 Alexander Varzari1,2*
Alexander Varzari1,2* Igor V. Deyneko3Gitte Hoffmann Bruun4,5
Igor V. Deyneko3Gitte Hoffmann Bruun4,5 Maja Dembic4,5,6,7Winfried Hofmann8Victor M. Cebotari9Sergei S. Ginda10
Maja Dembic4,5,6,7Winfried Hofmann8Victor M. Cebotari9Sergei S. Ginda10 Brage S. Andresen4,5
Brage S. Andresen4,5 Thomas Illig2
Thomas Illig2- 1Laboratory of Human Genetics, Chiril Draganiuc Institute of Phthisiopneumology, Kishinev, Moldova
- 2Hannover Unified Biobank, Hannover Medical School, Hannover, Germany
- 3Laboratory of Functional Genomics, Timiryazev Institute of Plant Physiology Russian Academy of Sciences, Moscow, Russia
- 4Department of Biochemistry and Molecular Biology, University of Southern Denmark, Odense M, Denmark
- 5The Villum Center for Bioanalytical Sciences, University of Southern Denmark, Odense, Denmark
- 6Department of Clinical Genetics, Odense University Hospital, Odense, Denmark
- 7Department of Mathematics and Computer Science, University of Southern Denmark, Odense, Denmark
- 8Department of Human Genetics, Hannover Medical School, Hannover, Germany
- 9Municipal Hospital of Phthisiopneumology, Department of Pediatrics, Kishinev, Moldova
- 10Laboratory of Immunology and Allergology, Chiril Draganiuc Institute of Phthisiopneumology, Kishinev, Moldova
Inborn errors of immunity are known to influence susceptibility to mycobacterial infections. The aim of this study was to characterize the genetic profile of nine patients with mycobacterial infections (eight with BCGitis and one with disseminated tuberculosis) from the Republic of Moldova using whole-exome sequencing. In total, 12 variants in eight genes known to be associated with Mendelian Susceptibility to Mycobacterial Disease (MSMD) were detected in six out of nine patients examined. In particular, a novel splice site mutation c.373–2A>C in STAT1 gene was found and functionally confirmed in a patient with disseminated tuberculosis. Trio analysis was possible for seven out of nine patients, and resulted in 23 candidate variants in 15 novel genes. Four of these genes - GBP2, HEATR3, PPP1R9B and KDM6A were further prioritized, considering their elevated expression in immune-related tissues. Compound heterozygosity was found in GBP2 in a single patient, comprising a maternally inherited missense variant c.412G>A/p.(Ala138Thr) predicted to be deleterious and a paternally inherited intronic mutation c.1149+14T>C. Functional studies demonstrated that the intronic mutation affects splicing and the level of transcript. Finally, we analyzed pathogenicity of variant combinations in gene pairs and identified five patients with putative oligogenic inheritance. In summary, our study expands the spectrum of genetic variation contributing to susceptibility to mycobacterial infections in children and provides insight into the complex/oligogenic disease-causing mode.
Introduction
Tuberculosis (TB) is a serious infectious disease usually caused by Mycobacterium tuberculosis (M. tuberculosis), but also by M. africanum, M. bovis and M. canettii species (Lawn and Zumla, 2011). About one-quarter of the world’s population has a TB infection (WHO, 2020), but most people who are infected with TB bacteria do not develop symptoms. Immunocompromised adults and young children are particularly at risk of progressing to active disease. They also develop more severe forms of TB (e.g., disseminated/milliary TB, TB meningitis). Therefore, the vaccination with attenuated M. bovis Bacille Calmette-Guérin (BCG) substrains is recommended to prevent severe TB disease (Roy et al., 2014). The vaccine is given routinely at birth or during childhood in countries with a high burden of TB and it is generally recognized as a safe and effective measure. Serious complications (or BCG infection) are, however, known to occur in children receiving BCG (Kourime et al., 2016).
Certain conditions may cause a child to become more vulnerable/susceptible to mycobacterial infections. It has been estimated that up to 30% of disseminated TB cases are due to monogenic/Mendelian errors (Alcaïs et al., 2005). Consistent with this estimation, several primary immunodefficiency (PID) disorders were systematically associated with mycobacterial disease (Boisson-Dupuis et al., 2015). These PIDs are heterogeneous in terms of clinical manifestations (Fekrvand et al., 2020; Tangye et al., 2020). Classical PIDs, such as combined immunodeficiency (CID) and severe combined immunodeficiency (SCID), chronic granulomatous disease (CGD) and GATA2 deficiency, are associated with increased susceptibility to diseases caused by diverse infectious pathogens, including mycobacteria. In contrast, Mendelian Susceptibility to Mycobacterial Disease (MSMD) constitutes an atypical PID caused by defects in IFN-γ-mediated immunity, leading to the selective predisposition to intermacrophagic infections, mostly Mycobacteria and Salmonella (Boisson-Dupuis et al., 2015; Bustamante, 2020). To date, over 200 disease-associated variants in 17 autosomal (IFNGR1, IFNGR2, IL12B, IL12RB1, IL12RB2, STAT1, TYK2, IRF8, SPPL2A, IL23R, ISG15, JAK1, RORC, ZNFX1, TBX21, IFNG and USP18) and two X-linked (IKBKG and CYBB) genes have been described in patients with MSMD (Bustamante, 2020; Kerner et al., 2020; Tom Le Voye et al., 2021; Martin-Fernandez et al., 2022). These, however, account for only half of the MSMD cases studied, suggesting that mutations in other genes may be involved in genetic susceptibility to MSMD (Bustamante, 2020; Tom Le Voye et al., 2021).
The use of next-generation sequencing (NGS) offers a rapid and efficient approach to find disease-causing mutations in affected individuals and to discover new disease genes (King et al., 2018). In this study, we implemented whole-exome sequencing (WES) in an attempt to identify risk genes and rare variants in a cohort of seven case-parent trios and two single pediatric cases with mycobacterial infections (BCGitis and disseminated/generalised TB) from the Republic of Moldova. The functional effect of identified candidate variants was investigated through experimental assessment of splicing.
Material and methods
Patient description
Nine children (six girls and three boys aged 2–24 months) with M. tuberculosis complex infection were recruited through the department of Paediatrics at Municipal Hospital in Chisinau, Moldova, during 2016–2018. Among these patients, eight were diagnosed with BCGitis and one with generalised M. tuberculosis infection. The diagnosis relied on the assessment of clinical features and for six cases was confirmed bacteriologically and/or histologically. The clinical, demographic and immunologic patient data presented in this study are based on medical records and summarized in Table 1. Immunologic values were within the reference range for most parameters studied in most patients. We note only decreased levels of IgA in patients P2 and P7 and diminished counts of B-lymphocytes (CD20+) in P5. These may indicate an immune system defect. However, overall clinical/immunologic phenotypes in these cases are inconsistent with those reported for BCG-effected patients with SCID or CGD (Fekrvand et al., 2020). Also, other known PIDs associated with BCG complications, such as GATA2 deficiency, which are even rarer than SCID and CGD, were excluded for the same reasons (i.e., inconsistency with the provided clinical/immunologic phenotypes), and thus a diagnosis of MSMD was suspected.
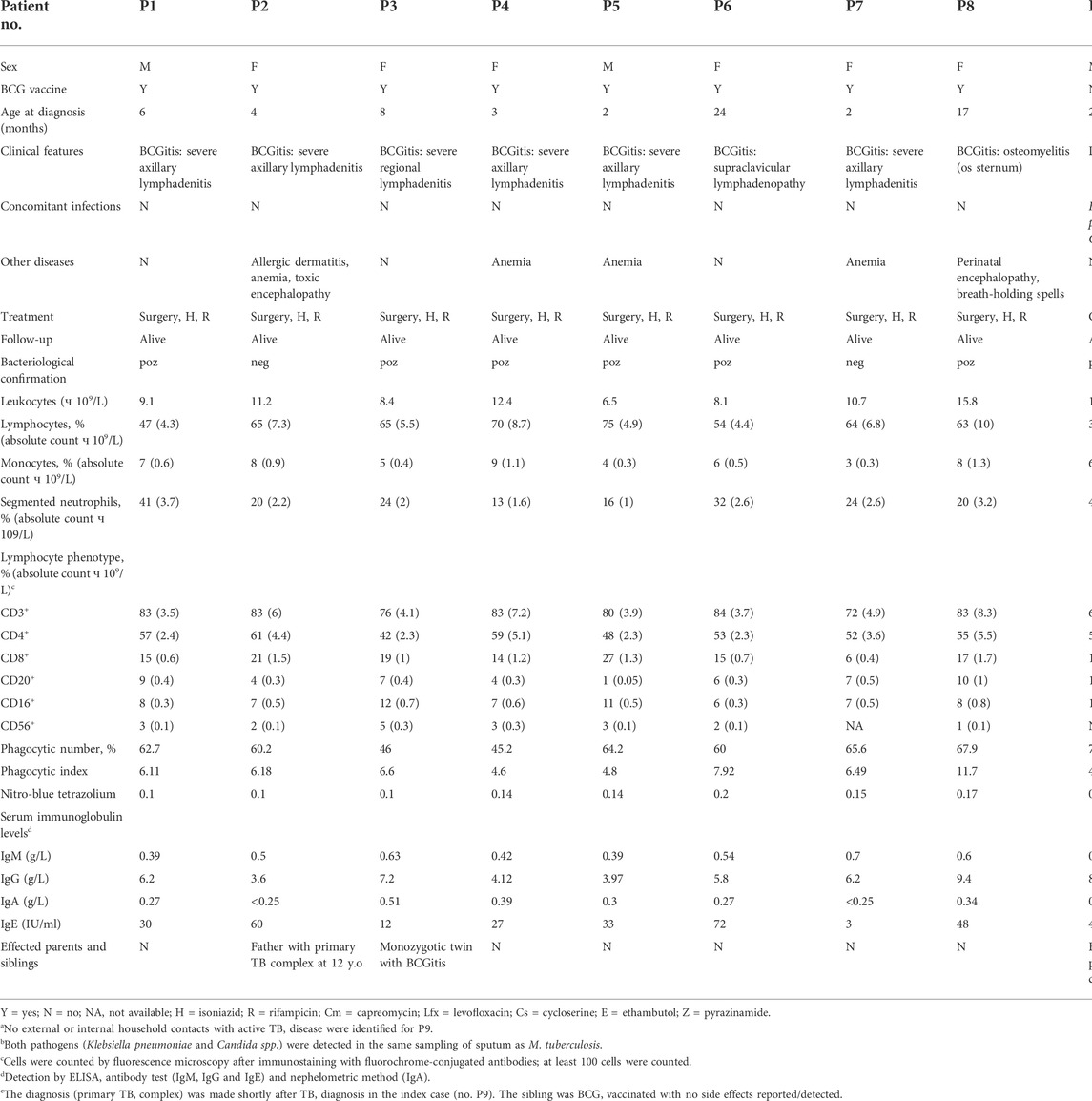
TABLE 1. Patients’ clinical characteristics and laboratory findings.
Both parents were present for the interviews and all of them, except one, denied mycobacterial disease during his/her lifetime. For the only father (P2) who announced TB during childhood, the diagnosis of primary (Ghon) complex of pulmonary TB at the age 12 years has been witnessed/confirmed by the medical record. No parents were consanguineous. DNA from both parents was available for seven probands (P1-P7). Therefore, a total of seven trios and two singletons (P8 and P9) were included in the study.
Three probands were selected based on sequencing results to experimentally validate the effects of the identified variants on RNA splicing. Two of them (P6 and P9) were available for direct RNA analysis and were invited and sampled again (both at the age 3 years), while the third proband (P2) was unavailable, thus an RNA sample was collected and used from the father instead, who harbors as well the suspected genetic variant. Additionally, four healthy children (all girls aged 3 years) were selected as controls for RNA analysis and sampled by the Municipal Hospital’s pediatric team.
Written informed consent for clinical and genetic analyses was obtained from all the parents or guardians of the subjects participating in the study. This study was approved by the Ethics Committee of the Chiril Draganiuc Institute of Phthisiopneumology (Reference # CE-20.2).
2.2 Exome sequencing and variant detection
DNA was extracted from blood leukocytes of all patients and their parents. Exome capturing was performed using the xGen Exome Research Panel v1.0 Kit (Integrated DNA Technologies, Coralville, Iowa), and sequenced on Illumina NextSeq 500 (Illumina, San Diego, CA) with 150-bp paired-end reads. Demultiplexing/conversion of the sequencing data was done using bcl2fastq, version 2.18. The resulting fastq files were aligned to the human reference genome (GRCh37) by bwa mem, version 0.7.17, the local realignment was done using abra, version 2.05. After the variant calling by freebayes, version 1.1.0, the annotation of the resulting vcf file was done by SnpSift/SnpEff, version 4.3t, and vcfannotate, version 1.0.0-rc1.
Variant filtering
The filtering of the resulting variants was performed using GSvar, Version 2018_03-12-gbe02570, following steps and criteria outlined in Figure 1. First, only variants with acceptable quality scores (MQM >20 and QUAL >10) were put through the filtering process. Variants were further annotated with population databases (1000G, ESP, ExAC, plus in-house database of variants, n = 900) and only rare variants (MAF cutoff of ≤0.01) with in-house occurrence ≤20 in heterozygous state and ≤2 in homozygous state were considered. We set up a MAF threshold of 0.01 (in the overall world population), because this value consistently explains the incidence of BCG-itis in the Republic of Moldova—15-20 cases per 30000-35000 newborns, assuming autosomal recessive inheritance of the disease. Of note, we primarily relied on the predicted pathogenicity of the variants rather than their frequencies, although we realize that the lower is the MAF the higher is the expected clinical effect.
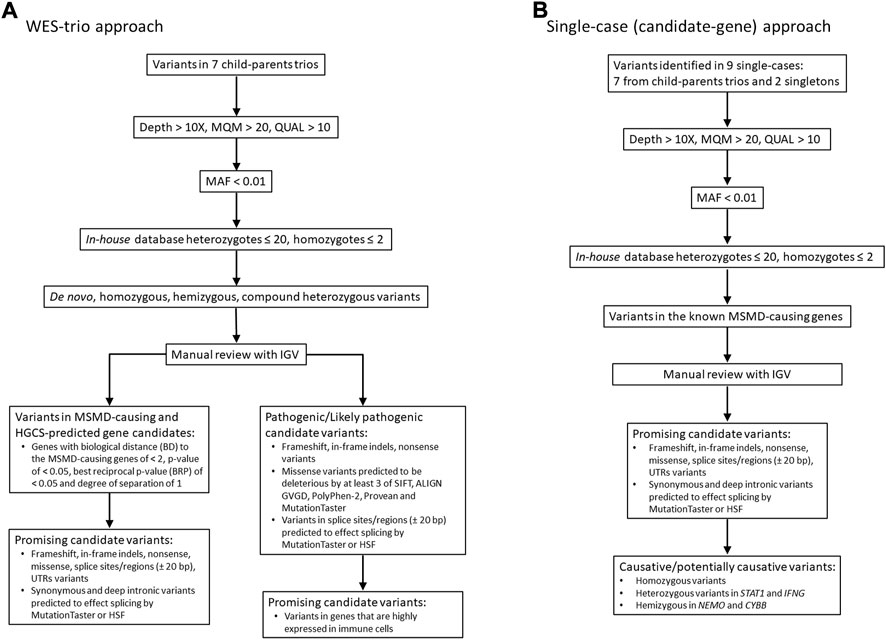
FIGURE 1. Workflow of WES analysis in 7 case-parental trios (A) and nine single cases (B). The filtered candidate rare variants in single-case and trio-based approaches are shown in Table 2 and Table 3, respectively. See also supplementary material for filtering query results in each trio/single case (Supplementary Tables S2, S3). MQM = mean mapping quality; QUAL = variant quality.
Two filtering approaches were used for the analysis of WES data in this study (Figure 1). In seven of nine cases in which both parents were available (family cases), WES analysis was trio-based (an affected proband with unaffected parents). In this approach, the variants were filtered according to compound heterozygous, homozygous recessive, de novo dominant and X-linked inheritance models. All detected variants were then manually checked using the Integrative Genomics Viewer (IGV) to confirm their quality. Subsequently, genes were prioritized based on predicted pathogenicity of mutations and/or their biological proximity to the known MSMD-causing genes as described in next subsections.
In the single-case (or index-based) analysis, variants in 19 known MSMD-causing genes (IL12B, IL12RB1, IL12RB2, ISG15, SPPL2A, IRF8, TYK2, FNGR1, IFNGR2, STAT1, KBKG, CYBB, JAK1, RORC, IL23R, ZNFX1, TBX21, IFNG and USP18) were considered by using a filter for the MSMD gene panel. Additionally, a panel consisting of 26 pattern recognition receptor (PRR) genes (TLR1, TlR2, TLR3, TLR4, TLR5, TLR6, TLR7, TLR8, TLR9, TLR10, CD14, NOD2, NLRP3, FCRLB, CLEC4E, CLEC7A, MRC1, CD209, MSR1, SCARB1, MARCO, CD36, STING1, AIM2, CGAS, and MBL2), which are promising hereditary candidates affecting susceptibility to mycobacterial infections (Randhawa et al., 2011; Pöyhönen et al., 2015; Varzari et al., 2019; Dubé et al., 2021), was evaluated for the occurrence of rare variants. This index-case approach increases sensitivity, but with a loss of specificity. We therefore applied it to all cases (singletons and those from case-parents trios). All variants identified in MSMD and PRR gene panels were validated by visual inspection using IGV.
In-silico prediction
Detailed in silico predictions for identified coding non-synonymous (missense) mutations were made with tools Align GVGD (Tavtigian et al., 2008), SIFT (Sim et al., 2012), MutationTaster (Schwarz et al., 2010), Provean (Choi and Chan, 2015) and Polyphen2 (Adzhubei et al., 2010) implemented in ALAMUT visual (interactive biosoftware, Version 2.7 rev.1, Rouen, France). The missense variants which were predicted as deleterious by at least three out of five methods were assumed likely as pathogenic. For predicting splicing effects, MutationTaster and the Human Splicing Finder (HSF) software (http://www.umd.be/HSF3/) were used (Desmet et al., 2009), and the overall effect was considered deleterious if at least one tool predicts it to be deleterious. Information from the variant genetics database ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), which adhere to the guidance of the American College of Medical Genetics (ACMG), was also employed for all types of variants.
The effects of mutations on protein structure were assessed using ExPASy translate tool (https://www.swissmodel.expasy.org/) (Waterhouse et al., 2018). In addition, I-Mutant 2.0 software (http://folding.biofold.org/i-mutant/i-mutant2.0.html) was used to predict the impact of mutations on protein structure stability based on the change in Gibbs free energy (ΔΔG) (Capriotti et al., 2006); the predictions were performed starting from the protein 3D structure with the temperature of 36°C and pH of 7.0 as default parameters. Finally, the Oligogenic Resource for Variant AnaLysis (ORVAL) platform was used to predict clinical consequences of intergenic interactions (https://orval.ibsquare.be/). Both heterozygous and homozygous rare variants identified through candidate genes and trio-based approaches were analyzed in ORVAL in accordance with recommendations of the developers (Renaux et al., 2019).
Functional annotation and categorization of genes
We used DAVID classification system (https://david.ncifcrf.gov/) and PubMed (https://pubmed.ncbi.nlm.nih.gov/) for information about gene function and their involvement in diseases.
Biological affinity/similarity between candidate genes identified by the trio-based prioritization analysis and 19 known MSMD-causing genes was evaluated using the Human Gene Connectome Server (HGCS) (https://hgc.rockefeller.edu/) (Itan et al., 2014). The level of protein-protein/gene-gene interaction was also analyzed and graphically visualized as nodes (genes/proteins) and edges (the relationship between proteins) using STRING software (https://string-db.org/). In this analysis, the novel and known MSMD-causing genes were considered as the seed molecules from which direct and indirect protein-protein/gene-gene interactions were obtained in accordance with the developers’ recommendations (Szklarczyk et al., 2019).
The genes were also categorized based on their expression pattern as reported in the GTEx database (GenotypeTissue Expression Project database, https://gtexportal.org/home/). Genes with the higher expression levels in whole blood or immune-related cells (i.e., spleen or EBV-transformed lymphocytes) were given the highest priority in this study. Additionally, RD-Match (http://genetics.bwh.harvard.edu/rdmatch/) was used to estimate probability of finding mutation by chance in a given gene (Akle et al., 2015).
Whole blood RNA extraction and RT-PCR
Peripheral blood samples were collected via PAXgene RNA tubes (Qiagen, Valencia, CA) and stored at - 80°C until RNA extraction. RNA was extracted via PreAnalytiX PAXgene blood RNA kit from PreAnalytiX/Qiagen. DNase I treatment was performed at the same time as the RNA isolation using the reagents from the PreAnalytiX PAXgene blood RNA kit. Quantity and quality of the extracted RNA was verified by the NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE. United States) and the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA. United States).
RNA was reverse transcribed into cDNA using SuperScript® VILO Master mix (Invitrogen, Massachusetts, United States). Polymerase chain reactions (PCR) of cDNA sequences flanking variants IL12B c.877A>G (rs1177184657), IL12B c.89–14T>C, HEATR3 c.395_396delAA, STAT1 c.373–2A>C and GBP2 c.1149+14T>C were performed using Tempase Hot Start DNA Polymerase (Ampliqon, Odense, DK) and specific primers: 5′-AGAGCAAGATGTGTCACCAG-3′ and 5′-TGATTGTCGTCAGCCACCAG-3′ for IL12B c.89–14T>C, 5′-CATCAGGGACATCATCAAACC-3′ and 5′-TCCATACATCCTGGCAGACA-3′ for IL12B c.877A>G, 5′-TCTGGAAAACGCCCAGAGATT-3′ and 5′-ATTGGTCTCGTGTTCTCTGTTCT-3′ for STAT1 c.373–2A>C, 5′-TGCTGGAAAAGCTCCAGCACC-3′ and 5′-TCCACAGCACGTTCACAGTC-3′ for HEATR3 c.395_396delAA, and 5′-AGGAGGAAGAGCTGAACCCT-3′ and 5′-GTCATCTCGCCTTGCTTCC-3′ for GBP2 c.1149+14T>C. The PCR products were separated and verified by electrophoresis on agarose gel. In case alternative splice variants were identified, the bands were purified by gel extraction using Qiagen Gel Extraction kit (Qiagen, Hilden, DE) and sequenced using the dideoxy chain-terminator method of Sanger.
GBP2 minigene reporter assay
A minigene splicing assay was performed to verify/validate whether the candidate mutation c.1149+14T>C in GBP2 affected splicing products. Briefly, a 1,157 bp fragment harboring exon 7 (281 bp) and part of the flanking introns (246 bp of intron 6 and 581 bp of intron 7) was amplified by standard PCR with patient genomic DNA as template employing a proofreading polymerase and primers: GBP2S-NotI (5′-AATCGGGCGGCCGCTTGACCTAGTATCTCAGCCCCA) and GBP2AS-BamH (5′-CGATTCGGATCCCCAGAACCTCTGACTGTAGCA). The fragment was cloned into the pSPL3 reporter vector between the NotI and BamHI sites (Life Technologies).
Sanger sequencing was used to evaluate whether the wild-type (WT) and mutant-type (MT) expression vectors had been successfully constructed without errors. The minigenes were transfected in duplicates into HepG2 cells using X-tremeGENE transfection reagent (Roche Life Science) according to the manufacturer’s instructions and our previous publications (Palhais et al., 2015; Palhais et al., 2016). Forty 8 hours after transfection cells were harvested and RNA was isolated with QIAzol (Qiagen) and chloroform extraction. cDNA synthesis was performed using SuperScript® VILO Master mix (Invitrogen). Splicing of GBP2 exon seven was examined by PCR using Tempase Hot Start DNA Polymerase (Ampliqon) and vector specific primers SD6 (5′-TCTGAGTCACCTGGACAACC-3′) and SA2 (5′-ATCTCAGTGGTATTTGTGAGC-3′). The PCR products were separated and verified by electrophoresis in an agarose gel and characterized by direct Sanger sequencing. The relative quantity of the bands corresponding to exon inclusion/exon skipping was estimated by capillary electrophoresis using the Fragment Analyzer™ (Agilent Technologies, Santa Clara, United States) instrument and software and reported as percent exon skipping (relative to the total intensity of the signal in each lane).
Real-time qPCR
The expression of GBP2 was analyzed by quantitative real time PCR in five RNA samples: two independently isolated RNA samples from the carrier of the mutation c.1149+14T>C (i.e., the father of the patient P2) and three matched control samples. All reactions were run in duplicate on the Roche LightCycler 480 with SYBR-green premix FastStart Essential DNA Green Master (Roche Diagnostics, CH) according to manufacturer instructions employing the following primer pairs: GBP2 forward 5′-CAATTACGCAGCCTGTGGTG-3′, GBP2 reverse 5′- GAGCCTAGAGAGAAGCCGTTT and RPL13A forward 5′- AAGGTCGTGCGTCTGAAG, RPL13A reverse 5′-GAGTCCGTGGGTCTTGAG. RPL13A was used as a normalization control because it is stably expressed across different tissue types, including blood cells (Ledderose et al., 2011; Sabatino et al., 2013; Xiao et al., 2017; Dembic et al., 2019).
Statistical analysis
Data from RT-qPCR are expressed as mean ± standard error (s.e.m). Samples were normalized to RPL13A. Means of different groups were compared and analyzed using the Student’s t-test.
Surface plasmon resonance imaging (SPRi)
SPRi was carried out as previously described (Holm et al., 2022). Briefly, biotinylated RNA-oligonucleotides (GBP2-WT: 5′-UAUUUGGUGUUCUACGUCAUGA-teg-bio and GBP2-MUT: 5′-UAUUUGGUGUCCUACGUCAUGA-teg-bio) were immobilized onto a G-strep sensorchip (SSENS) for 20 min. The hnRNPA1 recombinant protein were injected for 8 min, followed by dissociation for 4 min; 6.25–200 nM hnRNPA1 (Abcam, ab123212). A continuous flow of SPR buffer (10 mM Tris-HCl, ph 7.9, 150 mM KCl, 3.4 mM EDTA, 0.005% tween-20). Binding was fitted to a 1:1 kinetics model with Scrubber2 (v. 2.1.1.0; Biologics inc.). For hnRNPA1 a biphasic 1:2 model was used in ClampXP (version 3.50; Biosensor Data Analysis).
Results
Summary of exome sequencing data
Overall, 100 M raw reads per sample (range 76M–229 M) were generated, of which 98.7% (range 97.8%–99.5%) and 81.1% (range 74.11%–85.93%) were aligned to the human reference genome (hg19) with a mapping qualities of Q20 and Q30, respectively (Supplementary Table S1). On average, 97.9% of the targeted regions (range 95.3%–98.8%) were covered at least 10 times, and 97.0% of the targeted regions (range 90.6%–98.5%) were covered at least 20 times. The average read depth in the targeted region was 129x (range 96.7x–244.9x) reads per base. Above all, these data fully reflect the reliability of our sequencing, thus providing a solid basis for the follow-up analysis.
Two approaches (trio-based exonic screening and single cases candidate genes analysis) were employed to identify disease-associated variants (Figure 1). The filtering query results for each sample are given in Supplementary Tables S2, S3.
Variants in MSMD and PRR gene panels
Twelve qualifying low frequency variants within eight known MSMD-causing genes were identified in six of the nine patients investigated, including six missense, three splice site/region (±20 bp), two deep intronic and one synonymous mutations (Table 2 and Supplementary Table S3). All detected mutations were in a single heterozygous state. Three of them were found in genes STAT1 (c.373-2A>C in P9 and c.541+50T>C in P7) and IFNG (c.40G>A/p.(Val14Met) in P3), responsible for both autosomal dominant and autosomal recessive forms of mycobacterial disease. Three variants were found in the IL12B gene in two patients, with variants c.877A>G/p.(Lys293Glu) and c.89-14T>C present in the same patient (P6) in a compound heterozygous state (according to the trio-based analysis). Noteworthy, five variants, namely TYK2 c.2441C>T, TYK2 c.157G>A, IFNGR1 c.40G>A, IL23R c.257G>A, IL12B c.961G>A and IL12RB1 c.16196C>T, have been previously reported in patients with MSMD, but it is unclear whether they are actually connected to the disease (https://www.ncbi.nlm.nih.gov/clinvar/).
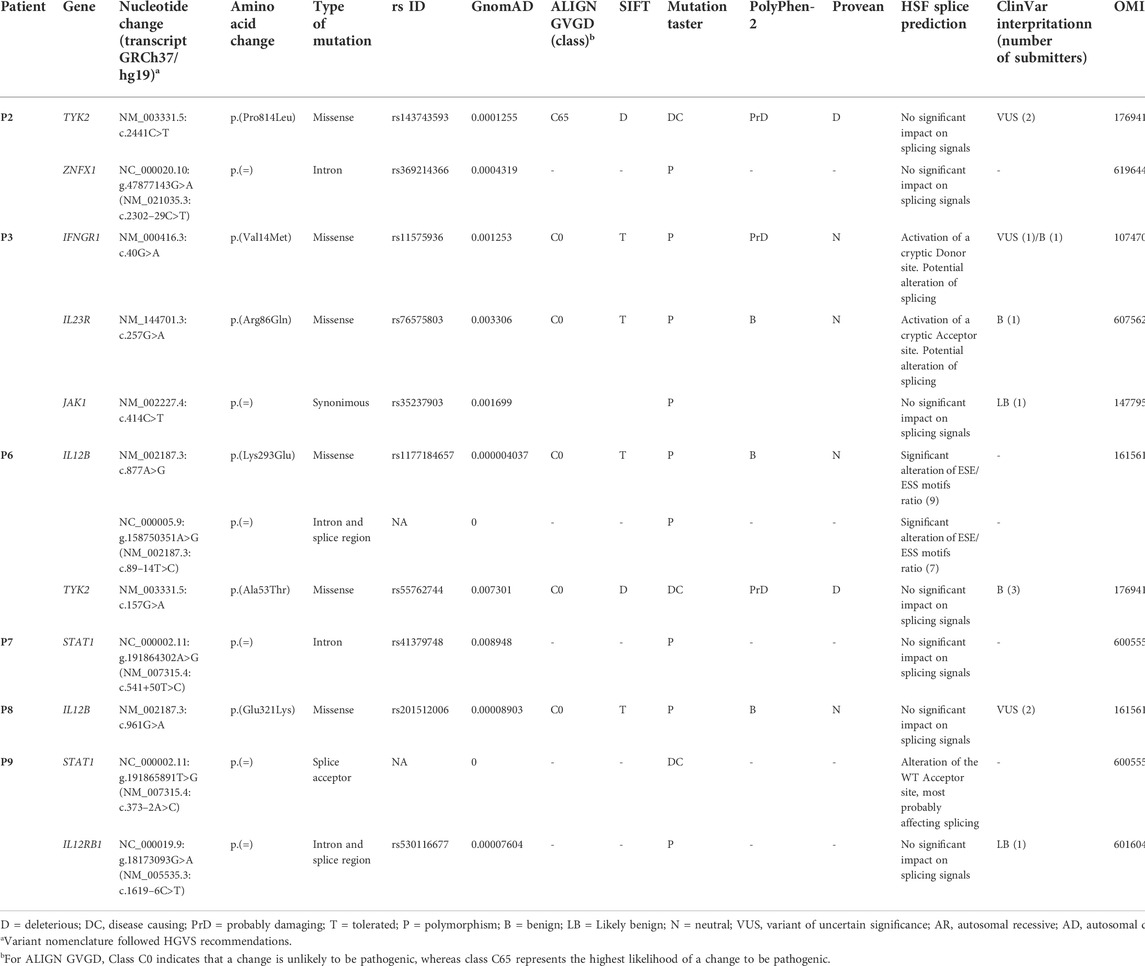
TABLE 2. Rare variants (MAF <0.01) identified in the known MSMD-associated genes.
The study also identified twelve variants in nine PRR genes in six patients (Supplementary Table S4). All were single heterozygous changes, inherited (as seen from the case-parents trio analysis), reported in databases with little or no functional consequences (i.e., intronic, synonymous, benign missense variants).
Trio-based exonic screening of rare variants and phenotype-based gene prioritization
Using trio-based WES, a total of 455 high-confidence rare variants in 201 genes that fit the proposed inheritance criteria (i.e., de novo, homozygous, hemizygous, compound heterozygous variants) were detected in seven trios. These variants included 389 compound heterozygous mutations in 135 genes, 20 homozygous (6 missense, four splice region intronic, one UTR, three intronic, five synonymous, one in non-coding transcript), 32 X-linked hemizygous (12 missense, one in-frame insertion, four splice region intronic, seven UTRs, two intronic, six synonymous), and 14 de novo (4 missense, one frame shift, one in-frame deletion, one splice region, two intronic, five synonymous) variants. After in-silico assessment of pathogenicity, 20 most promising variants in 13 genes were finally selected as top candidates for susceptibility to mycobacterial disease (Tables 3 and 4; Supplementary Table S5).

TABLE 3. Candidate rare variants (MAF <0.01) identified in the seven MD trio probands.
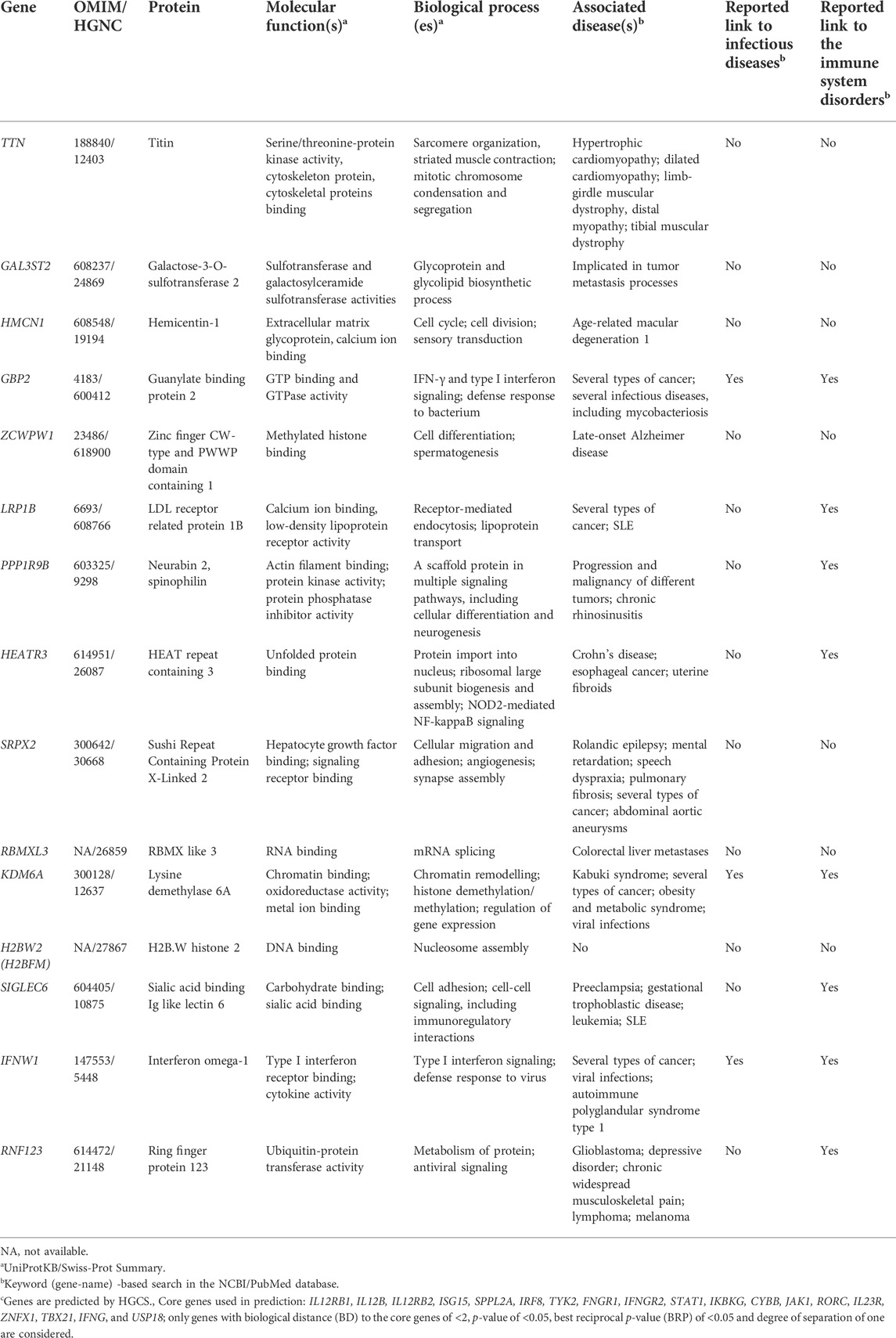
TABLE 4. Summary of the novel genes with rare variants detected in the trio-based WES analysis.
By analysis of biological distances between the identified 201 genes and MSMD core genes using HGCS software, two genes–GBP2 and IFNW1 were ranked as the closest to the MSMD genes. Two heterozygous variants in GBP2 (c.412G>A/p.(Ala138Thr) and c.1149+14T>C) and one homozygous variant in IFNW1 (c.58G>A/p.(Gly20Arg)) were therefore prioritized (Table 3). Similar results were obtained by analysis of gene network using String software (Figure 2). In this analysis, genes GBP2 and IFNW1 are closely interconnected with other known MSMD-associated genes, forming a densely connected module, which implies their direct functional involvement in the disease-associated (i.e., IFN-γ signaling) pathway. Also, KDM6A showed some affinity with the MSMD-associated gene TBX21. Other genes found in this study locate outside the network, suggesting either their affiliation to different pathways or indirect interactions via yet unknown elements.
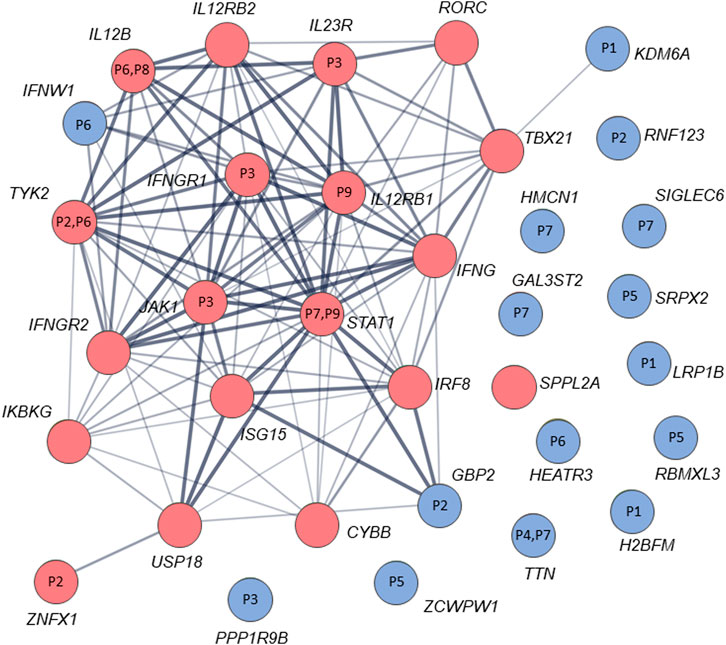
FIGURE 2. STRING interaction (network) analysis (https://string-db.org/) between 34 genes/proteins. Each circle represents a protein/gene: red nodes are known MSMD-causing genes and the blue nodes are novel candidate genes identified in this study via trio-based approach. Numbers inside the nodes denote codes for patients with mutations in the corresponding genes. Interactions between proteins/genes are indicated by lines with variable thickness showing the strength of data support. The confidence cutoff for showing interaction links has been set to ‘medium’ (0.400).
The significance of new candidate genes was also assessed by their expression in immune related tissues using the GTEx database. Of 15 prioritized in this study genes, four genes, namely GBP2, HEATR3, PPP1R9B and KDM6A, exhibited an elevated transcriptional level in whole blood, spleen or EBV-transformed lymphocytes (Supplementary Table S6), thus lending additional support to their role in increasing susceptibility to mycobacterial infections. Remarkably, genes HEATR3 and PPP1R9B harbor de novo variants, both with predicted damaging effects–a frameshift (Lys132Argfs*3) and a disruptive in frame deletion (Pro252_Pro254del), respectively. Assessment with RD-Match did not reveal true-positive matches between immunodeficiency and either our or known MSMD-associated genes (Supplementary Table S7), thus indicating its insufficient power for mitigating false-positive findings in this study.
Analysis of mRNA transcripts
Two patients were available for the analysis of mRNA, including one with BCG-tis (P6) and another with disseminated TB infection (P9). For the four candidate mutations identified in a patient with BCGitis, effects on splicing were predicted for two compound heterozygous variants c.89-14T>C and c.877A>G in the known MSMD-associated gene IL12B and one de-novo frameshift mutation c.395_396delAA in HEATR3. To examine the functional impact of these variants on mRNA splicing, we performed RT-PCR experiments on total RNA extracted from peripheral leukocytes. Agarose gel electrophoresis showed only single wild-type bands across four loci in all samples (the carriers and controls), indicating no visible effect of these mutations on the pre-mRNA splicing (Supplementary Figure S1).
A novel heterozygous STAT1 intron five splice site mutation, c.373-2A>C, abolishing the splice acceptor site of exon 6, was found in a patient with disseminated TB infection (P9). The mutation causes a dramatic reduction in the MaxEnt score for the mutant 3’ splice site (http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html). RT-PCR with primers located in exons five and seven of STAT1 revealed three products in the mutation carrier sample, while only one middle-length band was observed in all samples used as controls (Figure 3A). Sanger sequencing of the respective PCR products confirmed that the middle band (205 bp) corresponds to a wild type variant, the upper band (385 bp) corresponds to aberrant splicing where an upstream splice acceptor site is used, resulting in retention of 174 bp of intronic sequence, and the lower band (115 bp) corresponds to the product in which exon six is skipped (Figure 3B). Thus, we conclude that the mutation STAT1 c.373-2A>C affects the pre-mRNA splicing in vivo (Figure 3C).
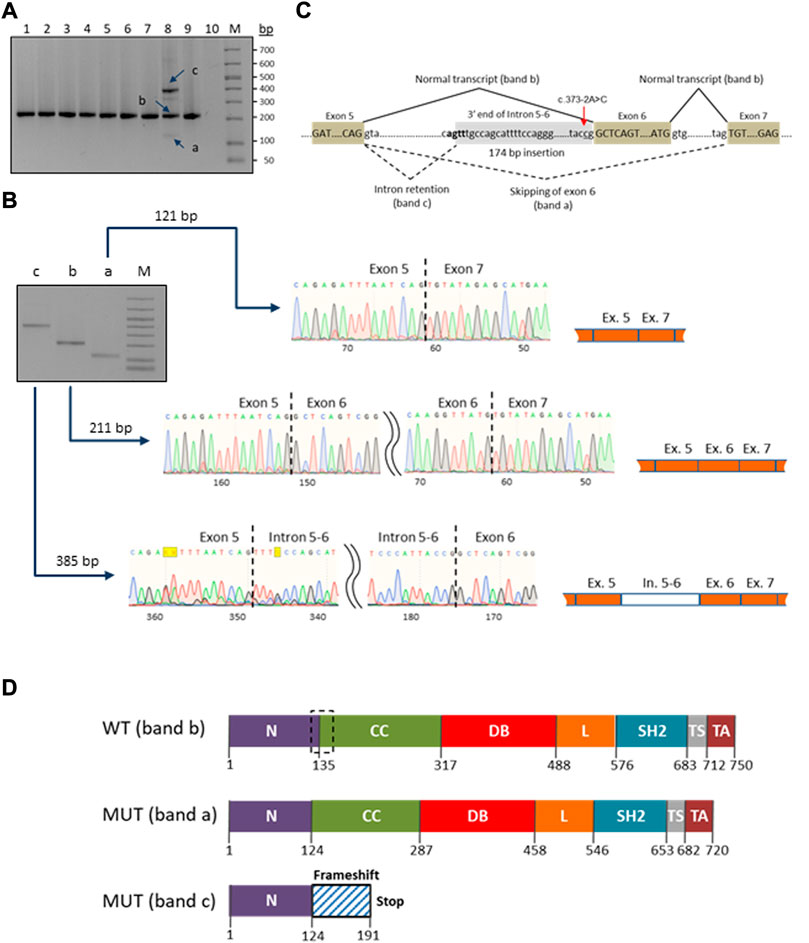
FIGURE 3. Characterization of the novel c.373–2A>C mutation in the STAT1 gene. (A) RT-PCR electrophoresis products on 2% agarose gel. The carrier patient (lane 8) showed three cDNA fragments (a, b and c) compared to one in the controls (lanes 1-7 and 9). (B) Purification and Sanger sequencing of RT-PCR products. Sanger sequencing revealed correctly spliced STAT1 transcript of 211 bp (lane b) and two mutated transcripts: an aberrant short fragment (121 bp, lane a) corresponding to the in-frame skipping of exon six and the aberrant long fragment (385 bp, lane c) corresponding to the out-of-frame retention of the intronic fragment. (C) Schematic representation of the splicing for the splice site mutation c.373-2A>C (red arrow). The newly generated splice signal between exons 5-6 is shown in bold. Exons are shown as boxes. (D) Prediction of impact of the splice site mutation c.373-2A>C on the STAT1 protein structure. Wild-type STAT1 is a multidomain protein of 750 amino acids. The mutant sequences are of 720 and 191 ammino acids length, corresponding to the short (exon six skipping/band a) and long (174 bp intronic fragment insertion/band c) transcripts/bands, respectively. The structural domains within the protein are the coiled-coil domain (CC), DNA binding domain (DB), linker domain (L), SH2 domain (SH2), tail segment domain (TS), and trans-activator domain (TA) are indicated, together with their amino acid boundaries. Dotted box in the wild type sequence delimits the boundaries of the deleted region (30 amino acids) in the long mutant isoform (MUT band a).
Functional characterization of GBP2 c.1149+14T>C variant
Patient P2 was found to be compound heterozygous for two mutations in the GBP2 gene, namely, a missense mutation c.412G>A/p.(Ala138Thr) and an intronic mutation c.1149+14T>C immediately downstream of the 5′ splice site of exon 7. As RNA was not available from this patient, in vitro functional experiments (i.e., mini-gene assay and SPRi analysis) were designed and used for testing the effect of c.1149+14T>C on splicing. In the mini-gene assay, two constructed minigenes, covering exon seven of the GBP2 gene and carrying either a T (wild type) or a C (mutant) at position c.1149+14T>C, were transfected into HepG2 cells and splice products were subsequently analyzed by RT-PCR (Figure 4A). The analysis showed a slightly decreased exon seven inclusion in HepG2 cells transfected with the MUT minigene as compared to the WT minigene. Quantitative analysis of exon seven inclusion/exclusion showed a statistically significant difference (p = 0.0028) between WT and MUT lanes, suggesting that c.1149+14T>C has an effect on splicing (Figure 4A). In particular, the mutation may increase the ability of negative splicing regulatory proteins, such as heterogeneous nuclear ribonucleoproteins (hnRNPs), to bind the pre-mRNA. To investigate this, the kinetics of RNA–protein interactions was analyzed by SPRi using recombinant hnRNP A1 protein and biotin-labeled wild-type or mutant (i.e., harboring c.1149+14T>C) oligonucleotides. The analysis revealed binding of the splicing inhibitory protein hnRNP A1 to oligonucleotides with wild type sequence and the binding was increased for the oligonucleotides carrying mutant sequence (Figure 4B). Thus, the results from the SPRi analysis supports the effect of the c.1149+14T>C variant on the exon seven skipping via increased binding of hnRNP A1.
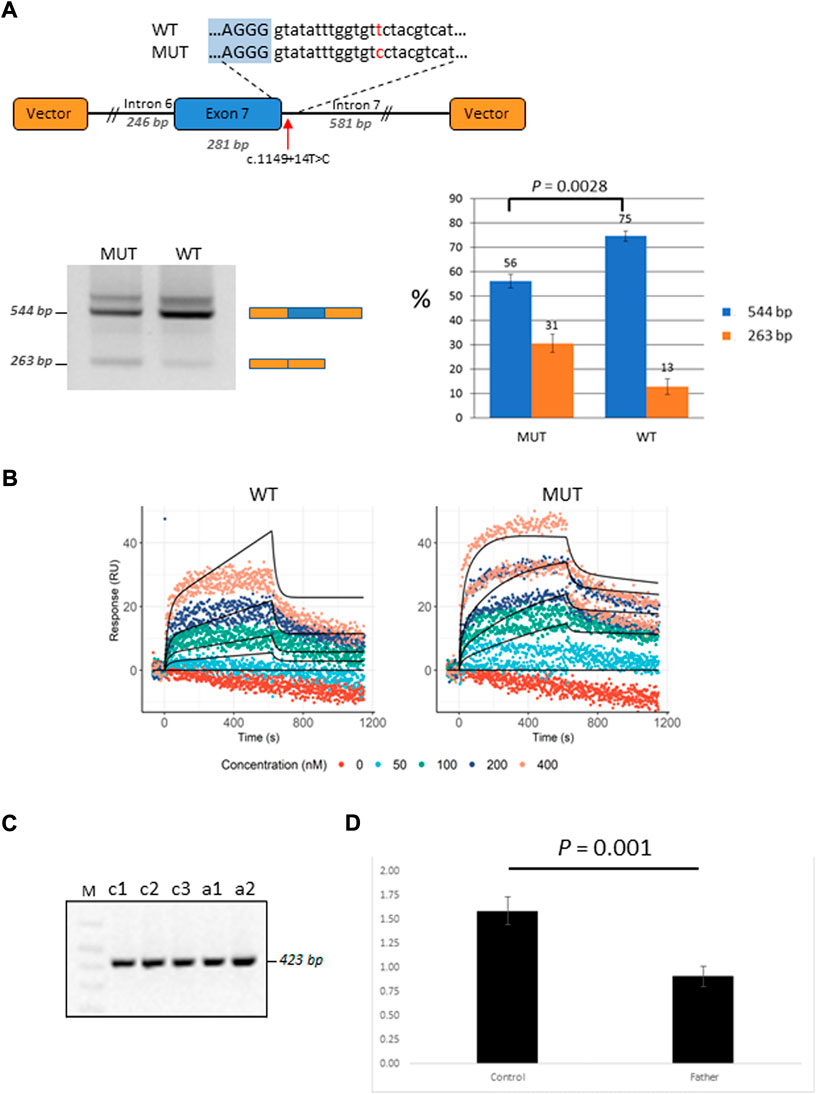
FIGURE 4. Functional characterization of the GBP2 intronic variant c.1149+14T>C. (A) Minigene analysis of c.1149+14T>C. Illustration of the GBP2 minigene. The 5′ splice site of exon seven and the downstream sequence covering the c.1149+14T>C variation is enlarged (top). RT-PCR analysis of minigene splicing in HepG2 cells. The analysis shows that the minigene carrying the mutant sequence (MUT) has a higher degree of exon seven skipping than the minigene carrying the wild type sequence (WT) (bottom, left). Quantification of the exon seven inclusion and exclusion rates (blue and orange columns, respectively). The inclusion/exclusion is quantified on a Fragment Analyzer instrument and represent the intensity of the 544 bp or 263 bp bands over the total intensity in the lane (bottom, right). (B) SPRi measurements of hnRNP A1 (splicing silencer factor) binding to the WT (the left hand side) and MUT (the right hand side) oligonucleotides. Dots correspond to the raw SPRi measurements while black lines correspond to the model-implied fits. Protein (hnRNP A1) was injected in increasing 2-fold concentrations from 50 to 400 nM shown in color dots. (C) RT-PCR analysis of c.1149+14T>C from the mRNA of the mutation carrier (the father of P2; samples: c1 and c2) and three healthy controls (samples: a1, a2 and a3). (D) qRT-PCR analysis of total GBP2 mRNA from whole blood in the carrier (the father of P2) and three healthy controls. GBP2 transcript levels were normalized by RPL13A expression. The values presented are the medians of duplicate determinations ±SD.
The father of the child (P2) who is a heterozygous carrier of the c.1149+14T>C variant was available for in vivo mRNA analysis of GBP2. Reverse transcriptase PCR (RT-PCR) analysis of splice variants showed no structural difference in the cDNA between the mutant and control samples for c.1149+14T>C, with only one wild-type band detected (Figure 4C). The absence of alternative transcript variants and inconsistency with the results from in-vitro mini-gene assay may be due to nonsense mediated decay (NMD), which recognizes and eliminates abnormal mRNAs making them virtually undetectable. To test this hypothesis, we compared the levels of GBP2 expression in the carrier of the c.1149+14T>C mutation (i.e., the father of the affected child) with the GBP2 expression levels in three matched healthy controls. As is shown in Figure 4D, the mutation carrier had significantly lower GBP2 expression compared to controls (p = 0.001). This finding is consistent with the hypothesis of mRNA degradation by NMD and supports the impact of the intronic mutation c.1149+14T>C in GBP2 on alternative splicing.
Protein structure prediction
Swiss ExPASy (Expert Protein Analysis System) Molecular Biology Server was used to predict the effects of identified mutations in HEATR3, STAT1 and GBP2 genes on the protein structure. The de novo deletion (-AA) in the third exon of HEATR3 is a frameshift mutation predicted to cause a severely truncated non-functional HEATR3 protein (Supplementary Figure S2). The skipping of exon six from the STAT1 gene transcript leads to an in-frame deletion of 90 nucleotides and the removal of 30 amino acids from a stretch linking the N-Terminal to the Coiled-Coil domain of STAT1, while the retention of a 174 bp intronic fragment leads to an out-of-frame change and generates a premature termination codon at position 191 (Figure 3D).
The missense mutation c.412G>A/p.(Ala138Thr) in GBP2 gene is located in an essential functional domain and was predicted to be deleterious by four out of five in silico tools used (Table 2). Noteworthy, the alanine (Ala) at position 138 is one of four hydrophobic residues located along one side of the α-helix (Figure 5). While the structural analysis revealed no essential differences between the wild-type and mutant models, the substitution of Ala to the more hydrophilic threonine (Thr) leads to the reduction in hydrophobicity of the whole helix from -0.68 to -0.92 (QQAMDQLHYV vs. QQTMDQLHYV, http://www.gravy-calculator.de), that may affect the stability of the protein. Indeed, the predicted folding free energy change determined using the I-Mutant2.0 software was negative (DDG = -1.64) with a sufficient reliability (RI) of 7.0, indicating a destabilizing effect of the above substitution on the protein structure and possibly function.
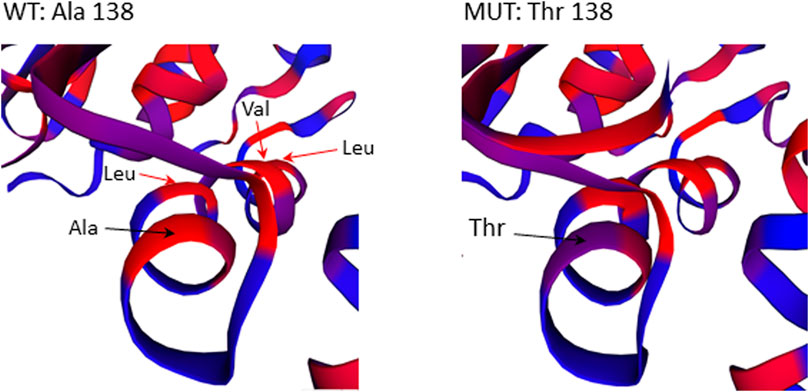
FIGURE 5. Close-up view of residues surrounding Ala138Thr in the 3D structure of the GBP2 protein. Substitution of Ala with more hydrophilic Thr reduces the stretch of hydrophobicity along one side of the α-helix.
Identification of an oligogenic etiology in patients with mycobacteriosis
In seven out of nine patients investigated (P1, P2, P3, P5, P6, P7 and P9) the identified variants occurred concomitantly in two or more different candidate genes (Tables 2 and 3). We next used the ORVAL platform to explore their potential digenic or oligogenic impact (i.e., epistatic interactions) in increased predisposition to mycobacterial disease. The analysis identified five patients with at least one pathogenic combination (cutoff >0.532 for CS and ≥50% for SS) between genes (Figure 6). In particular, the variant combinations in the gene pairs H2BW2-LRP1B, H2BW2-KDM6A (both in P1), GBP2-TYK2 (in P2), SRPX2-ZCWPW1 and ZCWPW1-RBMXL3 (both in P5) were predicted as disease-causing candidates with 95% confidence, while the variant combinations in the gene pairs GAL3ST2-HMCN1, GAL3ST2-TTN and HMCN1-TTN (all in P7) were within the 99% confidence zone for pathogenicity.
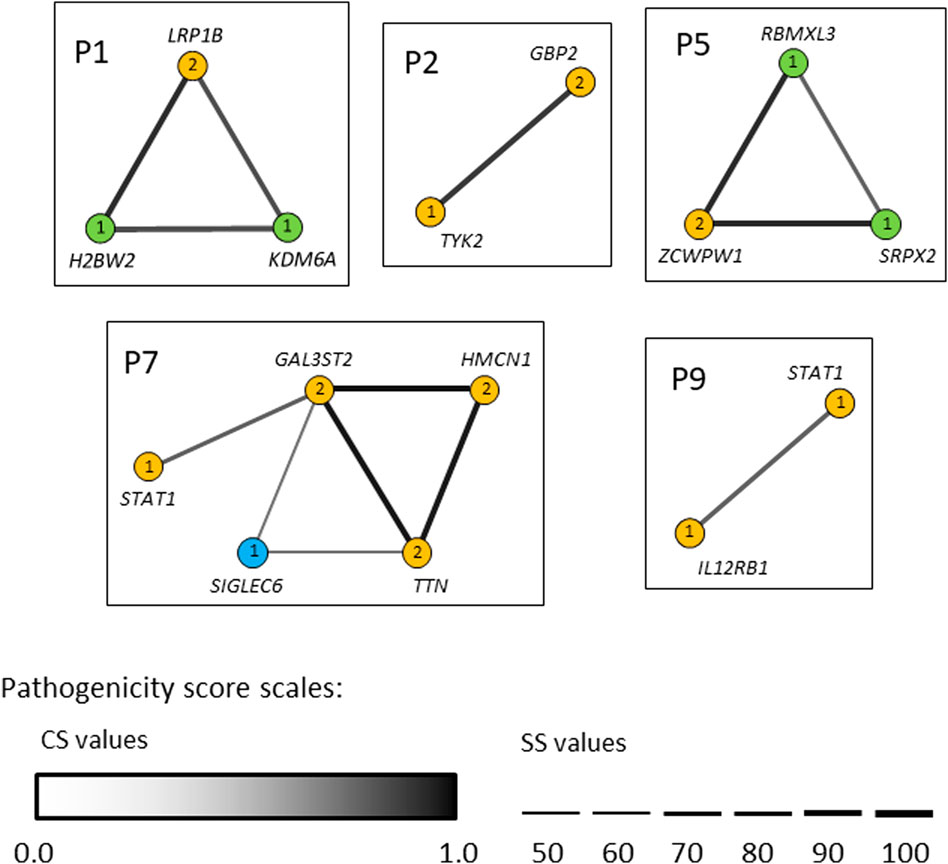
FIGURE 6. Gene networks showing the epistatic interactions between the genes in which rare variants have been identified. Nodes are individual genes and edges represent pair-wise interactions between genes/variants. Numbers inside the nodes denote number of variants. Yellow, blue and light-green nodes represent heterozygous, homozygous and hemizygous variants, respectively. Edge color intensity is proportional to the pathogenicity classification score (CS), whereas edge thickness is proportional to the pathogenicity support score (SS) for a combination of variants. In case two variants within one gene (i.e., compound heterozygotes), the highest pathogenicity scores computed for variant combinations are depicted. Only disease-causing variant combinations/networks (cutoff >0.532 for CS and ≥50% for SS) are illustrated.
Discussion
In this study, the genetic etiology of susceptibility to mycobacterial infections was explored in nine pediatric cases by whole-exome sequencing, bioinformatic analysis and functional assessment. As result, several potentially causative rare variants (mostly SNVs) in the MSMD-known and newly discovered genes were identified via candidate gene (singleton WES) and child-parents trio approaches.
Using singleton WES, 12 heterozygous (monoallelic) mutations in the eight MSMD-causing genes, namely TYK2, ZNFX1, IFNGR1, IL23R, JAK1, IL12B, STAT1 and IL12RB, were detected in six patients. As mutations in STAT1 and IFNGR1 are known to be responsible for both AD and AR forms of mycobacteriosis, the heterozygous variants identified in these genes (STAT1 c.373–2A>C and IFNGR1 c.40G>A/p.(Val14Met) are the most promising causal candidates in this study.
The novel splice-site mutation (c.373-2A>C) reported here in the gene STAT1 in a patient with disseminated TB leads to both partial intron five retention and exon six skipping. The retention of the intronic fragment leads to a frameshift, resulting in early termination of translation and a nonfunctional protein. The in-frame exon skipping event causes a deletion in the protein which would lead to the loss of connection between the N-Terminal and Coiled-Coil domains, which are known to play a role in the dimerization and DNA binding (Mertens et al., 2006). Variants with similar molecular outcomes have been reported previously in several cases with mycobacterial infections (Kong et al., 2010; Kristensen et al., 2011; Vairo et al., 2011; Sakata et al., 2020). These however were in biallelic homozygous or monoallelic compound heterozygous states and were inherited according to an AR mode, while in our case the finding of only a single monoallelic variant is more consistent with an AD inheritance pattern. A simple haploinsufficiency at the STAT1 locus may provide an explanation for our case, as the dosage of normal STAT1 protein generated by the single wild-type allele could be insufficient for complete protection against the highly virulent M. tuberculosis infection in this unprotected (i.e., BCG-unvaccinated) infant. Alternatively, a compound combination with an additional monoallelic variant located in a deep intronic or regulatory region of STAT1 not captured by our analysis may be casual. This second scenario may also be a cause for BCGitis in five patients with mutations in the TYK2, IL23R, JAK1, IL12B and IL12RB genes responsible for AR MSMD. Further genetic and functional studies should be carried out to evaluate the exact biological effects and inheritance pattern of the identified mutants. Regarding IFNGR1, although the identified variant IFNGR1 c.40G>A/p.(Val14Met) has a relatively high allele population frequency (0.002–0.003) in Europe, it was proven in previous studies to be damaging and was enriched in patients with eczema herpeticum (ADEH+)—a rare and serious skin infection caused by the herpes simplex virus (Gao et al., 2015).
In a trio approach, 25 variants in 15 unknown genes were bioinformatically prioritized. We then critically revised these genes by examining their expressions in immune related tissues. As a result, genes GBP2, HEATR3, PPP1R9B and KDM6A, showing elevated expressions in whole blood, spleen or EBV-transformed lymphocytes, were selected as top-ranked candidates for susceptibility to BCG infection in this study. Of these new candidate genes, GBP2 displays the closest biological relationship with the known MSMD-causing genes. The encoded by GBP2 protein (Guanylate Binding Protein 2) is a member of interferon-gamma induced guanylate-binding proteins (Ramsauer et al., 2007; Yan et al., 2016). Following induction, GBPs confer protective immunity against a variety of microbial pathogens (Kim et al., 2011; Praefcke, 2018; Tretina et al., 2019). Previous experimental studies using animal models and also functional gene-expression studies demonstrated its important role in the protection against mycobacteria (Berry et al., 2010; Kim et al., 2011; Marquis et al., 2011; Tretina et al., 2019; Perumal et al., 2021). In this study, two compound heterozygous mutations in GBP2 gene (c.412G>A/p.(Ala138Thr) and c.1149+14T>C) in a patient with BCG infection (P2) were identified. The maternally inherited missense mutation p.Ala138Thr was predicted to be deleterious by the majority of in silico tools, possibly having an impact on the protein folding and stability. Functional analysis of the second, paternally inherited intronic mutation c.1149+14T>C, demonstrated that this variant is likely to affect splicing of the GBP2 mRNA transcript, leading to decreased levels of functional transcript.
A recently discovered MSMD-causing gene SPPL2A was an outlier compared to the other MSMD-causing genes in our gene networking and connectome server analyses (Figure 2). Likewise, prioritized novel genes in the present study, although not directly linked to the core MSMD-causing genes, may also be involved in the disease pathways. Indeed, HEATR3 is a component of the NOD2-mediated NF-kappaB signaling, which plays a crucial role in the control of Mycobacterium within host macrophages (Deng and Xie, 2012; Zhang et al., 2013). Here, we identified a deletion frameshift mutation c.395_396delAA/p.(Lys132Argfs*3) in HEATR3 in a patient with BCG-related complications. The variant introduces an early premature termination codon, which could either be associated with no protein due to nonsense-mediated mRNA decay or a severely C-terminally truncated protein missing all functional domains. Moreover, the de novo status of this variant in our patient increases it is likelihood for being a real cause of BCGitis. Also, undescribed de novo disruptive in-frame deletion was found in PPP1R9B, which encodes a scaffolding protein involved in multiple signaling pathways and cytoskeletal rearrangement in NK cells (Meng et al., 2009).
Other genes in this study, including TTN and LRP1B, are secondary candidates, because of their insignificant expression in immune cells, although their impact cannot be completely excluded.
With the increasing use of next generation sequencing, it has become clear that oligogenic inheritance is more common than previously thought for hereditary disorders, including PIDs (Rigaud et al., 2011; Chong et al., 2015; de Valles-Ibáñez et al., 2018). Here, we identified five patients with potential oligogenic inheritance. Noteworthy, two of these cases carried trans heterozygous (monoallelic) variants in two genes belonging to the IL-12/IL-23/IFN-γ axis, namely, IL12RB1 and STAT1 in the patient P9, and TYK2 and GBP2 (a novel candidate-gene) in the patient P2. Since IL12RB1, STAT1 and TYK2 separately are known to cause autosomal recessive MSMD and GBP2 is a novel plausible candidate for MSMD, reduced activity of both proteins in the same close interacting network is a likely disease model. This disease mechanism, termed ‘synergistic heterozygosity’ (Vockley et al., 2000), could also explain differences in the penetrance of mycobacterial disease in some families and the above-mentioned inconsistency with previously published results in the inheritance of STAT1 c.373–2A>C. Also, H2BW2 (a histone protein) and KDM6A (a histone H3 demethylase protein) belong to the same chromatin remodeling pathway regulating gene expression. Although experimental validation of the oligogenic model still needs to be conducted, our data provide insight into the complex disease-causing mode of mycobacterial disease and suggest the combined effect of mutations in IL-12/IL-23/IFN-γ mediated and other pathways genes as an important mechanism in the susceptibility to mycobacterial infections.
Conclusion
The analysis presented here expand the spectrum of genetic variation underlying mycobacterial infections in children. Indeed, we identified 12 heterozygous variants, two of which were novel, in eight known MSMD-causing genes and found pathogenic or likely-pathogenic variants in 15 new candidate genes. Besides this, our study provides insights into a potential oligogenic disease-causing mode for conferring susceptibility to mycobacterial infections. The obtained results rise several topics for further research, including methodological application of WES to identify novel genes for susceptibility to mycobacterial and other infections, experimental and population-based validations of the identified variants and genes, role of oligogenic inheritance in susceptibility to infectious diseases, haploinsufficiency of STAT1 and other MSMD causing genes, relevance of intronic variants, compound heterozygous inheritance of MSMD, etc. Also, CNV and epigenetic modifications that were not tested are assumed for causality in this study and in general. Revealing novel genes and molecular mechanisms underlying mycobacterial infections in children might facilitate the development of therapeutic strategies to prevent and treat mycobacterial disease.
Data availability statement
Variants with functional assessments in this study are deposited in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/; submission SUB12127021, accession numbers SCV002581932 - SCV002581934). All other relevant data supporting conclusions are included in the article/Supplementary Material or available by request.
Ethics statement
The studies involving human participants were reviewed and approved by The Research Ethic Committee of the Institute of Phthisiopneumology (Republic of Moldova). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
TI acquired funding and supervised the study. AV, TI and BA conceived and designed the research. VC and SG collected patient samples and performed clinical assessments. AV, GB and MD performed the experiments. ID and WH provided bioinformatical expertise and support. AV drafted the manuscript. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Funding
This study was supported by the Hannover Unified Biobank. Alexander Varzari was sponsored by the Alexander von Humboldt Foundation (Georg Forster Research Fellowship) and by the Moldovan State Program Particularităţile recidivei tuberculozei pulmonare (theme No. 20.80009.8007.23). The research of Igor Deyneko was carried out within the assignment of Ministry of Science and Higher Education (theme No. 122042700043-9) and development of bioinformatics methods by RSF grant (project No. 22-14-00057).
Acknowledgments
We sincerely thank all the patients, their family members and control subjects for participating in this study. We are deeply grateful to Elena Privalova, Valentina Chirosca, Elena Tudor, Inna Suruceanu, Nelly Ciobanu, Maximilian Schieck, Gunnar Schmidt, Oliver Dittrich-Breiholz and Lise Lolle Holm for their contribution to this project. We would also like to thank Jean-Laurent Casanova and Jacinta Bustamante for their helpful advices and suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.969895/full#supplementary-material
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 (4), 248–249. doi:10.1038/nmeth0410-248
Akle, S., Chun, S., Jordan, D. M., and Cassa, C. A. (2015). Mitigating false-positive associations in rare disease gene discovery. Hum. Mutat. 36 (10), 998–1003. doi:10.1002/humu.22847
Alcaïs, A., Fieschi, C., Abel, L., and Casanova, J. L. (2005). Tuberculosis in children and adults: Two distinct genetic diseases. J. Exp. Med. 202 (12), 1617–1621. doi:10.1084/jem.20052302
Berry, M. P., Graham, C. M., McNab, F. W., Xu, Z., Bloch, S. A., Oni, T., et al. (2010). An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466 (7309), 973–977. doi:10.1038/nature09247
Boisson-Dupuis, S., Bustamante, J., El-Baghdadi, J., Camcioglu, Y., Parvaneh, N., El Azbaoui, S., et al. (2015). Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol. Rev. 264 (1), 103–120. doi:10.1111/imr.12272
Bustamante, J. (2020). Mendelian susceptibility to mycobacterial disease: Recent discoveries. Hum. Genet. 139 (6-7), 993–1000. doi:10.1007/s00439-020-02120-y
Capriotti, E., Fariselli, P., and Casadio, R. (2005). I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310. Web Server issue). doi:10.1093/nar/gki375
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31 (16), 2745–2747. doi:10.1093/bioinformatics/btv195
Chong, J. X., Buckingham, K. J., Jhangiani, S. N., Boehm, C., Sobreira, N., and Smith, J. D.Centers for Mendelian Genomics (2015). The genetic basis of mendelian phenotypes: Discoveries, challenges, and opportunities. Am. J. Hum. Genet. 97, 199–215. doi:10.1016/j.ajhg.2015.06.009
de Valles-Ibáñez, G., Esteve-Solé, A., Piquer, M., González-Navarro, E. A., Hernandez-Rodriguez, J., Laayouni, H., et al. (2018). Evaluating the genetics of common variable immunodeficiency: Monogenetic model and beyond. Front. Immunol. 9, 636. doi:10.3389/fimmu.2018.00636
Dembic, M., Andersen, H. S., Bastin, J., Doktor, T. K., Corydon, T. J., Sass, J. O., et al. (2019). Next generation sequencing of RNA reveals novel targets of resveratrol with possible implications for Canavan disease. Mol. Genet. Metab. 126 (1), 64–76. doi:10.1016/j.ymgme.2018.10.004
Deng, W., and Xie, J. (2012). NOD2 signaling and role in pathogenic mycobacterium recognition, infection and immunity. Cell. Physiol. biochem. 30 (4), 953–963. doi:10.1159/000341472
Desmet, F. O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., and Béroud, C. (2009). Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37 (9), e67. doi:10.1093/nar/gkp215
Dubé, J. Y., Fava, V. M., Schurr, E., and Behr, M. A. (2021). Underwhelming or misunderstood? Genetic variability of pattern recognition receptors in immune responses and resistance to Mycobacterium tuberculosis. Front. Immunol. 12, 714808. doi:10.3389/fimmu.2021.714808
Fekrvand, S., Yazdani, R., Olbrich, P., Gennery, A., Rosenzweig, S. D., Condino-Neto, A., et al. (2020). Primary immunodeficiency diseases and Bacillus calmette-guérin (BCG)-Vaccine-Derived complications: A systematic review. J. Allergy Clin. Immunol. Pract. 8 (4), 1371–1386. doi:10.1016/j.jaip.2020.01.038
Gao, L., Bin, L., Rafaels, N. M., Huang, L., Potee, J., Ruczinski, I., et al. (2015). Targeted deep sequencing identifies rare loss-of-function variants in IFNGR1 for risk of atopic dermatitis complicated by eczema herpeticum. J. Allergy Clin. Immunol. 136 (6), 1591–1600. doi:10.1016/j.jaci.2015.06.047
Holm, L. L., Doktor, T. K., Hansen, M. B., Petersen, U. S. S., and Andresen, B. S. (2022). Vulnerable exons, like ACADM exon 5, are highly dependent on maintaining a correct balance between splicing enhancers and silencers. Hum. Mutat. 43 (2), 253–265. doi:10.1002/humu.24321
Itan, Y., Mazel, M., Mazel, B., Abhyankar, A., Nitschke, P., Quintana-Murci, L., et al. (2014). Hgcs: An online tool for prioritizing disease-causing gene variants by biological distance. BMC Genomics 15, 256. doi:10.1186/1471-2164-15-256
Kerner, G., Rosain, J., Guérin, A., Al-Khabaz, A., Oleaga-Quintas, C., Rapaport, F., et al. (2020). Inherited human IFN-γ deficiency underlies mycobacterial disease. J. Clin. Invest. 130 (6), 3158–3171. doi:10.1172/JCI135460
Kim, B. H., Shenoy, A. R., Kumar, P., Das, R., Tiwari, S., and MacMicking, J. D. (2011). A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science 332 (6030), 717–721. doi:10.1126/science.1201711
King, J. R., and Hammarström, L. (2018). Newborn screening for primary immunodeficiency diseases: History, current and future practice. J. Clin. Immunol. 38 (1), 56–66. doi:10.1007/s10875-017-0455-x
Kong, X. F., Ciancanelli, M., Al-Hajjar, S., Alsina, L., Zumwalt, T., Bustamante, J., et al. (2010). A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood 116 (26), 5895–5906. doi:10.1182/blood-2010-04-280586
Kourime, M., Akpalu, E. N., Ouair, H., Jeddane, L., Benhsaien, I., Ailal, F., et al. (2016). BCGitis/BCGosis in children: Diagnosis, classification and exploration. Arch. Pediatr. 23 (7), 754–759. doi:10.1016/j.arcped.2016.04.003
Kristensen, I. A., Veirum, J. E., Møller, B. K., and Christiansen, M. (2011). Novel STAT1 alleles in a patient with impaired resistance to mycobacteria. J. Clin. Immunol. 31 (2), 265–271. doi:10.1007/s10875-010-9480-8
Lawn, S. D., and Zumla, A. I. (2011).Tuberculosis Lancet 378 (9785), 57–72. doi:10.1016/S0140-6736(10)62173-3
Le Voyer, T., Neehus, A. L., Yang, R., Ogishi, M., Rosain, J., Alroqi, F., et al. (2021). Inherited deficiency of stress granule ZNFX1 in patients with monocytosis and mycobacterial disease. Proc. Natl. Acad. Sci. U. S. A. 118 (15), e2102804118. doi:10.1073/pnas.2102804118
Ledderose, C., Heyn, J., Limbeck, E., and Kreth, S. (2011). Selection of reliable reference genes for quantitative real-time PCR in human T cells and neutrophils. BMC Res. Notes 4, 427. doi:10.1186/1756-0500-4-427
Marquis, J. F., Kapoustina, O., Langlais, D., Ruddy, R., Dufour, C. R., Kim, B. H., et al. (2011). Interferon regulatory factor 8 regulates pathways for antigen presentation in myeloid cells and during tuberculosis. PLoS Genet. 7 (6), e1002097. doi:10.1371/journal.pgen.1002097
Martin-Fernandez, M., Buta, S., Le Voyer, T., Li, Z., Dynesen, L. T., Vuillier, F., et al. (2022). A partial form of inherited human USP18 deficiency underlies infection and inflammation. J. Exp. Med. 219 (4), e20211273. doi:10.1084/jem.20211273
Meng, X., Kanwar, N., Du, Q., Goping, I. S., Bleackley, R. C., and Wilkins, J. A. (2009). PPP1R9B (neurabin 2): Involvement and dynamics in the NK immunological synapse. Eur. J. Immunol. 39 (2), 552–560. doi:10.1002/eji.200838474
Mertens, C., Zhong, M., Krishnaraj, R., Zou, W., Chen, X., and Darnell, J. E. (2006). Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. 20 (24), 3372–3381. doi:10.1101/gad.1485406
Palhais, B., Dembic, M., Sabaratnam, R., Nielsen, K. S., Doktor, T. K., Bruun, G. H., et al. (2016). The prevalent deep intronic c. 639+919 G>A GLA mutation causes pseudoexon activation and Fabry disease by abolishing the binding of hnRNPA1 and hnRNP A2/B1 to a splicing silencer. Mol. Genet. Metab. 119 (3), 258–269. doi:10.1016/j.ymgme.2016.08.007
Palhais, B., Præstegaard, V. S., Sabaratnam, R., Doktor, T. K., Lutz, S., Burda, P., et al. (2015). Splice-shifting oligonucleotide (SSO) mediated blocking of an exonic splicing enhancer (ESE) created by the prevalent c.903+469T>C MTRR mutation corrects splicing and restores enzyme activity in patient cells. Nucleic Acids Res. 43 (9), 4627–4639. doi:10.1093/nar/gkv275
Perumal, P., Abdullatif, M. B., Garlant, H. N., Honeyborne, I., Lipman, M., McHugh, T. D., et al. (2021). Validation of differentially expressed immune biomarkers in latent and active tuberculosis by real-time PCR. Front. Immunol. 11, 612564. doi:10.3389/fimmu.2020.612564
Pöyhönen, L., Nuolivirta, K., Vuononvirta, J., Kröger, L., Huhtala, H., Mertsola, J., et al. (2015). Toll-like receptor 2 subfamily gene polymorphisms are associated with Bacillus Calmette-Guérin osteitis following newborn vaccination. Acta Paediatr. 104 (5), 485–490. doi:10.1111/apa.12927
Praefcke, G. J. K. (2018). Regulation of innate immune functions by guanylate-binding proteins. Int. J. Med. Microbiol. 308 (1), 237–245. doi:10.1016/j.ijmm.2017.10.013
Ramsauer, K., Farlik, M., Zupkovitz, G., Seiser, C., Kröger, A., Hauser, H., et al. (2007). Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2 gene. Proc. Natl. Acad. Sci. U. S. A. 104 (8), 2849–2854. doi:10.1073/pnas.0610944104
Randhawa, A. K., Shey, M. S., Keyser, A., Peixoto, B., Wells, R. D., de Kock, M., et al. (2011). South African Tuberculosis Vaccine Initiative TeamAssociation of human TLR1 and TLR6 deficiency with altered immune responses to BCG vaccination in South African infants. PLoS Pathog. 7 (8), e1002174. doi:10.1371/journal.ppat.1002174
Renaux, A., Papadimitriou, S., Versbraegen, N., Nachtegael, C., Boutry, S., Nowé, A., et al. (2019). Orval: A novel platform for the prediction and exploration of disease-causing oligogenic variant combinations. Nucleic Acids Res. 47 (W1), W93-W98–W98. doi:10.1093/nar/gkz437
Rigaud, S., Lopez-Granados, E., Sibéril, S., Gloire, G., Lambert, N., Lenoir, C., et al. (2011). Human X-linked variable immunodeficiency caused by a hypomorphic mutation in XIAP in association with a rare polymorphism in CD40LG. Blood 118 (2), 252–261. doi:10.1182/blood-2011-01-328849
Roy, A., Eisenhut, M., Harris, R. J., Rodrigues, L. C., Sridhar, S., Habermann, S., et al. (2014). Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: Systematic review and meta-analysis. BMJ 349, g4643. doi:10.1136/bmj.g4643
Sabatino, L., Cabiati, M, Caselli, C., and Del Ry, S. (2013). Adenosine receptor expression and gene reference evaluation in human leukocytes. Clin. Lab. 59 (5-6), 571–577. doi:10.7754/clin.lab.2012.120219
Sakata, S., Tsumura, M., Matsubayashi, T., Karakawa, S., Kimura, S., Tamaura, M., et al. (2020). Autosomal recessive complete STAT1 deficiency caused by compound heterozygous intronic mutations. Int. Immunol. 32 (10), 663–671. doi:10.1093/intimm/dxaa043
Schwarz, J. M., Rödelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7 (8), 575–576. doi:10.1038/nmeth0810-575
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. Web Server issue). doi:10.1093/nar/gks539
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 (D1), D607-D613–D613. doi:10.1093/nar/gky1131
Tangye, S. G., Al-Herz, W., Bousfiha, A., Chatila, T., Cunningham-Rundles, C., Etzioni, A., et al. (2020). Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J. Clin. Immunol. 40 (1), 24–64. doi:10.1007/s10875-019-00737-x
Tavtigian, S. V., Byrnes, G. B., Goldgar, D. E., and Thomas, A. (2008). Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum. Mutat. 29 (11), 1342–1354. doi:10.1002/humu.20896
Tretina, K., Park, E. S., Maminska, A., and MacMicking, J. D. (2019). Interferon-induced guanylate-binding proteins: Guardians of host defense in health and disease. J. Exp. Med. 216 (3), 482–500. doi:10.1084/jem.20182031
Vairo, D., Tassone, L., Tabellini, G., Tamassia, N., Gasperini, S., Bazzoni, F., et al. (2011). Severe impairment of IFN-gamma and IFN-alpha responses in cells of a patient with a novel STAT1 splicing mutation. Blood 118 (7), 1806–1817. doi:10.1182/blood-2011-01-330571
Varzari, A., Deyneko, I. V., Vladei, I., Grallert, H., Schieck, M., Tudor, E., et al. (2019). Genetic variation in TLR pathway and the risk of pulmonary tuberculosis in a Moldavian population. Infect. Genet. Evol. 68, 84–90. doi:10.1016/j.meegid.2018.12.005
Vockley, J., Rinaldo, P., Bennett, M. J., Matern, D., and Vladutiu, G. D. (2000). Synergistic heterozygosity: Disease resulting from multiple partial defects in one or more metabolic pathways. Mol. Genet. Metab. 71 (1-2), 10–18. doi:10.1006/mgme.2000.3066
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2009). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296-W303–W303. doi:10.1093/nar/gky427
Xiao, J., Li, X., Liu, J., Fan, X., Lei, H., and Li, C. (2017). Identification of reference genes in blood before and after entering the plateau for SYBR green RT-qPCR studies. PeerJ 5, e3726. doi:10.7717/peerj.3726
Yan, M., Wang, H., Sun, J., Liao, W., Li, P., Zhu, Y., et al. (2016). Cutting edge: Expression of IRF8 in gastric epithelial cells confers protective innate immunity against Helicobacter pylori infection. J. Immunol. 196 (5), 1999–2003. doi:10.4049/jimmunol.15007663rd
Keywords: disseminated tuberculosis, BCG-itis, primary immunodeficiency (PID) disorders, whole-exome sequencing (WES), trio analysis, pre-mRNA splicing, candidate genes approach, oligogenic inheritance
Citation: Varzari A, Deyneko IV, Bruun GH, Dembic M, Hofmann W, Cebotari VM, Ginda SS, Andresen BS and Illig T (2022) Candidate genes and sequence variants for susceptibility to mycobacterial infection identified by whole-exome sequencing. Front. Genet. 13:969895. doi: 10.3389/fgene.2022.969895
Received: 15 June 2022; Accepted: 04 October 2022;
Published: 20 October 2022.
Edited by:
Maxim B. Freidin, Queen Mary University of London, United KingdomReviewed by:
Irina Saltykova, Baylor College of Medicine, United StatesJared C. Roach, Institute for Systems Biology (ISB), United States
Copyright © 2022 Varzari, Deyneko, Bruun, Dembic, Hofmann, Cebotari, Ginda, Andresen and Illig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Varzari, YWxleGFuZGVyLnZhcnphcmlAbWFpbC5ydQ==