 Christian Vogeley
Christian Vogeley Thach Nguyen
Thach Nguyen Selina Woeste
Selina Woeste Thomas Haarmann-Stemmann
Thomas Haarmann-Stemmann Andrea Rossi
Andrea Rossi- IUF-Leibniz Research Institute for Environmental Medicine, Düsseldorf, Germany
Transcriptome analysis experiments enable researchers to gain extensive insights into the molecular mechanisms underlying cell physiology and disease. Oxford Nanopore Technologies (ONT) has recently been developed as a fast, miniaturized, portable, and cost-effective alternative to next-generation sequencing (NGS). However, RNA-Seq data analysis software that exploits ONT portability and allows scientists to easily analyze ONT data everywhere without bioinformatics expertise is not widely available. We developed DuesselporeTM, an easy-to-follow deep sequencing workflow that runs as a local webserver and allows the analysis of ONT data everywhere without requiring additional bioinformatics tools or internet connection. DuesselporeTM output includes differentially expressed genes and further downstream analyses, such as variance heatmap, disease and gene ontology plots, gene concept network plots, and exports customized pathways for different cellular processes. We validated DuesselporeTM by analyzing the transcriptomic changes induced by PCB126, a dioxin-like PCB, and a potent aryl hydrocarbon receptor (AhR) agonist in human HaCaT keratinocytes, a well-characterized model system. DuesselporeTM was specifically developed to analyze ONT data, but we also implemented NGS data analysis. DuesselporeTM is compatible with Linux, Microsoft, and Mac operating systems and allows convenient, reliable, and cost-effective analysis of ONT and NGS data.
Introduction
In the past decade, RNA-sequencing (RNA-Seq) has become the leading method for analyzing whole-genome transcriptomes (Ashley, 2016). RNA-Seq is used in modern medicine for diagnosis, prognosis, and therapeutic selection in the fields of infectious disease, fetal monitoring, and cancer (Ku and Roukos, 2014; Ashley, 2016). The ability to sequence DNA and RNA quickly and inexpensively is essential to scientific research (Wheeler et al., 2008). This necessity has prompted the development of many sequencing techniques beyond the original Sanger sequencing (Shendure and Ji, 2008). The rise of next-generation sequencing (NGS) has considerably improved the output of data generated by Sanger sequencing (first-generation sequencing) (Buermans et al., 2017). NGS approaches have become widespread in research and diagnostic laboratories and have greatly increased our knowledge of many genetic disorders (Schuster, 2007; Shendure and Ji, 2008; Rivas et al., 2011; Jamuar et al., 2014; König et al., 2015). Nevertheless, these technologies remain expensive, laborious, time-consuming, and affected by the limitations of short-read sequencing. As some kind of a new generation, new sequencing methods were established, which aim to sequence long DNA or RNA molecules (Slatko et al., 2018), including Oxford Nanopore Technologies (ONT).
ONT sequencing is based on transmembrane proteins (nanopores) embedded into a lipid membrane and measures changes in electric current across these pores. These changes are caused by nanosized molecules, such as DNA or RNA (or even amino acids), as they occupy a volume that interferes with the ion flow and can be recorded by a semiconductor-based electronic detection system and afterward translated into sequences (Hornblower et al., 2007; Branton et al., 2008). In the last few years, ONT sequencing has proven to be a fast and cost-effective alternative to other sequencing techniques. Even if the read quality does not reach the high standard of other techniques, it offers other advantages such as fast and easy library preparation, real-time sequencing, PCR-free, direct RNA, and ultra-long read sequencing (Liu et al., 2019; Xu and Seki, 2019). The latter is especially useful for improving de novo assembly, mapping certainty, transcript isoform identification, and detection of structural variants (Jain et al., 2018).
Additionally, some ONT sequencers are small, granting portability, and enabling sequencing experiments outside the laboratory. Nevertheless, sequencing analysis produces large raw datasets that require bioinformatics skills, familiarity with statistics, and a Linux-based environment to decode this information. Performing multiple RNA-Seq analysis workflows requires the handling of dozens of dependent libraries and third-party software, and individual data preparation. It would be ideal to circumvent this issue by integrating multiple workflows without requiring additional data preparation steps.
Available software allows robust analysis of those data, but they usually require an internet connection and basic bioinformatic skills, lack a friendly user interface (Ewels et al., 2020; Williams, 2022), or are mainly limited to basic analysis (Epi2me).
Furthermore, scientists might be unwilling or even not allowed (e.g., human datasets) to upload their unpublished data to an online platform to employ open-access web services. We believe that a client-based computational approach implemented in Javascript would represent an alternative solution that shifts the computation workload to the client-side. We recently deployed a light computational web server solution to analyze genome editing experiments from ONT data (Nguyen et al., 2022). However, RNA-Seq workflows are more complicated and have not been translated or compiled into lightweight Javascript-like software. Moreover, ONT offers mobile platforms that can work in environments where internet access is limited or absent.
In order to overcome these issues and help the community process ONT and other RNA sequencing datasets, we developed DuesselporeTM, a local web server that is particularly tailored to the portability of Nanopore sequencers. Instead of using a centralized system, we deploy an efficient local-based system that runs on a Docker container without the necessity of being connected to the internet. Running in a virtual environment has further advantages: portability, agility, scalability, and security, all empowered by Docker. Furthermore, the pipeline (Figure 1) runs in an isolated virtual environment and avoids the incompatibility of our workflow with the host’s software.
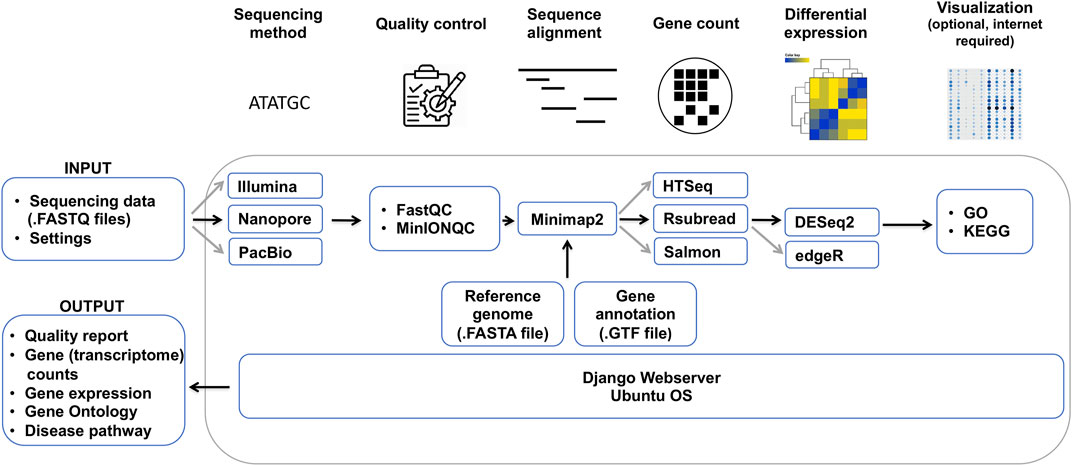
FIGURE 1. Bioinformatical data analysis pipeline of DuesselporeTM. Depicted are the required data, which need to be provided by the user, and the different steps of the data analysis, including quality control, sequence alignment, gene count, differential expression and visualization, and the output. Black arrows indicate the main workflow optimized for the analysis of ONT data, whereas grey arrows show the variations of the workflow that can be selected by the user.
Method and system description
HaCaT cell culture and stimulation
HaCaT keratinocytes were cultivated at 37°C and 5% CO2 in DMEM medium (PAN Biotech, Aidenbach, Germany) with low glucose (1 g/L) supplemented with 10% FBS and 1% antibiotics/antimycotics (PAN Biotech). HaCaT keratinocytes were stimulated in full growth medium for 24 h with 1 µM PCB126 or were solvent treated using 0.1% DMSO.
Library preparation
Total RNA was isolated using the GenUP Total RNA Kit (Biotechrabbit, Hennigsdorf, Germany). The High Sensitivity RNA ScreenTape System (Agilent Technologies, Santa Clara, CA) was used to assess the quality of isolated RNA. 50 ng of total RNA was reverse transcribed, and samples were barcoded with the PCR cDNA Barcoding Kit (SQK-PCB109, Oxford Nanopore Technologies, Oxford, United Kingdom). The quantity of amplified cDNA was then determined with the QubitTM 4 Fluorometer (Invitrogen, Carlsbad, CA), and the range of fragment size was assessed using the Agilent D1000 SreenTape assay (Agilent Technologies). The Flow Cell Priming Kit (EXP FLP002, Oxford Nanopore Technologies) was used to prime the flowcell (FLO-MIN106), and an equal amount of barcoded cDNA was loaded. Sequencing was carried out with a MinION (MN33710) using the MinKNOW software (v.21.02.1) over a period of 72 h.
Base-calling
Raw FAST5 reads were base-called and demultiplexed using Guppy (v4.5.4 + 66c1a7753), reads with a quality score below seven were excluded using the following command:
guppy_basecaller --min_qscore 7 --trim_barcodes --barcode_kits "SQK-PCB109" --compress_fastq -i {input.fast5} -s {output.folder} -c dna_r9.4.1_450bps_hac.cfg --x auto --chunks_per_runner 128
The resulting FASTQ files of each sample were concatenated into one file using the following command:
On linux and mac OS terminal
$ cat /path/to/fastq/files/*.fastq > /your/new/location/output.fastq
On Windows command prompt (NOTE: path symbol is different):
$ type \path\to\fastq\files\*.fastq>\your\new\location\output.fastq
Input data and run parameters
DuesselporeTM validates the input by automatic format detection. Input FASTQ files are compressed and structured in each subdirectory named by experimental condition. Each subdirectory contains multiple biological replicates for each condition. The default parameters in the webserver form are optimized for analyzing long-read RNA-Seq data obtained from ONT. However, the users can change the settings based on their experimental setup and sequencing method of choice.
Quality control
Our RNA-Seq workflow follows the standard data processing pipelines. First, we employ quality control with FastQC to quantify the sequence quality and export the quality into HTML files. Reads with a score below the predefined quality threshold (Q = 7) are filtered out.
Align and assign reads to gene/transcriptome
Minimap2 (Li, 2018) was used to map the quality-filtered ONT reads to the reference genome/transcriptome. Aligned BAM files were processed by Rsubread/featureCounts (Liao et al., 2019), HTSeq/htseq-count (Anders et al., 2015), or Salmon (Patro et al., 2017) to generate the raw count of read genes or transcriptome. Although Salmon can quantify transcripts directly from the FASTQ reads (Liao et al., 2019), Minimap2 displays a higher assigned ratio for handling noisy ONT data (Williams, 2022).
Differential expression analysis and gene ontology
The raw read count matrices are processed by the differential expression methods DESeq2 (Love et al., 2014) and edgeR/limma (Robinson et al., 2009; Ritchie et al., 2015). DESeq2 displays a high capability to process data with a small number of biological replicates, a common issue in NGS.
To analyze gene ontology, we integrated the gene ontology pipeline using Bioconductor. Results are clustered into various gene ontology pathways such as gene set enrichment analysis of gene ontology (gseGO), disease ontology, network of cancer genes (DOSE) (Yu et al., 2015), and pathview (Luo and Brouwer, 2013). Please note that whilst most of the analyses performed by DuesselporeTM do not require an internet connection, gene ontology and disease pathway (optional function) will need an internet connection (see supplementary information).
Web server and backend platform
DuesselporeTM is implemented in Python 3.8 with Django 3.2 web framework, which has long-term support from the open-source community. Bioconda, Biopython, and R/Bioconductor run on the Ubuntu 18.04 system encapsulated in the Docker container as backend. Docker can be configured depending on the host PC. All software is free and open-source. The detailed system architecture and manual are described in the Supplementary document.
Results
Validation of DuesselporeTM
DuesselporeTM is implemented in Python 3.8 with Django 3.2 web framework (Figure 1) and it encapsulates different pipelines including quality control, sequence alignment, gene count, differential expression, and visualization (Figure 1).
As input, DuesselporeTM takes multiple FASTQ files that are the output of base-called ONT or Illumina data. The output includes a quality report, gene counts and expression, and gene ontology and disease pathway optionally. DuesselporeTM can be installed in three steps (Figure 2A), and the user interface (Figure 2B) allows the user to fine-tune the sequencing analysis settings based on the experimental design of choice.
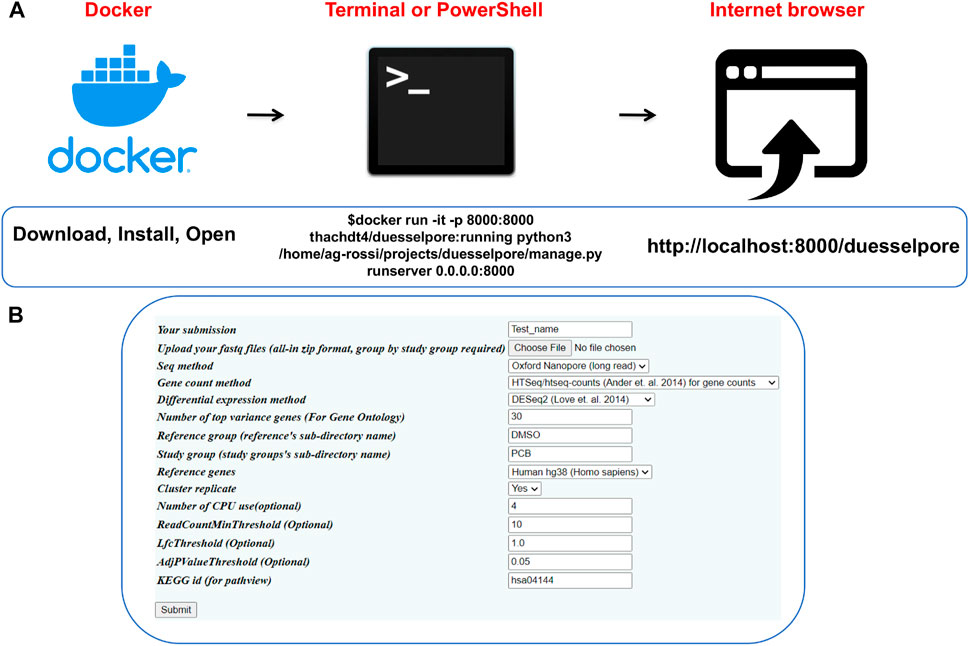
FIGURE 2. Operating Duesselpore. (A) Depicted are the required steps to launch DuesselporeTM on Linux/MacOS (Terminal) or Windows (PowerShell). (B) The interface can be accessed via the user’s browser.
To validate DuesselporeTM, immortalized HaCaT keratinocytes were stimulated with 3,3′,4,4′,5-pentachlorobiphenyl (PCB126) or solvent treated with 0.1% DMSO for 24 h. PCB126 was chosen as the mode of action because it is well understood how it regulates gene expression. According to their lipophilic properties, PCB126 molecules can easily pass through the cell membrane and bind the aryl hydrocarbon receptor (AHR), a cytosolic transcription factor trapped in a multiprotein complex. Upon ligand binding, the multiprotein complex dissociates, and the AHR translocates into the nucleus, where it dimerizes with the aryl hydrocarbon receptor nuclear translocator (ARNT) and binds to xenobiotic responsive elements (XRE) in the genome. This signaling pathway induces the expression of various genes encoding for Phase I and Phase II biotransformation enzymes (Bock and Köhle, 2006). Amongst these genes are the members of the superfamily cytochrome P450 (CYP) CYP1A1, CYP1B1, and aldehyde dehydrogenases (ALDH) ALDH3A1 as well as genes encoding for proteins involved in cell proliferation, differentiation, and apoptosis (Vogeley et al., 2019).
Total RNA was isolated and transcribed into complementary DNA (cDNA) as described. cDNAs were barcoded, loaded into the same flowcell, and sequenced over a period of 72 h to ensure a sufficient number of reads for further downstream analyses. The raw FAST5 output files were base-called with Guppy, ONTs base-calling algorithm, and demultiplexed (barcode sorted). Base-calling and barcode demultiplexing were not included in DuesselporeTM because these steps can be performed directly by the sequencer software (e.g., MinKNOW for ONT or Local Run Manager for Illumina). The generated FASTQ files were then concatenated and analyzed with DuesselporeTM.
DuesselporeTM provides a mapping summary, including the number of reads, the number of reads mapped, and the percentage of mapped reads (Table 1). Secondary mappings were excluded as those are not considered when these sequences are assigned to genetic features. For clarification, each replicate represents a different passage of cells analyzed on a different flow cell.
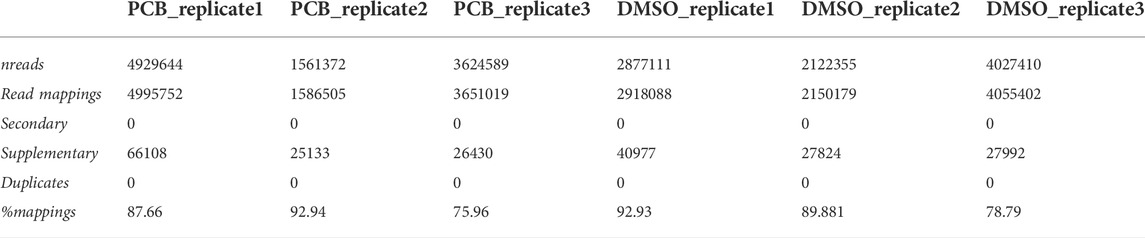
TABLE 1. Mapping summary of sequences aligned with minimap2.
A more detailed sample overview is provided in the sample summary table, which contains information about the read length, million base sequences (mbs), mean quality score (qval), and GC content (gc) (Figure 3A). Additionally, read length and quality score distributions are depicted in violin plots (Figures 3B,C).
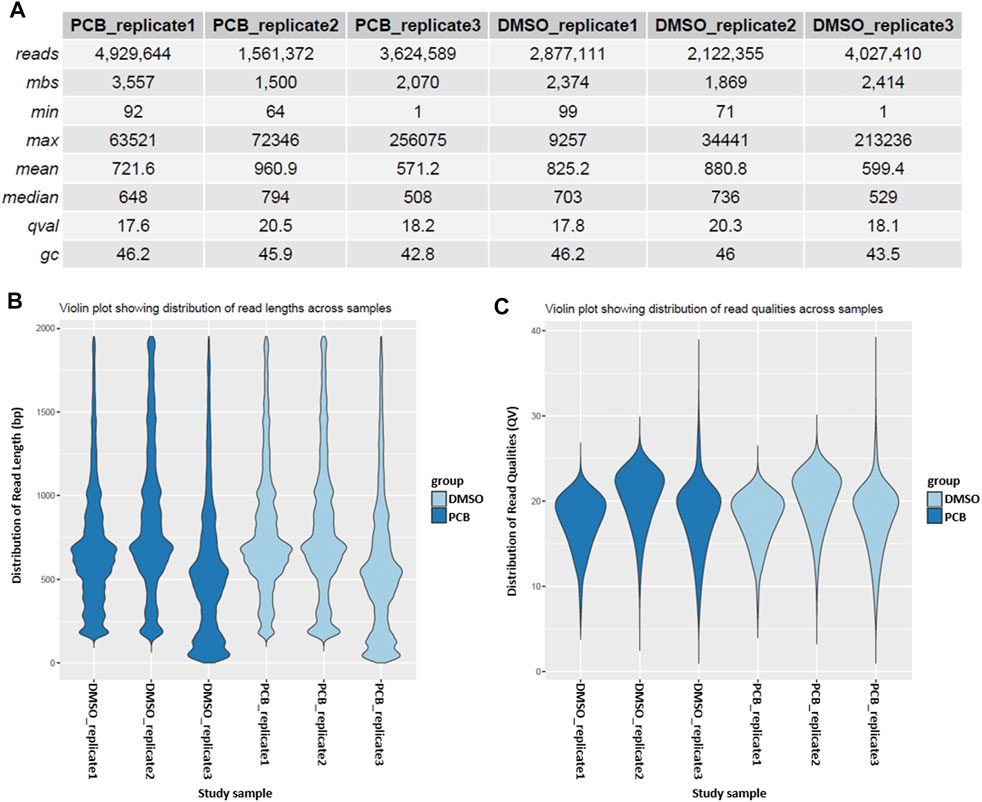
FIGURE 3. Sample summary and quality control. (A) Summary of the reads of every sample. The table provides information about the number of reads, million base sequences (mbs), minimal and maximum read length, mean and median read length, quality score (qval), and GC content (gc). Violin plots depict the distribution of read length (B) and the distribution of read quality (C) across different samples.
After carrying out sample quality control, DuesselporeTM performs differential gene expression (DGE) analysis. Therefore, the user is able to choose between the packages DESeq2 or edgeR. Both packages use statistical models based on the negative binomial distribution and are the most commonly used tools. It is recommended to use DESeq2 when the number of replicas is relatively low (below 5), as this tool shows the highest consistency in the identification of significantly differentially expressed (SDE) genes and shows the lowest false discovery rate (Seyednasrollah et al., 2015). The results of the differentially expressed gene (DEG) analysis are provided as an excel file, and in our data set, the genes CYP1A1, CYP1B1, ALDH3A1 as well as other established AHR target genes, such as ATP-binding cassette super-family G member 2 (ABCG2), plasminogen activator inhibitor-2 (SERPINB2), and TCDD-inducible poly [ADP-ribose] polymerase (TiPARP), are among the SDE genes. The regulation of CYP1A1, CYP1B1, and ALDH3A1 was confirmed by qRT-PCR (Supplementary Figures S1A–C). The top 30 genes with the highest variance are depicted in a variance heatmap (Figure 4A), also provided by DuesselporeTM. For this purpose, the counts per gene were normalized to the counts per million (CPM) scaling factor (Robinson et al., 2009). The number of depicted genes can be defined in the user interface under the menu item “Number of top variance genes.”
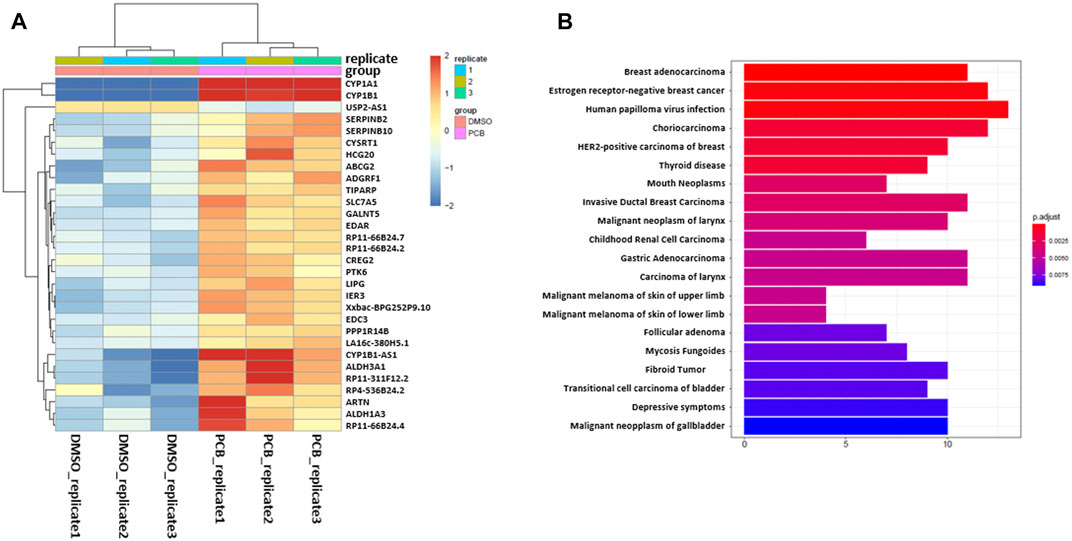
FIGURE 4. Variance of gene expression and enrichment analysis. (A) Heatmap showing the top 30 genes with the highest variance after normalizing for the CPM. (B) Enrichment analysis based on the DisGeNET database, revealing diseases, which are associated with the differentially expressed genes. X-axis shows the number of genes involved in each disease.
Besides the detection of differentially expressed genes, DuesselporeTM will also conduct gene set enrichment analyses (GSEA), enrichment analysis based on the DisGeNET (Piñero et al., 2015), and pathway-based data integration and visualization focusing on KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (Luo and Brouwer, 2013) (Supplementary Figure S2). These features will help the user to put the data into context and to identify potential signaling pathways and disease associations based on the SDE genes. To select a certain KEGG pathway, the user needs to provide a KEGG identifier and enter it into the webform under the menu item “KEGG id (for pathview).” PCB126 is highly toxic and exhibits mutagenic potential, which is not solely based on its capability to activate the AHR (Lauby-Secretan et al., 2013). Hence, genes regulated as a consequence of PCB126 exposure were associated with various malign neoplasias according to the DisGeNET database (Figure 4B). The relationship between genes and disease is depicted in a network plot either in a non-circular (Supplementary Figure S2A) or circular (Supplementary Figure S2B) format.
DuesselporeTM outputs were successfully validated by using other datasets (data not shown). Here, the ability to detect PCB126-induced gene expression proves the functionality of DuesselporeTM.
Discussion
Over the last decade, RNA-sequencing methods have become more affordable and advanced to a commonly used technique to analyze genome-wide transcriptomes. While each step of sequencing has been drastically simplified, i.e., sample preparation due to the implementation of preparation kits, users are still struck by the size and the amount of raw data that require further analysis. This type of analysis often needs bioinformatics expertise and a certain amount of computational resources. In order to reduce this burden, we developed DuesselporeTM, a local web server that delivers a solution without interfering with the mobile characteristics of ONT sequencers, including the absence of an internet connection. DuesselporeTM functionality was tested using ONT reads obtained by DMSO or PCB126 treated HaCaT keratinocyte samples. Among the DEGs, many prototypic genes that are regulated by the AHR, such as CYP1A1, CYP1B1, ALDH3A1, or SERPINB2, were upregulated in the test data set. The gene set enrichment analysis revealed an upregulation of gene patterns that are connected with different malign neoplasias. This observation is associated with the mutagenic potential of PCB126 and the fact that AHR activity is enhanced in many different tumor entities (Murray et al., 2014). These results were expected and thus validate the data obtained by DusselporeTM analysis (Bock and Köhle, 2006; Lauby-Secretan et al., 2013).
DuesselporeTM interface allows the selection of five commonly used genomes (human, rat, mouse, zebrafish, and C. elegans), provides different pipelines to analyze RNA-Seq data, and by using publicly available tools, it helps users to perform advanced bioinformatical data analysis and generates figures and tables that are suitable for publication. Most of these tools require various dependencies, which is the reason why we encapsulated and compiled this workflow into a Docker image, making DuesselporeTM independent from the host’s machine. All these properties grant the user strong flexibility in analyzing data even without profound bioinformatic knowledge. Furthermore, DuesselporeTM is not limited to the analysis of ONT-derived data, but it also allows the analysis of Illumina and PacBio data (Supplementary Material). Moreover, DuesselporeTM runs locally on the user’s machine, thus, data are not uploaded to the server. This is particularly helpful in avoiding risks associated with General Data Protection Regulation violations, security, and privacy.
In summary, we developed and successfully validated DuesselporeTM as a web tool, which is able to analyze in detail ONT and NGS (PacBio or Illumina) derived RNA-Seq data without the requirement of a profound bioinformatical background. This will further democratize the analysis of NGS- and ONT-derived transcriptome sequencing data.
Declarations
Code availability and data: please read Duesselpore manual for more information and software links:
https://github.com/thachnguyen/duesselpore.
Docker image:
https://hub.docker.com/repository/docker/thachdt4/duesselpore:running.
Test dataset (DMSO PCB standard and lightweight):
Sample result from ONT:
https://github.com/thachnguyen/duesselpore/blob/main/sample_result/nanopore/sample_result_ONT.zip
Sample result from Illumina:
https://github.com/thachnguyen/duesselpore/raw/main/sample_result/illumina/test_illumina.zip.
Sample result from PacBio:
https://github.com/thachnguyen/duesselpore/blob/main/sample_result/pacbio/Test_Pac_bio.zip.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
AR conceptualized this work; CV and TN programmed the web tool and performed data analysis; SW conducted library preparation and sequencing; TH-S provided resources; AR, CV, and TN wrote the manuscript; and JK and TH-S revised the manuscript.
Acknowledgments
The authors thank J. Dobner and Christiane Klasen for discussion and comments on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.931996/full#supplementary-material
References
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi:10.1093/bioinformatics/btu638
Ashley, E. A. (2016). Towards precision medicine. Nat. Rev. Genet. 17 (9), 507–522. doi:10.1038/nrg.2016.86
Bock, K. W., and Köhle, C. (2006). Ah receptor: Dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem. Pharmacol. 72, 393–404. doi:10.1016/J.BCP.2006.01.017
Branton, D., Deamer, D. W., Marziali, A., Bayley, H., Benner, S. A., Butler, T., et al. (2008). The potential and challenges of nanopore sequencing. Nat. Biotechnol. 26, 1146–1153. doi:10.1038/nbt.1495
Buermans, H. P. J., Vossen, R. H. A. M., Anvar, S. Y., Allard, W. G., Guchelaar, H. J., White, S. J., et al. (2017). Flexible and scalable full-length CYP2D6 long amplicon PacBio sequencing. Hum. Mutat. 38, 310–316. doi:10.1002/HUMU.23166
Ewels, P. A., Peltzer, A., Fillinger, S., Patel, H., Alneberg, J., Wilm, A., et al. (2020). The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38 (3), 276–278. doi:10.1038/s41587-020-0439-x
Hornblower, B., Coombs, A., Whitaker, R. D., Kolomeisky, A., Picone, S. J., Meller, A., et al. (2007). Single-molecule analysis of DNA-protein complexes using nanopores. Nat. Methods 4 (4), 315–317. doi:10.1038/nmeth1021
Jain, M., Koren, S., Miga, K. H., Quick, J., Rand, A. C., Sasani, T. A., et al. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36 (4), 338–345. doi:10.1038/nbt.4060
Jamuar, S. S., Lam, A.-T. N., Kircher, M., D’Gama, A. M., Wang, J., Barry, B. J., et al. (2014). Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 371, 733–743. doi:10.1056/NEJMOA1314432/SUPPL_FILE/NEJMOA1314432_DISCLOSURES
König, K., Peifer, M., Fassunke, J., Ihle, M. A., Künstlinger, H., Heydt, C., et al. (2015). Implementation of amplicon parallel sequencing leads to improvement of diagnosis and therapy of lung cancer patients. J. Thorac. Oncol. 10, 1049–1057. doi:10.1097/JTO.0000000000000570
Ku, C. S., and Roukos, D. H. (2014). From next-generation sequencing to nanopore sequencing technology: Paving the way to personalized genomic medicine. Expert Rev. Med. Devices 10, 1–6. doi:10.1586/ERD.12.63
Lauby-Secretan, B., Loomis, D., Grosse, Y., el Ghissassi, F., Bouvard, V., Benbrahim-Tallaa, L., et al. (2013). Carcinogenicity of polychlorinated biphenyls and polybrominated biphenyls. Lancet. Oncol. 14, 287–288. doi:10.1016/S1470-2045(13)70104-9
Li, H. (2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi:10.1093/bioinformatics/bty191
Liao, Y., Smyth, G. K., and Shi, W. (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47. doi:10.1093/NAR/GKZ114
Liu, Q., Fang, L., Yu, G., Wang, D., Xiao, C., Wang, K., et al. (2019). Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 10, 2449. doi:10.1038/s41467-019-10168-2
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi:10.1186/s13059-014-0550-8
Luo, W., and Brouwer, C. (2013). Pathview: An R/bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831. doi:10.1093/BIOINFORMATICS/BTT285
Murray, I. A., Patterson, A. D., and Perdew, G. H. (2014). Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 14, 801–814. doi:10.1038/NRC3846
Nguyen, T., Ramachandran, H., Martins, S., Krutmann, J., and Rossi, A. (2022). Identification of genome edited cells using CRISPRnano. Nucleic Acids Res. 50 (w1), W199–W203. doi:10.1093/NAR/GKAC440
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi:10.1038/nmeth.4197
Piñero, J., Queralt-Rosinach, N., Bravo, À., Deu-Pons, J., Bauer-Mehren, A., Baron, M., et al. (2015). DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Oxford, United Kingdom: Oxford University Press. doi:10.1093/database/bav028
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. doi:10.1093/nar/gkv007
Rivas, M. A., Beaudoin, M., Gardet, A., Stevens, C., Sharma, Y., Zhang, C. K., et al. (2011). Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073. doi:10.1038/NG.952
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2009). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi:10.1093/bioinformatics/btp616
Schuster, S. C. (2007). Next-generation sequencing transforms today’s biology. Nat. Methods 5, 16–18. doi:10.1038/nmeth1156
Seyednasrollah, F., Laiho, A., and Elo, L. L. (2015). Comparison of software packages for detecting differential expression in RNA-seq studies. Brief. Bioinform. 16, 59–70. doi:10.1093/BIB/BBT086
Shendure, J., and Ji, H. (2008). Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145. doi:10.1038/nbt1486
Slatko, B. E., Gardner, A. F., and Ausubel, F. M. (2018). Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 122, e59. doi:10.1002/CPMB.59
Vogeley, C., Esser, C., Tüting, T., Krutmann, J., and Haarmann-Stemmann, T. (2019). Role of the aryl hydrocarbon receptor in environmentally induced skin aging and skin carcinogenesis. Int. J. Mol. Sci. 20, E6005. doi:10.3390/IJMS20236005
Wheeler, D. A., Srinivasan, M., Egholm, M., Shen, Y., Chen, L., McGuire, A., et al. (2008). The complete genome of an individual by massively parallel DNA sequencing. Nature 452, 872–876. doi:10.1038/nature06884
Williams, J. (2022). CyVerse for reproducible research: RNA-Seq analysis. Methods Mol. Biol. 2443, 57–79. doi:10.1007/978-1-0716-2067-0_3
Xu, L., and Seki, M. (20192019). Recent advances in the detection of base modifications using the Nanopore sequencer. J. Hum. Genet. 65 (1), 25–33. doi:10.1038/s10038-019-0679-0
Keywords: Oxford nanopore sequencing, next-generation sequencing, RNA-seq analyses, NGS data analysis, Oxford Nanopore MinION device
Citation: Vogeley C, Nguyen T, Woeste S, Krutmann J, Haarmann-Stemmann T and Rossi A (2022) Rapid and simple analysis of short and long sequencing reads using DuesselporeTM. Front. Genet. 13:931996. doi: 10.3389/fgene.2022.931996
Received: 29 April 2022; Accepted: 28 June 2022;
Published: 11 August 2022.
Edited by:
Ka-Chun Wong, City University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Giuseppe Diego Puglia, National Research Council of Italy (CNR), ItalyDominik Strzalka, Rzeszów University of Technology, Poland
Miguel Pinheiro, University of Aveiro, Portugal
Copyright © 2022 Vogeley, Nguyen, Woeste, Krutmann, Haarmann-Stemmann and Rossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Rossi, QW5kcmVhLlJvc3NpQElVRi1EdWVzc2VsZG9yZi5kZQ==
†These authors have contributed equally to this work