematurely terminated codon in the transcribed mRNA, making C-terminal truncated proteins unable to bind myosin or titin and also leading to functional defects. About 70% of genetic variants in MYBPC3 represent C′’-truncations (Harris et al., 2011). Furthermore, C-terminal truncated proteins have never been detected by immunoblots in cardiac tissue of HCM patients (Marston et al., 2009; van Dijk et al., 2009). Importantly, cardiomyocytes from cMyBP-C mutants are markedly decreased from those of the wild-type protein (Yang et al., 1999). Altogether, these studies strongly suggest that mutant proteins and/or mRNAs are unstable and degraded accordingly. Therefore, it was proposed that frameshift and nonsense mutations might lead to cMyBP-C haploinsufficiency (van Dijk et al., 2009; Suay-Corredera et al., 2021). A non-functional or mutant protein incorporating into the sarcomere can cause filament disassembly, altered function, and, finally, HCM- or DCM-like phenotype (Schlossarek et al., 2012). While few studies have reported on MYBPC1 and MYBPC2, many studies have linked MYBPC1 mutations to distal arthrogryposis (Desai et al., 2020). For example, one study found that MYBPC1 mutations W236R and Y856H could cause distal arthrogryposis type 1 (Gurnett et al., 2010). Another study in a Chinese family found that E359K, R318X, and P319L mutations led to distal arthrogryposis type 2 (Li et al., 2015). Distal arthrogryposis is a skeletal muscle disorder characterized by joint contractures and deformities on distal body parts immobilizing muscle movements (Desai et al., 2020). Few MYBPC2 mutations have been explained clinically. However, a recent study from our laboratory shows that global knockout of MYBPC2 in mice results in reduced contractility and reduced myofilament calcium sensitivity and hypertrophic response to mechanical overload (Song et al., 2021).
Some disease-causing founder mutations are specific to ethnicity and limited to a geographic region (Dhandapany et al., 2009). In recent years, MYBPC3 has gained significant interest owing to its role in the regulation of contractility in the sarcomere machinery. MYBPC3 is known to regulate contraction upon its phosphorylation by various kinases (van Dijk et al., 2009). Studies have shown that C′-truncation of MYBPC3 causes cMyBP-C null and DCM in mice at the age of 3 months (McConnell et al., 1999), as well as significant epigenetic changes (Tabish et al., 2019), indicating the importance of normal cMyBP-C for regular cardiac function (Lynch et al., 2015). In comparison, much less is known about the two skeletal isoforms of MyBP-C (McNamara and Sadayappan, 2018).
Across the three genes, we observed the highest prevalence of coding variants and pathogenic coding variants in MYBPC3. While missense variants constitute most of the coding variants, a considerable prevalence of loss-of-function variants can be seen in the three genes represented by frameshift and truncation variants (Figure 2). From an evolutionary perspective, we also noted in all genes that the C5 domain is highly prone to variants, which could, therefore, be a potential therapeutic target in disease conditions. In MYBPC3, the cardiac specific N-terminal domain is also highly prone to variations, thus possibly crucial in the treatment of HCM (Figure 3). A stark change in the polarity of amino acids is observed in missense mutations, potentially altering protein binding and/or key post-translational modifications in MyBP-C proteins, thereby leading to a disease phenotype (Figure 4).
In our analyses, the most dominant variant among the MyBP-C paralogs was missense variants, followed by frameshift variants. For instance, a missense variant, like R403Q or R663H, ablates the binding of myosin with the C0-C7 fragment of cMyBP-C protein and causes hypercontractility (Sarkar et al., 2020). The N-terminal of cMyBP-C regulates contractility within the sarcomere machinery. Mutations in the domains comprising the N-terminal often lead to cardiac dysfunction. Our analysis revealed the C6 domain of cMyBP-C to have the most pathogenic variants, followed by C0 and C1. Nearly half of these variants localize to the C0-C4, comprising the N terminal of the protein. Mutations in the N -terminal can lead to either reduced or increased binding with the myosin region, depending on the mutation. This explains the loss- or gain-of-function in the case of mutations leading to cardiac dysfunction. As mentioned previously, MyBP-C has many conserved domains, and, over time, mutations can negatively impact contractile function. In our study, Glu > Lys (E/K) was most frequent amino acid substitution found in all MyBP-C paralogs. A switch from a negatively charged amino acid to a positively charged amino acid can lead to loss of binding to the neighboring amino acids in a protein and ultimately to reduced or increased function in the sarcomere. Arg was the most mutated amino acid across all three paralogs of MyBP-C missense variants. However, Arg was also the least susceptible to frameshift mutation, suggesting that the nucleotides coding for Arg do not often change the entire sequence of amino acids in the protein, another avenue for exploration in further studies. Among the pathogenic variants, exon 25 and 29 of MYBPC1 and MYBPC3 were the most likely to be mutated, suggesting the likelihood that these positions can destabilize the structure in a manner sufficient to alter the protein’s functionality. However, for MYBPC2, mutations in the exons 8, 10, and 27 were most likely associate with distal arthrogryposis (Desai et al., 2020).
In conclusion, our study demonstrates the evolutionary pattern of conserved variants in the MyBP-C family of proteins, potentially leading to complex genetic diseases. Overall, the results of our assessment can be used for genetic mapping and identifying genetic variants in individuals with a history of such mutations for the purpose of clinical diagnosis and prognosis.
Limitations of the Study
In this study, only the variants available on the gnomAD database were analyzed. Other variants may be present in other databases for MYBPC genes. Since the data in gnomAD represent aggregate data, phenotype and other individual-level data are not available. Follow-up studies can be undertaken using data from biobanks such as the UK Biobank. Additionally, gnomAD has an over-representation of data from European populations compared to participants from other communities (e.g., Middle Eastern, and Oceanian populations. Despite rigorous quality control, gnomAD may also contain sequencing and annotation artifacts (Gudmundsson et al., 2021). We used only one effect predictor, namely VEP, to annotate the variants. Current variant annotation tools, including VEP, annotate each variant independently and do not consider the potential compound effects of combining alternate alleles. In other words, two or more variants affecting the same codon are not considered when annotating. While VEP, or similar predictors, can predict the functional effects of genomic variants, without validation studies, the predicted deleterious variants cannot be claimed as absolutely pathogenic or cause a definite phenotype. With subjects’ samples (control and case datasets) underlying mechanisms of pathogenesis caused by these variants can be deduced.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
Conceptualization, DD, AJ, PD, and SS; methodology, DD, VR, and AJ; investigation, DD, VR, AJ, PD, and SS; data curation, DD, VR, AJ, PD, and SS; writing—original draft preparation, DD; writing—review and editing, DD, VR, AJ, PD, and SS; visualization, SS; supervision, AJ, PD, and SS; funding acquisition, SS. All authors have read and agreed to the published version of the manuscript.
Funding
DD was supported by an American Heart Association Predoctoral Fellowship (20PRE35120272). SS has received support from National Institutes of Health grants (R01 HL130356, R01 HL105826, R01 AR078001, and R01 HL143490), American Heart Association, Institutional Undergraduate Student (19UFEL34380251), Transformation (19TPA34830084) awards, the PLN Foundation (PLN crazy idea) awards, as well as Novo Nordisk, AstraZeneca, MyoKardia, Merck and Amgen. PD is supported by the Wellcome Trust-Indian Alliance (IA/I/16/1/502367), Rajiv Gandhi University of Health Sciences (RGUHS), Scientist Development Grant (15SDG23250005) from the American Heart Association (AHA), Department of Science and Technology (DST/CRG/2019/005401) and inStem core funding. VR is supported by ICMR-SRF (3/1/1 (8)/CVD/2020-NCD-1).
Conflict of Interest
SS provided consulting and collaborative research studies to the Leducq Foundation (CURE-PLAN), Red Saree Inc., Greater Cincinnati Tamil Sangam, AavantiBio, Pfizer, Novo Nordisk, AstraZeneca, MyoKardia, Merck and Amgen. AJ serves as a member of Scientific Advisory Board of GEn1E Lifesciences, Palo Alto, CA, United States.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.896117/full#supplementary-material
References
Al-Numair, N. S., Lopes, L., Syrris, P., Monserrat, L., Elliott, P., and Martin, A. C. R. (2016). The Structural Effects of Mutations Can Aid in Differential Phenotype Prediction of Beta-Myosin Heavy Chain (Myosin-7) Missense Variants. Bioinformatics 32 (19), 2947–2955. doi:10.1093/bioinformatics/btw362
Bamshad, M., Van Heest, A. E., and Pleasure, D. (2009). Arthrogryposis: a Review and Update. J. Bone Jt. Surg. Am. 91 (Suppl. 4), 40–46. doi:10.2106/JBJS.I.00281
Barefield, D., and Sadayappan, S. (2010). Phosphorylation and Function of Cardiac Myosin Binding Protein-C in Health and Disease. J. Mol. Cell. Cardiol. 48 (5), 866–875. doi:10.1016/j.yjmcc.2009.11.014
Bayram, Y., Karaca, E., Coban Akdemir, Z., Yilmaz, E. O., Tayfun, G. A., Aydin, H., et al. (2016). Molecular Etiology of Arthrogryposis in Multiple Families of Mostly Turkish Origin. J. Clin. Investigation 126 (2), 762–778. doi:10.1172/Jci84457
Bonne, G., Carrier, L., Bercovici, J., Cruaud, C., Richard, P., Hainque, B., et al. (1995). Cardiac Myosin Binding Protein-C Gene Splice Acceptor Site Mutation Is Associated with Familial Hypertrophic Cardiomyopathy. Nat. Genet. 11 (4), 438–440. doi:10.1038/ng1295-438
de Marvao, A., McGurk, K. A., Zheng, S. L., Thanaj, M., Bai, W., Duan, J., et al. (2021). Phenotypic Expression and Outcomes in Individuals with Rare Genetic Variants of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 78 (11), 1097–1110. doi:10.1016/j.jacc.2021.07.017
Desai, D., Stiene, D., Song, T., and Sadayappan, S. (2020). Distal Arthrogryposis and Lethal Congenital Contracture Syndrome - an Overview. Front. Physiol. 11, 689. doi:10.3389/fphys.2020.00689
Dhandapany, P. S., Sadayappan, S., Xue, Y., Powell, G. T., Rani, D. S., Nallari, P., et al. (2009). A Common MYBPC3 (Cardiac Myosin Binding Protein C) Variant Associated with Cardiomyopathies in South Asia. Nat. Genet. 41 (2), 187–191. doi:10.1038/ng.309
Flashman, E., Redwood, C., Moolman-Smook, J., and Watkins, H. (2004). Cardiac Myosin Binding Protein-C its Role in Physiology and Disease. Circulation Res. 94 (10), 1279–1289. doi:10.1161/01.RES.0000127175.21818.C2
Gagan, J., and Van Allen, E. M. (2015). Next-generation Sequencing to Guide Cancer Therapy. Genome Med. 7 (1), 80. doi:10.1186/s13073-015-0203-x
Geist Hauserman, J., Stavusis, J., Joca, H. C., Robinett, J. C., Hanft, L., Vandermeulen, J., et al. (2021). Sarcomeric Deficits Underlie MYBPC1-Associated Myopathy with Myogenic Tremor. JCI Insight 6 (19). e147612. doi:10.1172/jci.insight.147612
Gruen, M., Prinz, H., and Gautel, M. (1999). cAPK-phosphorylation Controls the Interaction of the Regulatory Domain of Cardiac Myosin Binding Protein C with Myosin-S2 in an On-Off Fashion. FEBS Lett. 453 (3), 254–259. doi:10.1016/s0014-5793(99)00727-9
Gudmundsson, S., Singer‐Berk, M., Watts, N. A., Phu, W., Goodrich, J. K., Solomonson, M., et al. (2021). Variant Interpretation Using Population Databases: Lessons from gnomAD. Hum. Mutat. 1. 1. doi:10.1002/humu.24309
Gurnett, C. A., Desruisseau, D. M., McCall, K., Choi, R., Meyer, Z. I., Talerico, M., et al. (2010). Myosin Binding Protein C1: a Novel Gene for Autosomal Dominant Distal Arthrogryposis Type 1. Hum. Mol. Genet. 19 (7), 1165–1173. doi:10.1093/hmg/ddp587
Harris, S. P., Bartley, C. R., Hacker, T. A., McDonald, K. S., Douglas, P. S., Greaser, M. L., et al. (2002). Hypertrophic Cardiomyopathy in Cardiac Myosin Binding Protein-C Knockout Mice. Circulation Res. 90 (5), 594–601. doi:10.1161/01.res.0000012222.70819.64
Harris, S. P., Lyons, R. G., and Bezold, K. L. (2011). In the Thick of it: HCM-Causing Mutations in Myosin Binding Proteins of the Thick Filament. Circ. Res. 108 (6), 751–764. doi:10.1161/CIRCRESAHA.110.231670
Helms, A. S., Thompson, A. D., Glazier, A. A., Hafeez, N., Kabani, S., Rodriguez, J., et al. (2020). Spatial and Functional Distribution of MYBPC3 Pathogenic Variants and Clinical Outcomes in Patients with Hypertrophic Cardiomyopathy. Circ Genomic Precis. Med. 13 (5), 396–405. doi:10.1161/CIRCGEN.120.002929
Jiang, J., Burgon, P. G., Wakimoto, H., Onoue, K., Gorham, J. M., O’Meara, C. C., et al. (2015). Cardiac Myosin Binding Protein C Regulates Postnatal Myocyte Cytokinesis. Proc. Natl. Acad. Sci. U.S.A. 112 (29), 9046–9051. doi:10.1073/pnas.1511004112
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Korte, F. S., McDonald, K. S., Harris, S. P., and Moss, R. L. (2003). Loaded Shortening, Power Output, and Rate of Force Redevelopment Are Increased with Knockout of Cardiac Myosin Binding Protein-C. Circulation Res. 93 (8), 752–758. doi:10.1161/01.RES.0000096363.85588.9A
Li, X., Zhong, B., Han, W., Zhao, N., Liu, W., Sui, Y., et al. (2015). Two Novel Mutations in Myosin Binding Protein C Slow Causing Distal Arthrogryposis Type 2 in Two Large Han Chinese Families May Suggest Important Functional Role of Immunoglobulin Domain C2. Plos One 10 (5), e0117158. doi:10.1371/journal.pone.0117158
Lin, B., Govindan, S., Lee, K., Zhao, P., Han, R., Runte, K. E., et al. (2013). Cardiac Myosin Binding Protein-C Plays No Regulatory Role in Skeletal Muscle Structure and Function. PloS One 8 (7), e69671. doi:10.1371/journal.pone.0069671
Lynch, T. L., Sivaguru, M., Velayutham, M., Cardounel, A. J., Michels, M., Barefield, D., et al. (2015). Oxidative Stress in Dilated Cardiomyopathy Caused byMYBPC3Mutation. Oxidative Med. Cell. Longev. 2015, 1–14. doi:10.1155/2015/424751
Markus, B., Narkis, G., Landau, D., Birk, R. Z., Cohen, I., and Birk, O. S. (2012). Autosomal Recessive Lethal Congenital Contractural Syndrome Type 4 (LCCS4) Caused by a Mutation in MYBPC1. Hum. Mutat. 33 (10), 1435–1438. doi:10.1002/humu.22122
Marston, S., Copeland, O. N., Jacques, A., Livesey, K., Tsang, V., McKenna, W. J., et al. (2009). Evidence from Human Myectomy Samples that MYBPC3 Mutations Cause Hypertrophic Cardiomyopathy through Haploinsufficiency. Circulation Res. 105 (3), 219–222. doi:10.1161/Circresaha.109.202440
McConnell, B. K., Jones, K. A., Fatkin, D., Arroyo, L. H., Lee, R. T., Aristizabal, O., et al. (1999). Dilated Cardiomyopathy in Homozygous Myosin-Binding Protein-C Mutant Mice. J. Clin. Invest. 104 (9), 1235–1244. doi:10.1172/JCI7377
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R. S., Thormann, A., et al. (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17 (1), 122. doi:10.1186/s13059-016-0974-4
McNamara, J. W., and Sadayappan, S. (2018). Skeletal Myosin Binding Protein-C: An Increasingly Important Regulator of Striated Muscle Physiology. Archives Biochem. Biophysics 660, 121–128. doi:10.1016/j.abb.2018.10.007
Méndez, I., Fernández, A. I., Espinosa, M. Á., Cuenca, S., Lorca, R., Rodríguez, J. F., et al. (2021). Founder Mutation in Myosin-Binding Protein C with an Early Onset and a High Penetrance in Males. Open Heart 8 (2), e001789. doi:10.1136/openhrt-2021-001789
Mooney, S. D., Krishnan, V. G., and Evani, U. S. (2010). Bioinformatic Tools for Identifying Disease Gene and SNP Candidates. Methods Mol. Biol. 628, 307–319. doi:10.1007/978-1-60327-367-1_17
Offer, G., Moos, C., and Starr, R. (1973). A New Protein of the Thick Filaments of Vertebrate Skeletal Myofibrils. Extractions, Purification and Characterization. J. Mol. Biol. 74 (4), 653–676. doi:10.1016/0022-2836(73)90055-7
Okagaki, T., Weber, F. E., Fischman, D. A., Vaughan, K. T., Mikawa, T., and Reinach, F. C. (1993). The Major Myosin-Binding Domain of Skeletal Muscle MyBP-C (C Protein) Resides in the COOH-Terminal, Immunoglobulin C2 Motif. J. Cell Biol. 123 (3), 619–626. doi:10.1083/jcb.123.3.619
Richard, P., Fressart, V., Charron, P., and Hainque, B. (2010). Génétique des cardiomyopathies héréditaires. Pathol. Biol. 58 (5), 343–352. doi:10.1016/j.patbio.2009.10.010
Sadayappan, S., and de Tombe, P. P. (2012). Cardiac Myosin Binding Protein-C: Redefining its Structure and Function. Biophys. Rev. 4 (2), 93–106. doi:10.1007/s12551-012-0067-x
Sarkar, S. S., Trivedi, D. V., Morck, M. M., Adhikari, A. S., Pasha, S. N., Ruppel, K. M., et al. (2020). The Hypertrophic Cardiomyopathy Mutations R403Q and R663H Increase the Number of Myosin Heads Available to Interact with Actin. Sci. Adv. 6 (14), eaax0069. doi:10.1126/sciadv.aax0069
Schlossarek, S., Schuermann, F., Geertz, B., Mearini, G., Eschenhagen, T., and Carrier, L. (2012). Adrenergic Stress Reveals Septal Hypertrophy and Proteasome Impairment in Heterozygous Mybpc3-Targeted Knock-In Mice. J. Muscle Res. Cell Motil. 33 (1), 5–15. doi:10.1007/s10974-011-9273-6
Schmitt, M. W., Kennedy, S. R., Salk, J. J., Fox, E. J., Hiatt, J. B., and Loeb, L. A. (2012). Detection of Ultra-rare Mutations by Next-Generation Sequencing. Proc. Natl. Acad. Sci. U.S.A. 109 (36), 14508–14513. doi:10.1073/pnas.1208715109
Shaffer, J. F., and Gillis, T. E. (2010). Evolution of the Regulatory Control of Vertebrate Striated Muscle: the Roles of Troponin I and Myosin Binding Protein-C. Physiol. Genomics 42 (3), 406–419. doi:10.1152/physiolgenomics.00055.2010
Shaffer, J. F., Kensler, R. W., and Harris, S. P. (2009). The Myosin-Binding Protein C Motif Binds to F-Actin in a Phosphorylation-Sensitive Manner. J. Biol. Chem. 284 (18), 12318–12327. doi:10.1074/jbc.M808850200
Song, T., McNamara, J. W., Ma, W., Landim-Vieira, M., Lee, K. H., Martin, L. A., et al. (2021). Fast Skeletal Myosin-Binding Protein-C Regulates Fast Skeletal Muscle Contraction. Proc. Natl. Acad. Sci. U.S.A. 118 (17), e2003596118. doi:10.1073/pnas.2003596118
Spirito, P., Seidman, C. E., McKenna, W. J., and Maron, B. J. (1997). The Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 336 (11), 775–785. doi:10.1056/NEJM199703133361107
Stavusis, J., Lace, B., Schäfer, J., Geist, J., Inashkina, I., Kidere, D., et al. (2019). Novel Mutations in MYBPC1 Are Associated with Myogenic Tremor and Mild Myopathy. Ann. Neurol. 86 (1), 129–142. doi:10.1002/ana.25494
Stelzer, J. E., Patel, J. R., Walker, J. W., and Moss, R. L. (2007). Differential Roles of Cardiac Myosin-Binding Protein C and Cardiac Troponin I in the Myofibrillar Force Responses to Protein Kinase A Phosphorylation. Circulation Res. 101 (5), 503–511. doi:10.1161/CIRCRESAHA.107.153650
Suay-Corredera, C., Pricolo, M. R., Herrero-Galán, E., Velázquez-Carreras, D., Sánchez-Ortiz, D., García-Giustiniani, D., et al. (2021). Protein Haploinsufficiency Drivers Identify MYBPC3 Variants that Cause Hypertrophic Cardiomyopathy. J. Biol. Chem. 297 (1), 100854. doi:10.1016/j.jbc.2021.100854
Tabish, A. M., Arif, M., Song, T., Elbeck, Z., Becker, R. C., Knöll, R., et al. (2019). Association of Intronic DNA Methylation and Hydroxymethylation Alterations in the Epigenetic Etiology of Dilated Cardiomyopathy. Am. J. Physiology-Heart Circulatory Physiology 317 (1), H168–H180. doi:10.1152/ajpheart.00758.2018
van Dijk, S. J., Dooijes, D., dos Remedios, C., Michels, M., Lamers, J. M. J., Winegrad, S., et al. (2009). Cardiac Myosin-Binding Protein C Mutations and Hypertrophic Cardiomyopathy: Haploinsufficiency, Deranged Phosphorylation, and Cardiomyocyte Dysfunction. Circulation 119 (11), 1473–1483. doi:10.1161/CIRCULATIONAHA.108.838672
Wang, T., Sun, J., Zhang, X., Wang, W.-J., and Zhou, Q. (2021). CNV-P: a Machine-Learning Framework for Predicting High Confident Copy Number Variations. PeerJ 9, e12564. doi:10.7717/peerj.12564
Watkins, H., Conner, D., Thierfelder, L., Jarcho, J. A., MacRae, C., McKenna, W. J., et al. (1995). Mutations in the Cardiac Myosin Binding Protein-C Gene on Chromosome 11 Cause Familial Hypertrophic Cardiomyopathy. Nat. Genet. 11 (4), 434–437. doi:10.1038/ng1295-434
Weber, F. E., Vaughan, K. T., Reinach, F. C., and Fischman, D. A. (1993). Complete Sequence of Human Fast-type and Slow-type Muscle Myosin-Binding-Protein C (MyBP-C). Differential Expression, Conserved Domain Structure and Chromosome Assignment. Eur. J. Biochem. 216 (2), 661–669. doi:10.1111/j.1432-1033.1993.tb18186.x
Yang, Q., Sanbe, A., Osinska, H., Hewett, T. E., Klevitsky, R., and Robbins, J. (1999). In Vivo modeling of Myosin Binding Protein C Familial Hypertrophic Cardiomyopathy. Circulation Res. 85 (9), 841–847. doi:10.1161/01.res.85.9.841
Yu, X., Yao, X., Wu, B., Zhou, H., Xia, S., Su, W., et al. (2021). Using Deep Learning Method to Identify Left Ventricular Hypertrophy on Echocardiography. Int. J. Cardiovasc Imaging 38, 759–769. Online ahead of print. doi:10.1007/s10554-021-02461-3
Dhoot, G. K., Hales, M. C., Grail, B. M., Perry, S. V., et al. (1985). The Isoforms of C Protein and Their Distribution in Mammalian Skeletal Muscle. J. Muscle Res. Cell Motil. 6 (4). 487–505. doi:10.1007/BF00712585
Glossary
cMyBP-C Cardiac myosin binding protein-C protein
fMyBP-C Fast skeletal myosin binding protein-C protein
sMyBP-C Slow skeletal myosin binding protein-C protein
MyBP-C Myosin binding protein-C protein
MYBPC1 Slow skeletal myosin binding protein-C gene
MYBPC2 Fast skeletal myosin binding protein-C gene
MYBPC3 Cardiac myosin binding protein-C gene
Ala (A) Alanine
Arg (R) Arginine
Asn (N) Asparagine
Asp (D) Aspartic acid
Cys (C) Cysteine
DCM Dilated cardiomyopathy
Gln (Q) Glutamine
Glu (E) Glutamic acid
Gly (G) Glycine
His (H) Histidine
HCM Hypertrophic cardiomyopathy
Ile (I) Isoleucine
Leu (L) Leucine
Lys (K) Lysine
Met (M) Methionine
Phe (F) Phenylalanine
Pro (P) Proline
Pyl (O) Pyrrolysine
Ser (S) Serine
Thr (T) Threonine
Trp (W) Tryptophan
Tyr (Y) Tyrosine
Val (V) Valine
SNP Single nucleotide polymorphism
NGS Next-generation sequencing
Keywords: MYBPC1, MYBPC2, MYBPC3, hypertrophic cardiomyopathy, distal arthrogryposis
Citation: Desai DA, Rao VJ, Jegga AG, Dhandapany PS and Sadayappan S (2022) Heterogeneous Distribution of Genetic Mutations in Myosin Binding Protein-C Paralogs. Front. Genet. 13:896117. doi: 10.3389/fgene.2022.896117
Received: 14 March 2022; Accepted: 07 June 2022;
Published: 27 June 2022.
Edited by:
Anastacia M. Garcia, University of Colorado Denver, United StatesReviewed by:
Hasan Orhan Akman, Columbia University Irving Medical Center, United StatesCharles K. Thodeti, University of Toledo, United States
Narasimman Gurusamy, University of Tennessee Health Science Center (UTHSC), United States
Copyright © 2022 Desai, Rao, Jegga, Dhandapany and Sadayappan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sakthivel Sadayappan, c2FkYXlhc2xAdWNtYWlsLnVjLmVkdQ==
†These authors have contributed equally to this work