 Vincenza Battaglia1†
Vincenza Battaglia1† Vincenzo Agostini1†
Vincenzo Agostini1† Elisabetta Moroni1
Elisabetta Moroni1 Giulia Colombo1
Giulia Colombo1 Gianluca Lombardo1
Gianluca Lombardo1 Nicola Rambaldi Migliore1
Nicola Rambaldi Migliore1 Paolo Gabrieli1,2
Paolo Gabrieli1,2 Maria Garofalo3
Maria Garofalo3 Stella Gagliardi3Ludvik M. Gomulski1Luca Ferretti1
Stella Gagliardi3Ludvik M. Gomulski1Luca Ferretti1 Ornella Semino1
Ornella Semino1 Anna R. Malacrida1Giuliano Gasperi1
Anna R. Malacrida1Giuliano Gasperi1 Alessandro Achilli1
Alessandro Achilli1 Antonio Torroni1
Antonio Torroni1 Anna Olivieri1*
Anna Olivieri1*- 1Dipartimento di Biologia e Biotecnologie “L. Spallanzani”, Università di Pavia, Pavia, Italy
- 2Department of Biosciences and Pediatric Clinical Research Center “Romeo ed Enrica Invernizzi”, University of Milan, Milan, Italy
- 3Molecular Biology and Transcriptomic Unit, IRCCS Mondino Foundation, Pavia, Italy
The tiger mosquito (Aedes albopictus) is one of the most invasive species in the world and a competent vector for numerous arboviruses, thus the study and monitoring of its fast worldwide spread is crucial for global public health. The small extra-nuclear and maternally-inherited mitochondrial DNA represents a key tool for reconstructing phylogenetic and phylogeographic relationships within a species, especially when analyzed at the mitogenome level. Here the mitogenome variation of 76 tiger mosquitoes, 37 of which new and collected from both wild adventive populations and laboratory strains, was investigated. This analysis significantly improved the global mtDNA phylogeny of Ae. albopictus, uncovering new branches and sub-branches within haplogroup A1, the one involved in its recent worldwide spread. Our phylogeographic approach shows that the current distribution of tiger mosquito mitogenome variation has been strongly affected by clonal and sub-clonal founder events, sometimes involving wide geographic areas, even across continents, thus shedding light on the Asian sources of worldwide adventive populations. In particular, different starting points for the two major clades within A1 are suggested, with A1a spreading mainly along temperate areas from Japanese and Chinese sources, and A1b arising and mainly diffusing in tropical areas from a South Asian source.
Introduction
Native to the tropical forests of Southeast Asia (Gratz, 2004), the tiger mosquito Aedes (Stegomyia) albopictus (Skuse, 1894) has colonized every continent except Antarctica over the last 50 years (Benedict et al., 2007; Medlock et al., 2015), and is one of the 100 most invasive species in the world, according to the Global Invasive Species Database (Global Invasive Species Database, 2010).
The initial spread began with the southeastern Asian zoophilic forest species’ move westwards to the Indian Ocean islands and eastwards to the Pacific Ocean Islands in the 19th century and in the first half of the 20th century, respectively (Knudsen, 1995). However, the species remained confined to those areas for the subsequent decades and its worldwide diffusion only started in the early 1980’s (Benedict et al., 2007; Medlock et al., 2015). Since then, Ae. albopictus has become established in the Mediterranean basin; the Americas, ranging from the United States to Argentina; Africa, where it first colonized South Africa and is now also present in the central and northern regions of the continent as well; the Middle East; Australia and the rest of Europe. In Europe, after its first occurrence in Albania in 1979 (Adhami and Murati, 1987; Benedict et al., 2007), the tiger mosquito started to expand across the continent from the 90s onward, when it was introduced into Italy through the port of Genoa (Sabatini et al., 1990; Scholte and Schaffner, 2007). Currently, it is established, or at least introduced, in most western and central European countries (European Center for Disease Prevention and Control, 2022).
This rapid and widespread expansion is due to the strong adaptability of the tiger mosquito to lay eggs in anthropized habitats, its diurnal biting habits and its preferential feeding on human blood (Benelli et al., 2020). Major roles are also played by the diapause as main strategy in enhancing overwinter survival, by desiccation-resistant eggs (Pasquali et al., 2020; Vega-Rùa et al., 2020), in addition to globalization, which favored rather frequent human-aided accidental transfer (mediated for example by commerce of plants and used vehicle tyres) (Ibáñez-Justicia et al., 2020), and a diffused lack of efficient surveillance and control methods. Moreover, climatic conditions, such as temperature and precipitations, strongly impact on its diffusion, and climate change is accelerating its spread towards the warming northern regions (Neteler et al., 2011; Roiz et al., 2011; Roiz et al., 2014). In Europe, recent climate changes have pushed Ae. albopictus to Moldova, where it was first detected in 2020 (Sulesco et al., 2021), with further introduction events been reported north of the currently colonized area, i.e. in Austria, Switzerland, and Belgium (Schoener et al., 2019).
Understanding and monitoring the tiger mosquito’s worldwide spread is of crucial importance not only due to its ecological and social impact, but also for global public health, since Ae. albopictus is a key competent vector for numerous arboviruses, like the yellow fever virus, dengue viruses, chikungunya virus, West Nile virus, Zika virus and Japanese encephalitis virus (Kraemer et al., 2015; Marconcini et al., 2021), and its recent expansion has caused severe outbreaks in many urban populations (Manni et al., 2017; Kraemer et al., 2019; Mariconti et al., 2019; Vega-Rùa et al., 2021).
Population genetics tools give access to a wealth of information on demography, geographic movements and links among populations, and the study of mitochondrial DNA (mtDNA) is best able to quickly reconstruct phylogenetic and phylogeographic relationships (Torroni et al., 2006; Gao et al., 2021). MtDNA is generally about 17 kb long in animals, thus given its size it may appear irrelevant compared to its nuclear counterpart. However, in contrast to autosomal markers, mtDNA is inherited along strict maternal lines of descent and its relatively fast molecular differentiation occurs only through the sequential accumulation of novel mutations, which often takes place during and after populations or species spread into different regions (Torroni et al., 2006; Goubert et al., 2016; da Silva et al., 2020). Thus, novel haplotypes or groups of related haplotypes (haplogroups) tend to be restricted to specific geographic areas and populations. MtDNA studies in (non-human) animals have now entered a new phase: from the analysis of short segments (generally 1 kb or less in length, either a portion of the control-region or of single protein-coding genes such as COI, ND2, ND5, or CYB) to the mitogenome-level analyses of a number of individuals (population mitogenomics) (Ruiling et al., 2018a; Fang et al., 2018; Lee et al., 2020). The mitogenome approach can be applied to all animal species, including wild invasive species such as mosquitoes. In 2016, Battaglia and co-workers (Battaglia et al., 2016) published the first entire mitogenome from a mosquito of the Italian (Rimini) laboratory-maintained strain, for which the nuclear genome is also available (Dritsou et al., 2015). This sequence, employed as reference sequence (GenBank accession number KX383916), was used to number and annotate the Ae. albopictus mitogenome. To identify the ancestral source(s) of Ae. albopictus adventive populations, Battaglia and co-workers analyzed the sequence variation at the level of the entire coding region of 27 mitogenomes, by using a phylogeographic approach. The analysis revealed that the mitogenomes clustered into five major haplogroups, namely A1a1, A1a2, A1b, A2, and A3. Among these, only the three A1 subclades were involved in the recent worldwide spread of the species, with the A1a1a1 sub-clade possibly having arisen in North America from a Japanese source. As for the other two clades, haplogroup A2 encompasses only samples from the Philippines, insular South-East Asia and the North Australian border area (Beebe et al., 2013). In light of this geographical distribution, it has been proposed that haplogroup A2 played a role in the human-mediated spread of Ae. albopictus from South-East Asia (in particular from Indonesia), towards northern Oceania (Žitko et al., 2011; Beebe et al., 2013; Zhong et al., 2013; Ismail et al., 2017; Tedjou et al., 2019). Whereas, haplogroup A3 includes only a single mitogenome from Taiwan, which suggests it might be rare and/or restricted to a specific geographic area, and that mosquitoes within this haplogroup are unlikely to be involved in the recent worldwide spread of the species.
Battaglia and co-workers’ study (2016) remained for years the only contribution to the analysis of mtDNA variability in the tiger mosquito at the mitogenome-level, while other studies continued to limit their survey to partial mtDNA sequences (Žitko et al., 2011; Beebe et al., 2013; Zhong et al., 2013; Ismail et al., 2017; Tedjou et al., 2019). In 2017, however, 17 novel mitogenomes from Algarve and Oporto, the two entry sites of the tiger mosquito into Portugal, were published, together with 31 COI sequences (Zé-Zé et al., 2020). With this addition, the mitogenome phylogeny improved considerably, but the novel sequences were from only two locations, thus with limited phylogeographic impact.
The present study analyzed, in the framework of available data, the mitogenome variation of 37 new specimens, 29 of which came from wild populations, mainly collected in countries or regions that were not included in previous studies. Our aim was to improve the mtDNA phylogeny of Ae. albopictus and better assess the geographic distributions of the previously identified haplogroups, with a particular focus on haplogroup A1, the one involved in the recent worldwide spread.
Materials and methods
Sample
A total of 37 novel mosquito samples were included in this study. Of these, 29 were from wild populations collected in Africa, the Americas, Asia and Europe (Table 1). In particular, four were from Cameroon, in Africa; 15 from the Americas: four from Florida (Vero Beach), six from California (Los Angeles), one from Mexico (Tapachula) and four from Brazil (Rio de Janeiro); four from Japan: two from Wakayama, one from Fukushima, and one from Tokyo; six from Europe: two from Italy (one from Reggio Emilia and one from Perugia) and four from France (all from Cuers, Var Department). The remaining eight were adults from laboratory-maintained strains: one from the Japanese Ikuta strain (Kanagawa Prefecture), three from the Chinese Foshan strain (Center for Disease Control and Prevention of Guangdong Province), three from the Foshan inbred strain Pavia A (FPA) and one from the Crema strain (Laboratory of Genomics and Biotechnology of Insects of Agricultural and Medical Importance, University of Pavia) (Palatini et al., 2020). These novel mitogenomes were analyzed together with those previously published (Battaglia et al., 2016; Zé-Zé et al., 2020) (Table 1). The study did not involve protected species and samples were not collected at protected sites.
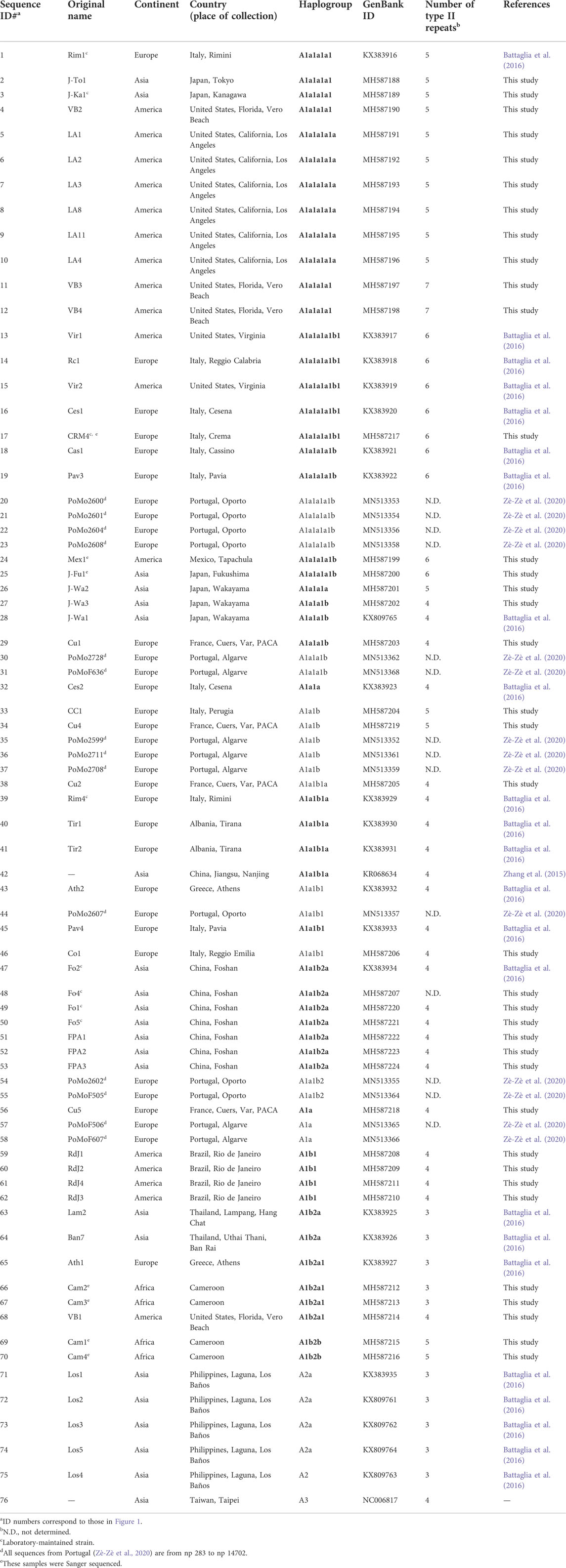
TABLE 1. Origin and haplogroup affiliation of Ae. albopictus mitogenomes analysed in this study. The 37 newly sequenced samples are included together with 39 from previous studies. Haplogroups indicated in bold in the Table are new or re-defined relative to (Battaglia et al., 2016).
DNA extraction, amplification and sequencing of Ae. albopictus mtDNAs
Genomic DNA from Ae. albopictus specimens was extracted from whole insect bodies, freshly collected or preserved in 80% ethanol at −20°C until extraction, with the Wizard® Genomic DNA Purification Kit (Promega) following the manufacturer’s standard protocol. Each mitogenome was amplified in two partially overlapping long range PCR fragments, L1 and L2 (Battaglia et al., 2016), covering the coding region comprised between nps 274–14717 (NC006817), whereas the coding-region segments between nps 1–273 and nps 14718–14893 (NC006817) and the entire control (AT-rich) region (nps 14894–16665; NC006817) were amplified in two short PCR fragments, S1 and S2, using previously reported primer sets (Battaglia et al., 2016) and amplification protocols (Battaglia et al., 2016).
Most of the samples were sequenced using a Next Generation Sequencing (NGS) platform (MiSeq system, Illumina) with the MiSeq Reagent Nano Kit, v2 (300 cycles). The sequencing library was set-up using the Nextera XT DNA sample preparation kit (Illumina), following the manufacturer’s protocol, starting from the two PCR products L1 and L2 (∼0.75 ng of each PCR product), purified with Wizard® SV Gel and PCR Clean-Up System (Promega) and quantified with a Quantus Fluorometer (Promega). On-board software created results in FASTQ format. Reads depth was generally >100X per each nucleotide position. The software Geneious v.8.1 (Kearse et al., 2012) was used to align FASTQ files to the tiger mosquito mitogenome #1 (KX383916, Figure 1) (Battaglia et al., 2016) obtained from the Italian Rimini strain (Bellini et al., 2007; Manni et al., 2015) in order to generate a single contig and a report of the sequence variants (nucleotide substitutions and indels). The threshold used for heteroplasmic positions was 20% and the average reads depth was ∼4,000X.
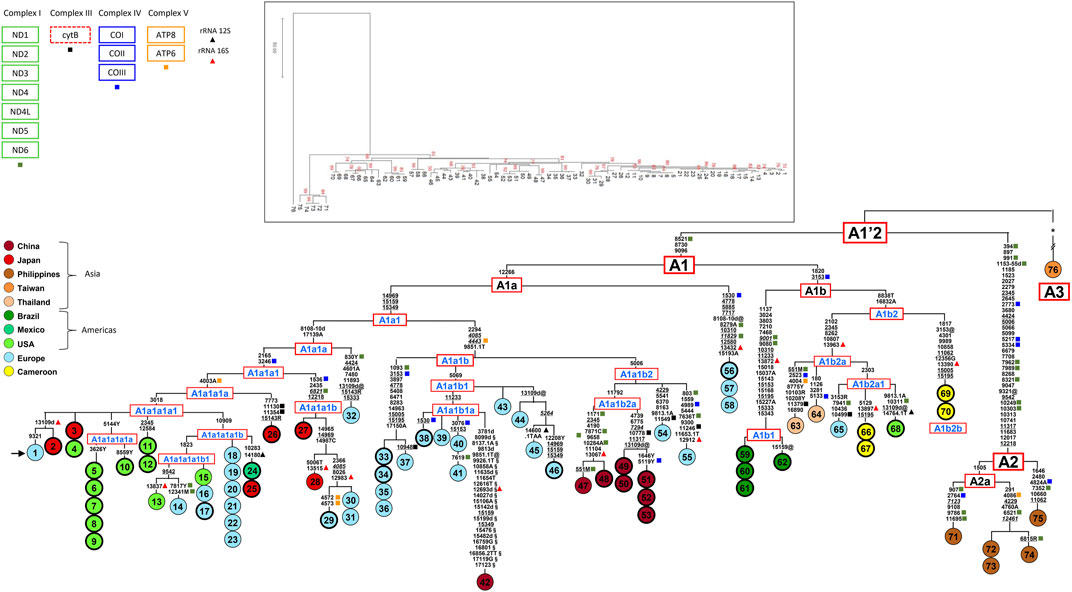
FIGURE 1. Phylogeny of Ae. albopictus mitogenomes. The tree encompasses 37 novel (circles marked by a thicker line) and 39 previously published sequences (24 from Battaglia et al., 2016, 1 from Zhang et al., 2015, 14 from Zé-Zé et al., 2020). The asterisk (*) refers to the 363 mutations (347 and 16 in the coding- and control-regions, respectively), listed in Supplementary Figure S1 (Battaglia et al., 2016), separating mitogenome #76 from the A1’2 node. For the phylogeny construction, the entire coding-region variation of all mitogenomes was included as well as some control-region mutations (from np 14897 to np 15350 and from np 16831 to np17150). The mitogenome from a mosquito of the Italian Rimini strain (#1, marked by the arrow) was employed to number the mutations shown on the branches. Mutations are transitions, unless a base is appended to indicate a transversion (to A, G, C, or T), or a suffix for indels (1, d). Heteroplasmic positions are indicated using the IUPAC nucleotide code. Recurrent mutations within the phylogeny are underlined (and in italics if present in mitogenome #76) and back mutations are marked with the suffix @. The numerous mutations shared only by the published mitogenomes #42 and #76 are marked with the suffix §. Sequences from Portugal (Zé-Zé et al., 2020) are from np 283 to np 14702; mutations out of this range were inferred according to their position in the tree. Colours illustrate geographic origins. Length variation (insertions/deletions) in polynucleotide stretches beginning at nps 3808, 8134, and 17140 were not considered. Haplogroups in blue are new relative to Battaglia et al., 2016. Non-synonymous mutations are indicated with squares, with colours matching the protein-coding gene complexes (top left in the figure); mutations in 12S and 16S rRNA genes are shown too, with blue and red diamonds. The MP tree is shown in the top inset; branch lengths are not proportional to molecular divergence.
The NGS method was used to sequence the two long range PCR fragments (L1 and L2), which covered almost the entire mitogenome coding-region. The missing coding-region segments (nps 1–273 and nps 14723–14896, KX383916) were Sanger sequenced, starting from the two short PCR fragments S1 and S2, and were assembled to the consensus sequence obtained in Geneious 8.1 with the software Sequencher 5.0 (Gene Codes Corporation), using mitogenome #1 as reference. A subset of the mitogenomes (i.e., #17, 24, 25, 66, 67, 69 and 70 in Table 1) were completely Sanger sequenced following a well-established protocol (Battaglia et al., 2016). Also in this case, they were aligned, assembled and compared to mitogenome #1, using Sequencher 5.0. The 37 Ae. albopictus mtDNA coding regions were submitted to GenBank (accession numbers MH587188–MH587224).
Phylogeny construction and phylogeographic analyses
A Maximum Parsimony (MP) tree encompassing a total of 76 mitogenomes, including 37 novel sequences from this study, 37 previously published (Battaglia et al., 2016; Zé-Zé et al., 2020), the reference sequence from Taiwan (NC006817) and one sequence from the Jiangsu Province (China) (Zhang et al., 2015), was built using mitogenome #1 as reference for numbering nucleotide positions. Two of the previously published mitogenomes (#9 and #13; Battaglia et al., 2016) were not included because of gaps in their sequences.
The tree structure was also assessed using MEGA11 (Tamura et al., 2021) employing the Tree-Bisection-Regrafting (TBR) algorithm (Nei and Kumar, 2000) with default parameters for a MP reconstruction and a number of 1000 bootstrap replications (Figure 1). To conduct this analysis, we aligned the 76 coding-region sequences by performing a Multiple-Sequence Alignment with the Clustal algorithm (Chenna et al., 2003) implemented by Sequencher 5.0. Mitogenomes taken from Battaglia et al., 2016 are 23, two are previously published Taiwanese and Chinese sequences (NC006817 and KR068634) (Chen et al., 2015), 14 are from Zè-Zè et al., 2020 and 37 from this study (only one of the three FPA mitogenomes was considered).
A Bayesian tree was also inferred using BEAST1.10.4 (Drummond and Rambaut, 2007), under the HKY substitution model (gamma-distributed rates plus invariant sites) and running 50,000,000 iterations, with samples drawn every 10,000 Markov chain Monte Carlo (MCMC) steps. It was visualized using FigTree v.1.4.2. Phylogeny reconstruction was performed considering all the nucleotide substitutions (excluding indels and heteroplasmies) in the coding region (from np 1 to np 14896, relative to mitogenome #1) and the five informative control-region mutations 14969, 15159, 15349, 16832A, and 17139A.
To reconstruct the spatial dynamics of the tiger mosquito across the world, we performed a continuous phylogeographic analysis with BEAST 1.10.4, as described by (Dellicour et al., 2021). We used the same alignment of the phylogenetic reconstruction; sampling dates and coordinates were associated to each sample and Bayesian SkyGrid Coalescence model was set up under the HKY substitution model. We summarized the information within the sampled trees with TreeAnnotator, while the continuous phylogeographic analysis was visualized with SpreaD3 (Bielejec et al., 2016).
To evaluate population size trends through time, we obtained a Bayesian Skyline Plot (BSP) from BEAST 1.10.4 under the same model used for phylogeny reconstruction and considering a mutation rate calculated with Maximum Likelihood (ML) analysis. PamlX 1.3.1 (Yang 1997) was used to obtain ML estimates, assuming the HKY85 mutation model with gamma-distributed rates (approximated by a discrete distribution with 32 categories). Mutational distances were converted into years assuming an age of 32 years for haplogroup A1a1a1a1 (Bonizzoni et al., 2013) as a prior. This age was attributed taking into account that 1) this haplogroup most likely expanded first into North America and then into Europe; 2) Ae. albopictus was first detected in the United States (Port of Houston) in 1985; and iii) most of the samples were collected in 2017. We visualized the BSP obtained in plot with Tracer v1.5 and then converted it to an Excel graph by using a generation time that was twice (40 days), because of diapause in temperate regions, of that previously reported (20 days) (Nur Aida et al., 2008) for populations living in tropical areas.
Results and discussion
To improve knowledge of Ae. albopictus mitogenome variation and acquire more information on the geographic sources of the recently established adventive populations, 37 novel mitogenomes were sequenced, many from areas not previously assessed (Battaglia et al., 2016; Zé-Zé et al., 2020). Table 1 reports the geographic origins of the 76 Ae. albopictus samples included in our study as well as their haplogroup/sub-haplogroup affiliations, which were re-defined, in some cases, on the basis of our more comprehensive phylogenetic survey.
The updated mitogenome phylogeny of Ae. albopictus
The 37 novel mitogenomes were all from adventive Ae. albopictus populations, and all clustered into haplogroup A1. The lack of A2 or A3 mitogenomes confirms A1 as the haplogroup involved in the worldwide spread of the species. Their inclusion in the global Ae. albopictus mitogenome phylogeny allowed for the identification of a rather large number of previously unidentified sub-branches within A1, improving knowledge of their geographic distributions. As shown in MP tree of Figure 1 and the Bayesian tree of Supplementary Figure S1, haplogroup A1 comprises two major sister clades, A1a and A1b, which in turn are subdivided into numerous sub-branches. The overall tree structure is virtually identical with the two approaches, indicating high degree of internal consistency for all major branches.
Regarding A1a, the newly sequenced mitogenome #56 from Southern France bears the same haplotype as two Portuguese mitogenomes (#57–58) from Algarve (Figure 1). This shared haplotype redefines the distinguishing mutations of the node from which A1a departs, which is now solely defined by the transition at nucleotide position (np) 12266. Haplogroup A1a1 is further subdivided into A1a1a and A1a1b (formerly A1a1 and A1a2 in Battaglia et al., 2016). Within haplogroup A1a1a, two mitogenomes from Japan (#27–28) and three from Europe (#29–31) form a new well-defined clade, here named A1a1a1b, characterized by the mutational motif 1536–2435–6821–12218. The introduction of new samples from Japan and Mexico modified the former node A1a1a1 (Battaglia et al., 2016), and the haplogroup affiliation of the previously published samples descending from that node. The original (A1a1a1) node is now split into an ancestral node defined solely by the transversion T to A at np 4003, which includes the mitogenome from Wakayama #26 (Japan) as well as the rather common derived clade (A1a1a1a1) marked by the transition at np 3018. The latter includes the mitogenomes from Italy, Portugal and the United States previously classified as A1a1a1 (Battaglia et al., 2016; Zé-Zé et al., 2020) as well as 14 of the novel mitogenomes: three from different regions of Japan (#2, 3 and 25), five from California (#5–10), three from Florida (#4, 11 and 12), one from Mexico (#24) and the mitogenome from the Italian Crema strain (#17). The updated phylogenetic tree also permitted the identification of two major sub-haplogroups within A1a1a1a1, named A1a1a1a1a and A1a1a1a1b, the latter further split into two novel sub-branches.
The nomenclature of haplogroup A1a2 (Battaglia et al., 2016), now re-named as A1a1b, was also updated. It encompasses samples from Europe, mostly collected in Mediterranean countries (Albania, Greece, Italy, Portugal and Southern France) as well as from China. The Chinese samples form a sub-haplogroup (A1a1b2a) made up of four haplotypes, all found in the Foshan strain: the virtually identical mitogenomes #47 (Battaglia et al., 2016) and #48, which differ only by a segregating heteroplasmic variant, and mitogenomes #49–53, which form a sister branch with two haplotypes differing for two segregating heteroplasmic mutations. As expected, the three mitogenomes (#51–53) from the Foshan inbred strain, Pavia A, harbored the same haplotype. These findings indicate that at least two females, with distinct but related A1a1b2a mitogenomes, founded the Foshan strain.
Haplogroup A1b is split into A1b1 and A1b2. The first comprises only mitogenomes from Brazil, either identical (#59–61) or differing for a single reversion (#62), sharing a mutational motif of ten coding-region mutations, which include the transition at np 7210 in ND5, already identified as distinguishing marker for Brazilian Ae. albopictus (Birungi and Munstermann, 2002; Battaglia et al., 2016; Vega-Rùa et al., 2020). The composition of A1b2 is more geographically heterogeneous, since it encompasses the four samples (#66–67 and #69–70) from Africa (Cameroon), which are characterized by two divergent haplotypes within the branches A1b2a and A1b2b, both newly defined here.
Tracing the recent spread of Ae. albopictus from its native range to new temperate and tropical areas
The 37 novel mitogenomes included in this study cluster into haplogroup A1, thus confirming this haplogroup as the only one involved in the worldwide spread of Ae. albopictus. It should be noted that A1 encompasses 70 of the 76 mitogenomes included in our phylogeny (Figure 1), but is made up by only 42 distinct haplotypes, with some differing from each other by only one or a few heteroplasmic variants, thus indicating the extremely recent occurrence of the distinguishing mutation(s).
The inclusion of the novel mitogenomes greatly improved the detail of the global mitogenome phylogeny, uncovering numerous previously unidentified branches and sub-branches, in particular within haplogroups A1a1a, A1a1b and A1b, thereby extending and improving previous results and conclusions.
It should be underlined that many of the tiger mosquitoes analyzed here (25 out of 37) were sampled in geographic areas that were not represented in previous studies, in particular California, Florida, Mexico, France, Cameroon, and different regions of Italy and Japan. This led to a better description of the geographic distributions of both the already identified haplogroups and the new ones.
Temperate areas
Haplogroup A1a encompasses the majority of the mitogenomes (#1–55) in our phylogeny and is now present in the temperate areas of all continents (Figure 1). The identification of many previously unknown branches and sub-branches within haplogroup A1a allows new inferences to be made concerning the Asian sources of adventive populations and the migration routes followed by the mosquitoes that founded these recently established populations (Figure 2). A1a1a1 (mitogenomes #1–31), which has been detected in Europe, North America and Asia, is likely to have begun its spread from Japan. Indeed, the six Asian A1a1a1 haplotypes are all from Japan, and some are identical, or extremely closely related, to those found in Europe and North America. The spread of A1a1a1 involved at least four females, as attested by the four different subclades bearing Japanese mitogenomes, which might have originated from different regions of Japan. The out-of-Asia spread of these mitogenomes probably followed a Pacific route, initially reaching the coastal areas of California and western Mexico, then the eastern coast of the United States (Virginia and Florida, haplogroup A1a1a1a1), and finally Europe (Italy, France, Portugal).
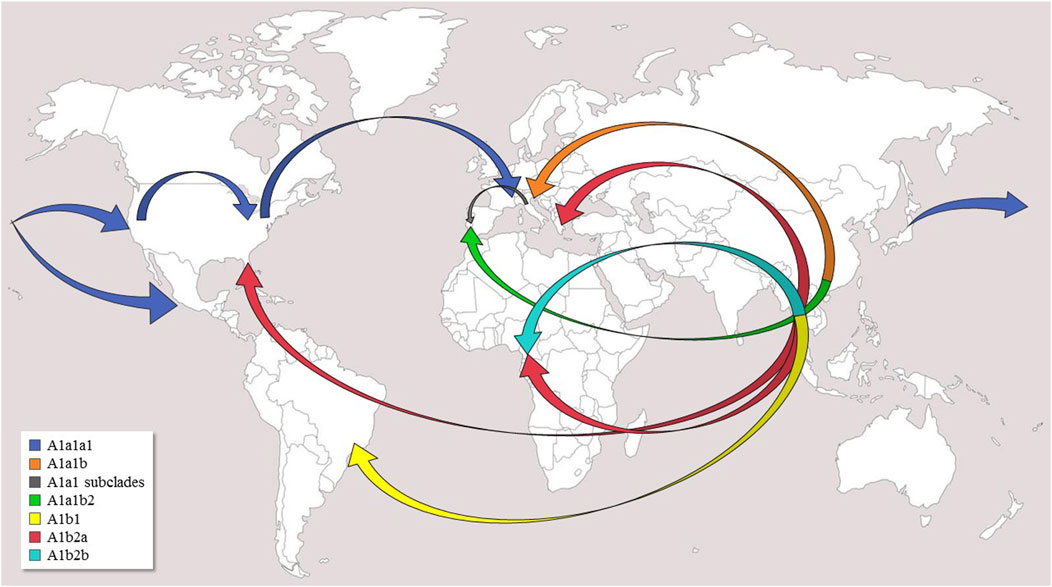
FIGURE 2. Possible worldwide diffusion routes of Ae. albopictus. Arrows indicate postulated diffusion routes from the native home-range (South-East Asia) and subsequent dispersals of major clades and subclades within haplogroup A1. The original world map is from the website (http://www.freeworldmaps.net).
Previous data had already identified genetic links between Japanese, North American and Italian populations, suggesting a Japanese source for the spread of Ae. albopictus to North America and later to Italy (Kambhampati et al., 1991; Urbanelli et al., 2000; Battaglia et al., 2016; Manni et al., 2017; Vega-Rùa et al., 2020), but our observations on the different subclades of A1a1a1 add further details to the overall picture. For instance, our Californian samples from Los Angeles all clustered into sub-haplogroup A1a1a1a1a, highlighting a single Japanese source. This result partially confirms previous data suggesting diversified Asian sources (Japan, South China, Singapore) for Californian tiger mosquito populations (Zhong et al., 2013).
A new important piece of information is provided by the single Mexican mitogenome (#24), which belongs to A1a1a1a1b. It harbors a sequence identical to the Japanese mitogenome #25. When considering that the Mexican mosquito was collected in Tapachula, a major trading site in the far south of the Mexican Pacific coast, this finding suggests that Ae. albopictus was introduced into Mexico either by land, from the United States (Pech-May et al., 2016; Vega-Rùa et al., 2020), or by sea directly from a Japanese source.
Another route of spread is possibly marked by the distribution of A1a1b, the sister branch of A1a1a (Figure 2). Taking into account that the A1a1b mitogenomes found in Europe cluster into at least four rather divergent subsets (Figure 1), and that so far all Asian members of A1a1b are from China, the most parsimonious scenario, at least with currently available data, is that A1a1b arose in China, possibly from multiple geographic sources, and from there it directly reached Mediterranean Europe (Albania, Greece, Italy, Southern France, and Portugal). However, an additional intermediate step through La Rèunion (as often suggested, Vega-Rùa et al., 2021) cannot be ruled out based on our phylogeny, since it lacks mitogenomes from this geographic area, which played a pivotal role in the demographic history of Ae. albopictus. Our data depict a scenario in which the Mediterranean basin was affected by multiple invasions of the tiger mosquito from different geographic areas and at different times (Vega-Rùa et al., 2021). In particular, our findings are in agreement with the scenario that Italian tiger mosquitoes have at least a dual origin: from North America, as discussed above for A1a1a, and from an eastern Asian source, although indirectly, i.e., reaching Italy possibly through other Mediterranean sources such as Albania and/or Greece, both being among the most important sea-trading partners with China (Enserink, 2008; Battaglia et al., 2016).
Also, the inclusion of four specimens from southeastern France further details the worldwide spread of Ae. albopictus. They were all collected in Cuers, close to the beaches of Hyère, in the Provence-Alpes-Côte d’Azur, between Toulon (a cargo port and French naval base) and Marseille, the second busiest port of the Mediterranean Sea, and they harbored four distinct haplotypes, falling into haplogroups A1a1a1b (#29), A1a1b (#34), A1a1b1a (#38), and A1a, which directly descends from the root (#56). Our phylogeny takes the Portuguese samples from Zé-Zè et al., 2020 into consideration as well. The first detection of tiger mosquitoes in Portugal dates back to no earlier than 2017, when most of the other Mediterranean countries had already been invaded. The Portuguese haplotypes fall within haplogroup A1a1 variability, belonging to at least seven different subclades. The overall phylogeography of haplogroup A1a1 points to multiple independent introduction events in both France and Portugal, from different geographic regions, via human-mediated transports. Some events were promoted by land transportations, mainly from Italy (as already suggested by Zé-Zé et al., 2020), probably passing through Southern France, towards both the Algarve and Oporto regions, as marked by the A1a1a1a1b (#20–23), A1a1a1b (#30–31), A1a1b (#35–37) and A1a1b1 (#44) haplotypes, and by the two mitogenomes from Algarve descending from the A1a root (#57–58) bearing the same haplotype of the previously mentioned French mitogenome #56. Other introduction events appear instead to have involved international connections through seaports. This seems particularly true for the Oporto region, where a direct link with China is testified by haplogroup A1a1b2, which for the moment includes only Chinese and Oporto mitogenomes (#47–55). However, our phylogeny lacks mitogenomes from Spain, a key geographic region whose inclusion could be vital for better depicting the geographic spread of this invasive species in Iberia. Overall, this pattern strongly hints to the role of both Mediterranean ports and land trade routes in the mosquito’s spread within the context of worldwide trade.
In brief, it appears that human-mediated trade activities triggered the diffusion of different A1a subclades in worldwide temperate regions. Thus, the model of this invasive species’ spread seems now consolidated, based on long inter-continental distances covered by sea-trade, followed by localized spread by land around cities where goods arriving from Asia are sorted and redirected for further distribution.
Tropical and sub-tropical areas
With the exception of one mitogenome (#65) from Athens (Greece), the two A1b sub-branches (A1b1 and A1b2) exclusively encompass samples from either tropical (Brazil, Thailand, Cameroon) or sub-tropical (Florida) areas (Figure 1).
A1b1 is made up exclusively of the four Brazilian mitogenomes from Rio de Janeiro, an urban context with a tropical climate. In the refined phylogeny they cluster tightly, virtually as a single haplotype. The mutational motif of this branch includes the transition at np 7210 in the ND5 gene, which was often surveyed in early studies. This mutation was previously reported as a marker of Brazilian Ae. albopictus (Birungi and Munstermann, 2002) and identified also in two other ND5 published haplotypes: one from Phuntsholing in Southern Bhutan (JQ436953) and one from Chiang Mai in Thailand (JQ436956) (Porretta et al., 2012; Battaglia et al., 2016). These findings suggest that Indochina might be the ancestral source of the A1b1 mitogenomes found in Brazil, pointing to a possible direct connection between the two geographic areas (Battaglia et al., 2016; Goubert et al., 2016). This observation is in contrast with the very first allozyme results (Kambhampati et al., 1991) claiming that both Brazilian and United States tiger mosquitoes had originated in Japan, suggesting, instead, separate introductions in Brazil and United States, from different sources (Birungi and Munstermann, 2002; Vega-Rùa et al., 2020). Such a scenario of distinct introduction events is supported also by the ability to induce diapause, absent in tropical areas (such as Indochina and Brazil) and present in temperate areas (such as Japan and United States) (Batz et al., 2020).
In contrast, A1b2 is extremely well-differentiated in terms of both haplotype variation and geographical distribution (Figure 1). Indeed, its sub-clades A1b2a and A1b2b include samples from geographically distant tropical countries (Cameroon and Thailand), the mitogenome (#65) from Athens and one (#68) from Florida, in temperate and sub-tropical contexts, respectively. The phylogenetic links between these mitogenomes suggest Thailand as the Asian ancestral source, or at least an intermediate one, for the now worldwide-diffused A1b2a branch. However, neighboring countries in Indochina are also strong candidates. The sub-branch A1b2a1 links closely the Greek and Florida samples, as well as two from Cameroon (#66 and #67, with the same haplotype). It should be underlined that Port Piraeus in Athens is a hub for shipping from Asia, including Thailand, to the Mediterranean region (Manni et al., 2017; Vega-Rùa et al., 2020).
An additional recent introduction in Cameroon can be hypothesized for mitogenomes #69 and #70, again with the same haplotype, which form A1b2b, whose very distinctive mutational motif has not been found elsewhere. However, a previous survey of the COI variation in 259 Ae. albopictus from Australasia, South-East Asia, islands of the Indian and Pacific Oceans as well as the United States (Maynard et al., 2007) revealed ten samples harboring the transitions at nps 1820 and 1817, located at the A1b and A1b2b nodes, respectively (Figure 1). These two transitions are part of the mutational motif shared by the Cameroonian mitogenomes #69 and #70. Interestingly, eight of the ten COI sequences harboring both mutations are from Malaysia (haplotype accession numbers KY907368-69 from Kuala Lumpur; KY907370-71 from Ipoh) and two from Singapore (haplotype accession number KY907372), thus strongly favoring the hypothesis that the Malaysian Peninsula was the second source of Cameroon tiger mosquitoes.
The overall scenario marked by the A1b sub-branches suggests that (a) haplogroup A1b2 derives from an Asian tropical area that definitely includes Thailand and Malaysia, but other as-yet unsurveyed geographical sources in Indochina are also possible; (b) the current (and limited) distribution of A1b2 in adventive populations might be restrained by climate conditions. Indeed, outside tropical areas, we have found A1b2 mitogenomes only in Athens (Greece) and Florida, both with climate conditions that are indeed quite close to those of sub-tropical areas.
Reconstruction of the dispersal history through a continuous phylogeographic analysis
To further assess the trajectory of tiger mosquito across its current distribution range, we also performed a continuous phylogeographic analysis (Dellicour et al., 2021), resulting from Bayesian molecular clock inference of sequence and trait evolutionary processes, by considering both sampling dates and coordinates as traits. The resulting dispersal map is depicted in Supplementary Figure S2. It confirms that 1) South East Asia is the native homeland of the tiger mosquito, as revealed by darker red shades indicating an older and prolonged presence in those geographic areas, and 2) supports the trajectory routes of Figure 2, which were obtained by taking into consideration historical data. However, it is worth mentioning that our samples were all collected in the last twelve years, despite tiger mosquitoes colonized the sampled regions earlier, in some cases several decades prior to twelve years ago, and at different times.
Population size trend
To investigate Ae. albopictus population size trends, a Bayesian Skyline Plot (BSP) was also obtained (Supplementary Figure S3), assuming that haplogroup A1a1a1a1 expanded in North America and taking into account that Ae. albopictus was first recorded in continental United States in 1985. An initial episode of population expansion started at ∼120 years ago in agreement with available information concerning the first diffusion of this species outside its ancestral East Asian homeland, which is dated back at the beginning of the last century. However, it should be underlined that the poor representation of Asian samples in our data set does not allow to identify how many Asian founder haplotypes gave rise to the A1a1a1a1 branches detected in Europe and North America. Thus, the assumption of an age of 32 years for the expansion of A1a1a1a1 needs to be reassessed in the future when additional Asian mitogenomes will become available.
Conclusion
The analysis of 37 novel mitogenomes of Ae. albopictus from numerous wild adventive populations and some laboratory strains, in the context of previously available data, allowed an in-depth refinement of the mitogenome phylogeny of Ae. albopictus to be extensively refined. This species is characterized by three major divergent haplogroups (A1, A2 and A3), but only haplogroup A1 with its numerous clades and sub-clades, largely re-defined in this study, is actively involved in the recent worldwide spread.
As expected in populations founded by a few females from the same geographic source, thus strongly affected by founder events and genetic drift, shared haplotypes characterize most if not all the mitogenomes deriving from the same lab strain, for instance mitogenomes #49–50 from the Chinese Foshan strain and mitogenomes #51–53 from the Foshan inbred strain Pavia A (FPA). However, the major role played by founder events in the current distribution of the tiger mosquito mitogenome variation is also clearly visible in many of the wild adventive populations. For instance, five of the six mosquitoes (#5–10) from Los Angeles (California) share the same haplotype, and the sixth (#10) harbors a closely related haplotype within the same local sub-haplogroup (A1a1a1a1a). A similar scenario, three identical mitogenomes and one differing by a single reversion, is highlighted by the four mitogenomes (#59–62) from Rio de Janeiro (Brazil), which form the highly divergent sub-haplogroup A1b1. However, in other cases the founder event is not restricted to a single country, but shared by multiple countries or even by distant continents, highlighting a series of clonal and sub-clonal founding events, which occurred sequentially and involved the same founder mitogenome or one of its immediate, often still heteroplasmic, derivatives. This observation also implies that the fast and extremely invasive spread of tiger mosquitoes often started and/or involved few individuals, at least from a female perspective, thus pointing to the crucial role of control strategies.
The connections between the haplotypes/branches detected in the different areas confirm the previously proposed scenario and provides fresh clues to the Asian sources of European, African and American adventive populations.
Haplogroup A1a harbors two main starting points, Japan (A1a1a) and China (A1a1b), and spread mainly along the temperate belt. From Japan, A1a1a most likely moved first to North America and from there to Europe, while A1a1b arrived directly in Europe from China. In contrast, A1b arose in the tropical areas of South East Asia, most likely Thailand and/or Malaysia. From there it expanded to the tropical areas of Africa (Cameroon), possibly with two separate invasion events, but also reached Greece and Florida, which are not in tropical areas, but are characterized by climatic conditions not too dissimilar to those of sub-tropical regions.
Our new insights into Ae. albopictus mitogenome variation could provide important information to control the current spread of this invasive species and limit its detrimental social, medical and economic effects.
Future studies should perform an assessment of the Ae. albopictus mitogenome sequence variation worldwide, also in comparison with both closely related and distant species, in order to identify candidate mtDNA mutations that might play a functional role and facilitate the spread of specific haplogroups into temperate regions. Moreover, functional evaluations/analyses involving living mosquitoes (from lab strains and possibly wild populations), cells or cellular extracts and larvae should be considered to confirm/dismiss the potential adaptive role of candidate mtDNA mutations.
Finally, further analyses are needed to clarify whether the extreme divergence of the single A3 mitogenome from all other mitogenomes is simply due to different arrival sources, or to local population divergences (Ruiling et al., 2018b) or to the presence in Taiwan of cryptic species, as already shown in some regions of China (Guo et al., 2018).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
VB, AT, and AO conceived and supervised the project; PG, LG, AM, and GG performed the sample collection; VB and PG conducted the experiments, with guidance from OS, AM, GG, AA, and AO; VB and MG performed sequencing runs, under the supervision of SG; VB, VA, EM, GC, GL, and NRM performed the analyses; LF, OS, AA, AT, and AO contributed reagents/materials/analytical tools; VB, VA, AT, and AO wrote the manuscript with inputs from LF, OS, and AA. All authors reviewed and approved the manuscript.
Funding
This research received support from: the Italian Ministry of Education, University and Research (MIUR): the project PRIN 2017 2017CWHLHY (to AT) and Dipartimenti di Eccellenza Program (2018–2022)—Department of Biology and Biotechnology “L. Spallanzani” University of Pavia (to AA, AO, AT, LF, and OS); and from the Fondazione Adriano Buzzati—Traverso for the Cavalli-Sforza fellowship (to NRM).
Acknowledgments
We thank Mariangela Bonizzoni for providing the samples from Mexico and the Foshan (FPA) and Crema (CRM) strains.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.931163/full#supplementary-material
References
Adhami, J., and Murati, N. (1987). Presence du moustique Aedes albopictus en Albanie. Rev. Mjekesore 1, 13–16.
Battaglia, V., Gabrieli, P., Brandini, S., Capodiferro, M. R., Javier, P. A., Chen, X.-G., et al. (2016). The worldwide spread of the tiger mosquito as revealed by mitogenome haplogroup diversity. Front. Genet. 7, 208. doi:10.3389/fgene.2016.00208
Batz, Z. A., Clemento, A. J., Fritzenwanker, J., Ring, T. J., Garza, J. C., and Armbruster, P. A. (2020). Rapid adaptive evolution of the diapause program during range expansion of an invasive mosquito. Evolution 74, 1451–1465. doi:10.1111/evo.14029
Beebe, N. W., Ambrose, L., Hill, L. A., Davis, J. B., Hapgood, G., Cooper, R. D., et al. (2013). Tracing the tiger: Population genetics provides valuable insights into the Aedes (stegomyia) albopictus invasion of the australasian region. PLoS Negl. Trop. Dis. 7, e2361. doi:10.1371/journal.pntd.0002361
Bellini, R., Calvitti, M., Medici, A., Carrieri, M., Celli, G., and Maini, S. (2007). “Use of the sterile insect technique against Aedes albopictus in Italy: First results of a pilot trial,” in Use of the sterile insect technique against Aedes albopictus in Italy: First results of a pilot trial” in area-wide control of insect pests. Editors M. J. B. Vreysen, A. S. Robinson, and J. Hendrichs (Dordrecht, Netherlands: Springer), 505–515. doi:10.1007/978-1-4020-6059-5_47
Benedict, M. Q., Levine, R. S., Hawley, W. A., and Lounibos, L. P. (2007). Spread of the tiger: Global risk of invasion by the MosquitoAedes albopictus. Vector-Borne Zoonotic Dis. 7, 76–85. doi:10.1089/vbz.2006.0562
Benelli, G., Wilke, A. B. B., and Beier, J. C. (2020). Aedes albopictus (Asian tiger mosquito). Trends Parasitol. 36, 942–943. doi:10.1016/j.pt.2020.01.001
Bielejec, F., Baele, G., Vrancken, B., Suchard, M. A., Rambaut, A., and Lemey, P. (2016). SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169. doi:10.1093/molbev/msw082
Birungi, J., and Munstermann, L. E. (2002). Genetic structure of Aedes albopictus (Diptera: Culicidae) populations based on mitochondrial ND5 sequences: Evidence for an independent invasion into Brazil and United States. an 95, 125–132. doi:10.1603/0013-8746(2002)095[0125:gsoaad]2.0.co;2
Bonizzoni, M., Gasperi, G., Chen, X., and James, A. A. (2013). The invasive mosquito species Aedes albopictus: Current knowledge and future perspectives. Trends Parasitol. 29, 460–468. doi:10.1016/j.pt.2013.07.003
Chen, X.-G., Jiang, X., Gu, J., Xu, M., Wu, Y., Deng, Y., et al. (2015). Genome sequence of the Asian Tiger mosquito, Aedes albopictus , reveals insights into its biology, genetics, and evolution. Proc. Natl. Acad. Sci. U.S.A. 112, E5907–E5915. doi:10.1073/pnas.1516410112
Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G., et al. (2003). Multiple sequence alignment with the clustal series of programs. Nucleic Acids Res. 31, 3497–3500. doi:10.1093/nar/gkg500
da Silva, A. F., Machado, L. C., de Paula, M. B., da Silva Pessoa Vieira, C. J., de Morais Bronzoni, R. V., de Melo Santos, M. A. V., et al. (2020). Culicidae evolutionary history focusing on the Culicinae subfamily based on mitochondrial phylogenomics. Sci. Rep. 10, 18823. doi:10.1038/s41598-020-74883-3
Dellicour, S., Gill, M. S., Faria, N. R., Rambaut, A., Pybus, O. G., Suchard, M. A., et al. (2021). Relax, keep walking - a practical guide to continuous phylogeographic inference with BEAST. Mol. Biol. Evol. 38, 3486–3493. doi:10.1093/molbev/msab031
Dritsou, V., Topalis, P., Windbichler, N., Simoni, A., Hall, A., Lawson, D., et al. (2015). A draft genome sequence of an invasive mosquito: An ItalianAedes albopictus. Pathogens Glob. Health 109, 207–220. doi:10.1179/2047773215Y.0000000031
Drummond, A. J., and Rambaut, A. (2007). Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214. doi:10.1186/1471-2148-7-214
European Centre For Disease Prevention and Control. (2022). Ecdc. Available online: http://Ecdc.Europa.Eu.
Fang, Y., Zhang, J., Wu, R., Xue, B., Qian, Q., and Gao, B. (2018). Genetic polymorphism study on Aedes albopictus of different geographical regions based on DNA barcoding. BioMed Res. Int. 2018, 1–10. doi:10.1155/2018/1501430
Gao, J., Zhang, H. D., Guo, X. X., Xing, D., Dong, Y.-D., Lan, C.-J., et al. (2021). Dispersal patterns and population genetic structure of Aedes albopictus (Diptera: Culicidae) in three different climatic regions of China. Parasites Vectors 14 (1), 12. doi:10.1186/s13071-020-04521-4
Global Invasive Species Database. (2010). Iucngisd. AvailableAt: http://Www.Iucngisd.Org/Gisd/100_worst.Php.
Goubert, C., Minard, G., Vieira, C., and Boulesteix, M. (2016). Population genetics of the Asian tiger mosquito Aedes albopictus, an invasive vector of human diseases. Heredity 117, 125–134. doi:10.1038/hdy.2016.35
Gratz, N. G. (2004). Critical review of the vector status of Aedes albopictus. Med. Vet. Entomol. 18, 215–227. doi:10.1111/j.0269-283X.2004.00513.x
Guo, Y., Song, Z., Luo, L., Wang, Q., Zhou, G., Yang, D., et al. (2018). Molecular evidence for new sympatric cryptic species of Aedes albopictus (Diptera: Culicidae) in China: A new threat from Aedes albopictus subgroup? Parasites Vectors 11, 228. doi:10.1186/s13071-018-2814-8
Ibáñez-Justicia, A., Smitz, N., den Hartog, W., van de Vossenberg, B., De Wolf, K., Deblauwe, I., et al. (2020). Detection of exotic mosquito species (Diptera: Culicidae) at international airports in Europe. Ijerph 17, 3450. doi:10.3390/ijerph17103450
Ismail, N.-A., Adilah-Amrannudin, N., Hamsidi, M., Ismail, R., Dom, N. C., Ahmad, A. H., et al. (2017). The genetic diversity, haplotype analysis, and phylogenetic relationship of Aedes albopictus (Diptera: Culicidae) based on the cytochrome oxidase 1 marker: A Malaysian scenario. J. Med. Entomol. 54, 1573–1581. doi:10.1093/jme/tjx126
Kambhampati, S., Black, W. C., and Rai, K. S. (1991). Geographic origin of the US and Brazilian Aedes albopictus inferred from allozyme analysis. Heredity 67, 85–94. doi:10.1038/hdy.1991.67
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi:10.1093/bioinformatics/bts199
Knudsen, A. B. (1995). Global distribution and continuing spread of Aedes albopictus. Parassitologia 37, 91–97.
Kraemer, M. U. G., Reiner, R. C., Brady, O. J., Messina, J. P., Gilbert, M., Pigott, D. M., et al. (2019). Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 4, 854–863. doi:10.1038/s41564-019-0376-y
Kraemer, M. U. G., Sinka, M. E., Duda, K. A., Mylne, A., Shearer, F. M., Brady, O. J., et al. (2015). The global compendium of Aedes aegypti and Ae. albopictus occurrence. Sci. Data 2, 150035. doi:10.1038/sdata.2015.35
Lee, E., Yang, S.-C., Kim, T.-K., Noh, B.-E., Lee, H. S., Kim, H., et al. (2020). Geographical genetic variation and sources of Korean Aedes albopictus (Diptera: Culicidae) populations. J. Med. Entomol. 57, 1057–1068. doi:10.1093/jme/tjz254
Manni, M., Gomulski, L. M., Aketarawong, N., Tait, G., Scolari, F., Somboon, P., et al. (2015). Molecular markers for analyses of intraspecific genetic diversity in the Asian Tiger mosquito, Aedes albopictus. Parasites Vectors 8, 188. doi:10.1186/s13071-015-0794-5
Manni, M., Guglielmino, C. R., Scolari, F., Vega-Rúa, A., Failloux, A.-B., Somboon, P., et al. (2017). Genetic evidence for a worldwide chaotic dispersion pattern of the arbovirus vector, Aedes albopictus. PLoS Negl. Trop. Dis. 11, e0005332. doi:10.1371/journal.pntd.0005332
Marconcini, M., Pischedda, E., Houé, V., Palatini, U., Lozada-Chávez, N., Sogliani, D., et al. (2021). Profile of small RNAs, vDNA forms and viral integrations in late chikungunya virus infection of Aedes albopictus mosquitoes. Viruses 13, 553. doi:10.3390/v13040553
Mariconti, M., Obadia, T., Mousson, L., Malacrida, A., Gasperi, G., Failloux, A.-B., et al. (2019). Estimating the risk of arbovirus transmission in Southern Europe using vector competence data. Sci. Rep. 9, 17852. doi:10.1038/s41598-019-54395-5
Maynard, A. J., Ambrose, L., Cooper, R. D., Chow, W. K., Davis, J. B., Muzari, M. O., et al. (2007). Tiger on the prowl: Invasion history and spatio-temporal genetic structure of the asian tiger mosquito Aedes albopictus (skuse 1894) in the indo-pacific. PLoS Negl. Trop. Dis. 11, e0005546. doi:10.1371/journal.pntd.0005546
Medlock, J. M., Hansford, K. M., Versteirt, V., Cull, B., Kampen, H., Fontenille, D., et al. (2015). An entomological review of invasive mosquitoes in Europe. Bull. Entomol. Res. 105, 637–663. doi:10.1017/S0007485315000103
Nei, M., and Kumar, S. (2000). Molecular evolution and phylogenetics. New York: Oxford University Press.
Neteler, M., Roiz, D., Rocchini, D., Castellani, C., and Rizzoli, A. (2011). Terra and aqua satellites track tiger mosquito invasion: Modelling the potential distribution of Aedes albopictus in North-eastern Italy. Int. J. Health Geogr. 10, 49. doi:10.1186/1476-072X-10-49
Nur Aida, H., Abu Hassan, A., Nurita, A. T., Che Salmah, M. R., and Norasmah, B. (2008). Population analysis of Aedes albopictus (Skuse) (Diptera:Culicidae) under uncontrolled laboratory conditions. Trop. Biomed. 25, 117–125.
Palatini, U., Masri, R. A., Cosme, L. V., Koren, S., Thibaud-Nissen, F., Biedler, J. K., et al. (2020). Improved reference genome of the arboviral vector Aedes albopictus. Genome Biol. 21, 215. doi:10.1186/s13059-020-02141-w
Pasquali, S., Mariani, L., Calvitti, M., Moretti, R., Ponti, L., Chiari, M., et al. (2020). Development and calibration of a model for the potential establishment and impact of Aedes albopictus in Europe. Acta Trop., 105228. doi:10.1016/j.actatropica.2019.105228
Pech-May, A., Moo-Llanes, D. A., Puerto-Avila, M. B., Casas, M., Danis-Lozano, R., Ponce, G., et al. (2016). Population genetics and ecological niche of invasive Aedes albopictus in Mexico. Acta Trop. 157, 30–41. doi:10.1016/j.actatropica.2016.01.021
Porretta, D., Mastrantonio, V., Bellini, R., Somboon, P., and Urbanelli, S. (2012). Glacial history of a modern invader: Phylogeography and species distribution modelling of the asian tiger mosquito Aedes albopictus. PLoS ONE 7, e44515. doi:10.1371/journal.pone.0044515
Roiz, D., Neteler, M., Castellani, C., Arnoldi, D., and Rizzoli, A. (2011). Climatic factors driving invasion of the tiger mosquito (Aedes albopictus) into new areas of Trentino, Northern Italy. PLoS ONE 6, e14800. doi:10.1371/journal.pone.0014800
Roiz, D., Ruiz, S., Soriguer, R., and Figuerola, J. (2014). Climatic effects on mosquito abundance in Mediterranean wetlands. Parasites Vectors 7, 333. doi:10.1186/1756-3305-7-333
Ruiling, Z., Peien, L., Xuejun, W., and Zhong, Z. (2018a). Molecular analysis and genetic diversity of Aedes albopictus (Diptera, Culicidae) from China. Mitochondrial DNA Part A 29, 594–599. doi:10.1080/24701394.2017.1325481
Ruiling, Z., Tongkai, L., Dezhen, M., and Zhong, Z. (2018b). Genetic characters of the globally spread tiger mosquito,Aedes albopictus(Diptera, Culicidae): Implications from mitochondrial gene COI. J. Vector Ecol. 43, 89–97. doi:10.1111/jvec.12287
Sabatini, A., Raineri, V., Trovato, G., and Coluzzi, M. (1990). Aedes albopictus in Italy and possible diffusion of the species into the Mediterranean area. Parassitologia 32, 301–304.
Schoener, E., Zittra, C., Weiss, S., Walder, G., Barogh, B. S., Weiler, S., et al. (2019). Monitoring of alien mosquitoes of the genus Aedes (Diptera: Culicidae) in Austria. Parasitol. Res. 118, 1633–1638. doi:10.1007/s00436-019-06287-w
Scholte, E. J., and Schaffner, F. (2007). “Waiting for the tiger: Establishment and spread of the Aedes albopictus mosquito in Europe,” in Emerging pests and vector-borne diseases in Europe. Editors W. Takken, and B. G. J. Knols (The Netherlands: Wageningen Academic Publishers).
Skuse, F. A. (1894). The banded mosquito of Bengal. Indian Museum Notes 3, 20. doi:10.5281/zenodo.163480
Șuleșco, T., Bușmachiu, G., Lange, U., Schmidt-Chanasit, J., and Lühken, R. (2021). The first record of the invasive mosquito species Aedes albopictus in Chişinӑu, Republic of Moldova, 2020. Parasites Vectors 14, 565. doi:10.1186/s13071-021-05060-2
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi:10.1093/molbev/msab120
Tedjou, A. N., Kamgang, B., Yougang, A. P., Njiokou, F., and Wondji, C. S. (2019). Update on the geographical distribution and prevalence of Aedes aegypti and Aedes albopictus (Diptera: Culicidae), two major arbovirus vectors in Cameroon. PLoS Negl. Trop. Dis. 13, e0007137. doi:10.1371/journal.pntd.0007137
Torroni, A., Achilli, A., Macaulay, V., Richards, M., and Bandelt, H. (2006). Harvesting the fruit of the human mtDNA tree. Trends Genet. 22, 339–345. doi:10.1016/j.tig.2006.04.001
Urbanelli, S., Bellini, R., Carrieri, M., Sallicandro, P., and Celli, G. (2000). Population structure of Aedes albopictus (skuse): The mosquito which is colonizing mediterranean countries. Heredity 84 (3), 331–337. doi:10.1046/j.1365-2540.2000.00676.x
Vega-Rúa, A., Marconcini, M., Madec, Y., Manni, M., Carraretto, D., Gomulski, L. M., et al. (2020). Vector competence of Aedes albopictus populations for chikungunya virus is shaped by their demographic history. Commun. Biol. 3, 326. doi:10.1038/s42003-020-1046-6
Yang, Z. (1997). Paml: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 13, 555–556. doi:10.1093/bioinformatics/13.5.555
Zé-Zé, L., Borges, V., Osório, H. C., Machado, J., Gomes, J. P., and Alves, M. J. (2020). Mitogenome diversity of Aedes (stegomyia) albopictus: Detection of multiple introduction events in Portugal. PLoS Negl. Trop. Dis. 14, e0008657. doi:10.1371/journal.pntd.0008657
Zhang, H., Xing, D., Wang, G., Li, C., and Zhao, T. (2015). Sequencing and analysis of the complete mitochondrial genome of Aedes albopictus (Diptera: Culicidae) in China. Mitochondrial DNA Part A 27, 2787–2788. doi:10.3109/19401736.2015.1053067
Zhong, D., Lo, E., Hu, R., Metzger, M. E., Cummings, R., Bonizzoni, M., et al. (2013). Genetic analysis of invasive Aedes albopictus populations in Los Angeles County, California and its potential public health impact. PLoS ONE 8, e68586. doi:10.1371/journal.pone.0068586
Keywords: Aedes albopictus spread, MtDNA variation, haplogroups, sources of adventive populations, mitogenome, phylogeny
Citation: Battaglia V, Agostini V, Moroni E, Colombo G, Lombardo G, Rambaldi Migliore N, Gabrieli P, Garofalo M, Gagliardi S, Gomulski LM, Ferretti L, Semino O, Malacrida AR, Gasperi G, Achilli A, Torroni A and Olivieri A (2022) The worldwide spread of Aedes albopictus: New insights from mitogenomes. Front. Genet. 13:931163. doi: 10.3389/fgene.2022.931163
Received: 28 April 2022; Accepted: 28 July 2022;
Published: 26 August 2022.
Edited by:
Horacio Naveira, University of A Coruña, SpainCopyright © 2022 Battaglia, Agostini, Moroni, Colombo, Lombardo, Rambaldi Migliore, Gabrieli, Garofalo, Gagliardi, Gomulski, Ferretti, Semino, Malacrida, Gasperi, Achilli, Torroni and Olivieri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Olivieri, YW5uYS5vbGl2aWVyaUB1bmlwdi5pdA==
†These authors have contributed equally to this work and share first authorship