 Clara Estela Díaz-Velásquez1†Rina Gitler2†Adriana Antoniano2
Clara Estela Díaz-Velásquez1†Rina Gitler2†Adriana Antoniano2 Ronny Kershenovich Sefchovich3
Ronny Kershenovich Sefchovich3 Aldo Hugo De La Cruz-Montoya4Héctor Martínez-Gregorio1
Aldo Hugo De La Cruz-Montoya4Héctor Martínez-Gregorio1 Ernesto Arturo Rojas-Jiménez1Ricardo Cortez Cardoso Penha5
Ernesto Arturo Rojas-Jiménez1Ricardo Cortez Cardoso Penha5 Luis Ignacio Terrazas1,4
Luis Ignacio Terrazas1,4 Talia Wegman-Ostrosky6Ephrat Levi-Lahad7
Talia Wegman-Ostrosky6Ephrat Levi-Lahad7 Jovanny Zabaleta8,9Sandra Perdomo5*
Jovanny Zabaleta8,9Sandra Perdomo5* Felipe Vaca-Paniagua1,4,6*
Felipe Vaca-Paniagua1,4,6*- 1Laboratorio Nacional en Salud, Diagnóstico Molecular y Efecto Ambiental en Enfermedades Crónico-Degenerativas, Facultad de Estudios Superiores Iztacala, Tlalnepantla, Estado DeMéxico, Mexico
- 2Fundación Alma, Mexico City, Mexico
- 3Instituto Nacional de Medicina Genómica, Mexico City, Mexico
- 4Unidad de Biomedicina, Facultad de Estudios Superiores Iztacala, UNAM, Tlalnepantla, Estado DeMéxico, Mexico
- 5Genomic Epidemiology Branch, International Agency for Research on Cancer (IARC/WHO), Lyon, France
- 6Subdirection of Basic Research, Instituto Nacional de Cancerología, Mexico City, Mexico
- 7Department of Medical Genetics, Shaare Zedek Medical Center, Jerusalem, Israel
- 8Departament of Interdisciplinary Oncology, School of Medicine, LSU Health New Orleans, New Orleans, LA, United States
- 9Stanley S. Scott Cancer Center, LSU Health New Orleans, New Orleans, LA, United States
Background: Individuals of Ashkenazi Jewish ancestry have been identified as having higher prevalence of specific pathogenic variants associated with susceptibility to specific rare and chronic diseases. In Mexico, the prevalence and composition of rare cancer predisposing germline variants in Ashkenazi Jewish individuals has not been evaluated.
Aim and methods: We aimed to evaluate the prevalence of pathogenic variants by massive parallel sequencing in a panel of 143 cancer-predisposing genes in 341 women from the Ashkenazi Jewish community of Mexico, who were contacted and invited to participate in the study through the ALMA Foundation for Cancer Reconstruction. Pre- and posttest genetic counseling was given and a questionnaire on personal, gyneco-obstetric, demographic and lifestyle variables was conducted. From peripheral blood DNA, the complete coding region, and splicing sites of a panel of 143 cancer susceptibility genes, including 21 clinically relevant genes, were sequenced. The Mexican founder mutation BRCA1 ex9-12del [NC_000017.10(NM_007294):c. (825+1–826-1)_(4,589+1–4,590-1)del] was also evaluated.
Results: Among study participants (mean age ±standard deviation: 47 ± 14) 15% reported a personal history of cancer (50/341). Fourteen percent of participants (48/341) were carriers of pathogenic and likely pathogenic variants distributed among seven high-risk genes (APC, CHEK2, MSH2, BMPR1A, MEN1, MLH1, and MSH6), whereas 18.2% (62/341) had variants of uncertain clinical significance in genes associated with breast and ovarian cancer susceptibility (list of genes with VUS). Pathogenic and likely pathogenic variants in 16 susceptibility genes with ambiguous or non-well-established risk association for cancer were detected in 17.6% (60/341) of participants. Sixty four percent of participants reported current alcohol consumption compared with the 39 percent prevalence of alcohol consumption in Mexican women. None of the participants carried the recurrent Ashkenazi and Mexican founder mutations in BRCA1 or BRCA2, but 2% (7/341) had pathogenic Ashkenazi Jewish founder variants in BLM.
Conclusion: Our findings show a diverse pathogenic variant composition among the recruited individuals of Ashkenazi Jewish ancestry in Mexico consistent with being a high-risk population for genetic diseases, which warrants further investigation to adequately assess the burden of hereditary breast cancer in this group and implement appropriate preventative programs.
Introduction
The Ashkenazi Jewish (AJ) community has been identified as a group with elevated risk for numerous genetic diseases including hematological, biochemical diseases, and various types of cancers, most likely because of a genetic bottleneck that preceded a period of rapid population growth (Behar et al., 2004). This historical trajectory fostered a high prevalence of genetic variants related with numerous dominant and recessive conditions (Gusev et al., 2012; Palamara et al., 2012). The Jewish community represents the third religious group in the Mexican population (INEGI, 2020) and constitutes the third largest Jewish community in Latin America (Avni et al., 2011).
In Mexico, breast cancer is the leading cause of cancer-related deaths among women aged 20 to 59, and 5%–10% of cases can be explained by hereditary genetic factors. Pathogenic and Likely Pathogenic (P/LP) genetic variants in the BRCA1/2 (Breast Cancer 1/2) genes are the main cause of hereditary breast cancer. Among individuals of AJ ancestry, most breast cancers are due to three BRCA1/2 founder mutations (BRCA1: 185delAG [c.68_69del], 5382insC [c.5266dup]; BRCA2: 6174delT [c.5946del]); while in the Mexican population the founder mutation BRCA1 ex9-12del has a high frequency. The prevalence of pathogenic variants in non-BRCA genes has not been widely investigated in the Latin American Region generally, or Mexico specifically (Cock-Rada et al., 2018; Quezada Urban et al., 2018; Adaniel et al., 2019; Oliver et al., 2019; Sandoval et al., 2021; Solano et al., 2021).
While various studies have identified germline pathogenic variants associated with hereditary cancer syndromes in the Mexican population, there is currently no information on genetic alterations specifically focused on Mexicans who self-identify as AJ (Torres-Mejía et al., 2015; Villarreal-Garza et al., 2015; Moreno-Ortiz et al., 2016; Quezada Urban et al., 2018; Gallardo-Rincón et al., 2020). The lack of this information remains an important limitation to further evaluate the implications of identifying high risk populations in genomic testing and public health policies in Mexico. To address this gap, we conducted a genetic analysis of 143 genes associated with a variety of cancers to ascertain the prevalence and composition of pathogenic genetic variants among individuals of AJ ancestry who were born and live in Mexico.
Materials and methods
Study population and data collection
Women were contacted and invited to participate in the study in community centers through the ALMA Foundation for Cancer Reconstruction (https://www.alma.org.mx/), a recognized charity organization within the AJ community in Mexico City. Inclusion criteria for recruitment included: all women self-identified as AJ and who have an AJ mother or father, as well as AJ maternal or paternal grandparents. Adopted women were excluded.
The protocol was approved by the Ethics Committee of the Faculty of Health Sciences of the Anahuac University (201923) and was conducted in accordance with the Declaration of Helsinki. A total of 341 women over the age of 18, of Mexican nationality accepted to participate in the study. All participants provided written informed consent for participation after a detailed explanation of the study, and their samples were anonymized and sent to the Laboratorio Nacional en Salud: Diagnóstico Molecular y Efecto Ambiental en Enfermedades Crónico-Degenerativas, Facultad de Estudios Superiores Iztacala, UNAM. All participants had pre-test and post-test genetic counseling provided by two clinical geneticists.
Sample preparation and DNA extraction
Four mL of whole blood was drawn from each participant and stored at −80°C locally. The time interval between collection and freezing of samples was never greater than 36 h. Peripheral blood DNA was extracted according to the manufacturer’s instructions using the DNeasy Blood & Tissue Kit (Qiagen). The DNA concentration was determined using the Invitrogen Qubit dsDNA HS Assay Kit, and the material’s integrity and purity were determined using agarose gel electrophoresis and spectrophotometry.
Epidemiological information on additional risk factors
All participants were invited to complete a lifestyle questionnaire including information on socioeconomic status, health, and reproductive history (age at menarche, pregnancy, number of births, age at each birth, breastfeeding), use of hormones (e.g., oral contraceptives), smoking and alcohol habits, maximum attained weight, and body mass index (BMI), and personal history of cancer. Information on family history of cancer was limited without precise details on degree of siblings, family arm or cancer types reported, and therefore was excluded from the analyses. Information was collected using REDCap electronic data capture tools (Harris et al., 2009; Harris et al., 2019).
Statistical analysis
Age and BMI were included as continuous variables, whereas the rest of the factors were considered as categorical variables. Differences in clinicopathological characteristics between individuals with and without pathogenic variants were analyzed using X2 test and a p-value <0.05 was considered statistically significant. All the statistical analyses were performed using R (www.r-project.org).
Library preparation and sequencing
Library preparation was performed as described previously (Quezada Urban et al., 2018). Briefly, The GeneRead Cancer Predisposition V2 Kit (Qiagen) was used to prepare the library, which targets 143 genes whose loss of function is a well-known mechanism associated with more than 80 inherited oncologic diseases based on data from the College of American Pathologists (CAP) guidelines, the National Comprehensive Cancer Network (NCCN) guidelines, and The Cancer Genome Atlas (TCGA). The amplification was carried out in four-pool PCR reactions and all libraries were barcoded and diluted equimolarly. Sequencing was performed in a HiSeq 4000 (Illumina) with pair-end (2x150) chemistry and to a theoretical coverage of 2000X.
Bioinformatic analyses and variant calling
Bioinformatic analyses were performed as previously described (Quezada Urban et al., 2018). Briefly, BWA (Li and Durbin, 2009) and GATK were used to perform alignment and variant calling (Van der Auwera et al., 2013). With BWA-MEM, fastq files were aligned to the human genome reference hg19; indels were realigned and bases were recalculated. Adaptors were soft-clipped and reads with a length of less than 20 bp were discarded. HaplotypeCaller was used to determine genetic variants (Van der Auwera et al., 2013). ANNOVAR and InterVar were used to annotate variants (Wang et al., 2010; Li and Wang, 2017). The Human Genome Variation Society (HGVS) nomenclature was used to describe the genetic variants (den Dunnen et al., 2016). The classification of variants followed the American College of Medical Genetics and Genomics’ (ACMG) five-tier criteria (Richards et al., 2015) and was manually curated. We excluded synonymous variants, those with a depth of <10X or a mutant allele fraction of <15%, those found in homopolymeric tracts (>8 bp) or spurious variants with conflicting patterns, including those in only one strand, within erroneous base tracts or at the end of only one of the amplicons (with no redundancy in additional amplicons). All splicing and null variants (stop-gain/loss, frameshift indels), as well as missense variants, were considered pathogenic (P) or likely pathogenic (LP) as defined by the ACMG guidelines and if reported as pathogenic in ClinVar (ACMG supporting evidence of pathogenicity PPP5) (Landrum et al., 2020). In addition, we used VarSome and InterVar to compare the ClinVar information and determine more precisely the variant classification. Null variants located at downstream than 50 bp of the final splice junction at the 3′extreme end of the gene were excluded. Minor allelic frequency 0.001 in non-Ashkenazi populations was used as a threshold to eliminate common human variation with the gnomAD, ExAC and the 1,000 Genomes (1000 G) project databases. Variants with higher Ashkenazi Jew specific population frequency were evaluated manually to account for bottleneck population effects in this population. All filtered variants were manually curated using the IGV software (Thorvaldsdóttir et al., 2013). The Leiden Open Variation Database (Fokkema et al., 2021) was also used to investigate variants in MLH1, MSH2, MSH6, and PMS2. Ninety six percent of the pathogenic and likely pathogenic variants in high-risk genes were validated by Sanger sequencing.
Detection of the Mexican founder mutation in BRCA1
PCR based sequencing limits the identification of large deletions including the highly prevalent Mexican founder mutation BRCA1 ex9-12del [NC_000017.10(NM_007294):c. (825+1-826-1)_(4,589+1–4,590-1)del]. Therefore, this pathogenic variant was evaluated by PCR amplification of the mutant and wildtype allele, using specific primers based on the method developed by Weitzel et al. (2007), with a positive control previously validated by multiplex ligation-dependent probe amplification (MLPA). PCR products were resolved in 1.5% agarose gels to identify the amplification of the truncated allele and sequenced.
Genetic ancestry component
To evaluate the ancestral genetic composition of the study participants DNA samples were genotyped at the University of Minnesota Genomics Center with the MassARRAY System (Agena Bioscience) with a panel of 104 ancestry informative markers (AIMs) that can be used to estimate Indigenous American, African, and European ancestry as previously shown (Fejerman et al., 2008; Serrano-Gomez et al., 2016). Single nucleotide polymorphisms with call rate <90% (1 polymorphism) or that deviated from Hardy–Weinberg equilibrium were removed from the analysis. We used PLINK with subsequent principal component analysis (PCA) to detect ancestral structure using 1000vG as reference population as an initial quality control step (Supplementary Table S1) (Tang et al., 2005). A total of 341 cases were genotyped, but only 327 cases remained after excluding samples with genotype call rates <90%. The final set available for analysis included 327 participants and 103 AIMs. We used a maximum likelihood (ML) approach for ancestry estimation and included available reference data from the 1000 G project. Genotype data were converted into PLINK pgen format using PLINKv.2.0 (Chang et al., 2015).
Of the 103 AIMs, 58 were found in the 1,000 Genomes dataset (Auton et al., 2015). To detect and remove variants in linkage disequilibrium, an independent pairwise test (LD window size of 50 kb and r2 threshold of 0.2) was performed using PLINK, remaining 57 independent variants that were used in the genetic ancestry analyses. The genotype data with 327 cases was then merged with the 1,000 Genomes genotype data for the reference populations (Europeans: British, Finnish, Iberian, Northern and Western European, and Toscani; Latin-Americans: Mexican, Peruvian, Colombian and, Puerto Rican; African:Yoruba).
Results
Demographic characteristics and risk factors of Mexican Ashkenazi Jewish
The demographic profile of participants was characterized by a normal BMI, a high level of education (university and postgraduate: 91%), high maternity rate and age of first pregnancy before the age of 30. More than 50% of participants were never smokers but current drinkers (Table 1). Fifteen percent (50/341) of individuals had a personal history of cancer, including 10 women previously diagnosed with breast cancer, 3 with ovarian cancer, 2 with colorectal cancer and 1 with pancreatic cancer.

TABLE 1. Demographic characteristics of women of Ashkenazi Jewish ancestry who reside in Mexico City (N = 341).
Genetic ancestry composition
Ancestry categories based on self-identity are more comprehensive because they account for individual knowledge of ancestry similarities including cultural and social components. In addition, we identified the genetic ancestry fractions of the participants using the allelic composition of 103 ancestry informative SNPs. We used principal component analysis which showed that 92.3% (302/327) of the individuals genotyped had more than 70% allelic composition matching those in the reference populations grouped in Europe (Figure 1; Supplementary Table S1). The allelic frequencies of those 25 individuals below the 70% cutoff ranged between 56%–69% and grouped with the reference Latin American populations from the 1000 G (CLM, MLX, PUR) (Figure 1).

FIGURE 1. Principal component analysis showing ancestral composition of the AJ women recruited in comparison with the reference populations from 1000 G. PJA, Population of Mexican Ashkenazi Jews represented by yellow dots. POP, Populations, GBR, British, FIN, Finnish, IBS, Iberian, CEU, Northern and Western European, TSI, Toscani, MXL, Mexican, PEL, Peruvian, CLM, Colombian, PUR, Puerto Rican, YRI, Yoruba.
High-risk pathogenic variants detected
Pathogenic and likely pathogenic variants detected in high and moderate risk genes
Overall, we identified 58 pathogenic (P) and 51 likely pathogenic (LP) variants in 26.7% of the individuals (91/341), distributed across 23 genes (Figure 2). Nevertheless, only 14% had P/LP variants in clinically relevant genes (48/341). Seven high-risk genes considered reportable by the ACMG SF v3.0 (Miller et al., 2021) and the NCCN Version 2.2021 guidelines for Breast, Ovarian and Colorectal cancers (Benson et al., 2021; Daly et al., 2021) were affected and included APC (8.5%; 29/341), CHEK2 (2.9%; 10/341), MSH2 (1.2%, 4/341), BMPR1A (0.9%; 3/341), MEN1 (0.3%; 1/341), MLH1 (0.3%; 1/341), and MSH6 (0.3%). We detected 10 pathogenic alleles in these seven genes (Table 2).
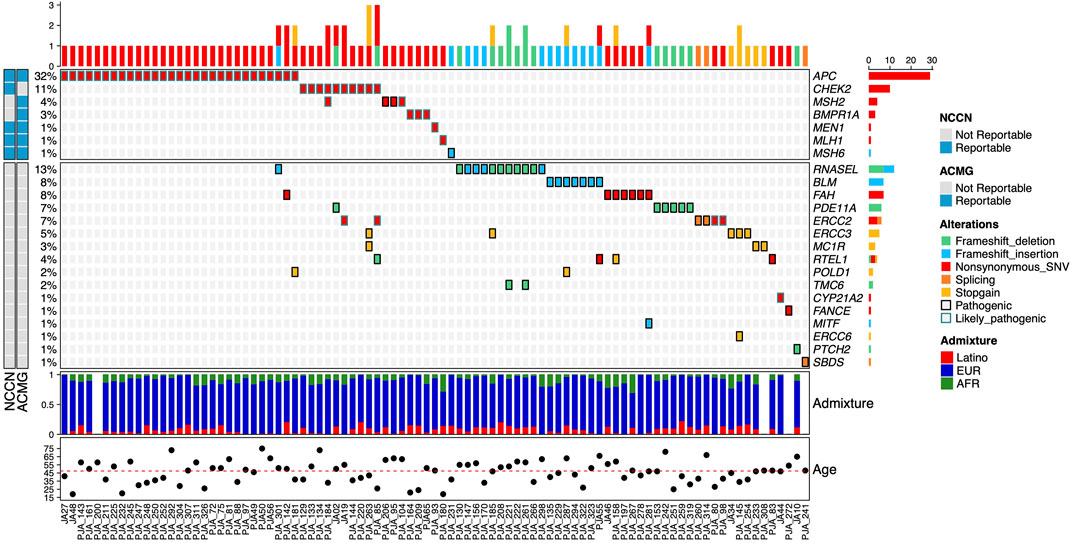
FIGURE 2. Allelic distribution of pathogenic variants in high-risk genes for HBOC and genes with unknown risk level. The relative frequencies, type of mutation, and NCCN and ACMG reportable genes are shown in the upper panel. The center panel illustrates pathogenic variants detected in genes with unknown or insufficiently documented risk of HBOC. The lower panel represents the three-component admixture distribution of the participants. The mutation type (alterations) and admixture component are color-coded. The relative frequency and total count for gene alterations are shown in the left and right.
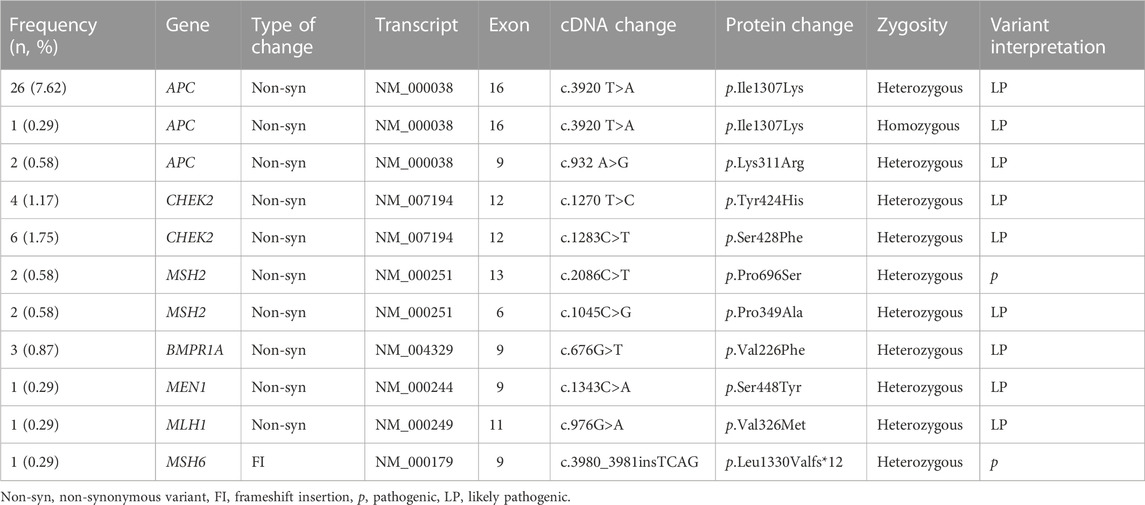
TABLE 2. Pathogenic and likely pathogenic alterations in high-risk genes detected in 341 participants.
Eight participants had more than one P or LP variant: seven participants had one LP variant in a high-risk gene (APC and CHEK2) and up to two additional p or LP variants in genes of unknown risk (RNASEL, FAH, PDE11A, ERCC2, ERCC3, MC1R, RTEL1, and POLD1). One participant had two LP variants in CHEK2 and MSH2 (Figure 2).
The P variant APC p.Lys311Arg initially classified as a VUS, was reclassified as LP based on the participants’ family history of adenomatous polyposis. This variant was present in two consanguineous members of the same family. Presence of P/LP variants was significantly associated with age of participants at recruitment (p = 0.043). We did not find significant associations between lifestyle factors variables (BMI, alcohol consumption, reproductive factors) or personal history of cancer and the presence of P or LP variants (Supplementary Table S2).
Pathogenic variants associated with hereditary breast and ovarian cancer (HBOC) syndrome
In comparison to other populations, the AJ community has a higher prevalence of founder mutations associated with HBOC. None of the participants analyzed had the recurrent Ashkenazi founder mutations in BRCA1 or BRCA2 (Levy-Lahad et al., 1997). We also tested participants for the high-frequency BRCA1 ex9-12del. Mexican native founder variant without any positive result. Other HBOC P and LP variants in moderate penetrance genes were found in 13 participants and were CHEK2 p.Ser428Phe and p.Tyr424His, detected in six and four participants, respectively and the variants p.Pro696Ser and p.Pro349Ala in MSH2, found in two participants. None of the participants harboring these variants had personal history of cancers associated with HBOC syndrome.
Variants of unknown clinical significance (VUS) associated with hereditary breast and ovarian cancer (HBOC) syndrome
In total, 18.2% (62/341) of the participants harbored monoallelic variants of uncertain significance (VUS) in genes of clinical relevance for HBOC (Supplementary Table S3). These variants were detected in BRIP1 (2.3%; 8/341), ATM (2%; 7/341), MSH6 (1.8%; 6/341), FANCI (1.5%; 5/341), KIT (1.2%; 4/341), RET (1.2%), MSH2 (0.9%; 3/341), BRCA2 (0.9%), BARD1 (0.9%), PALB2 (0.9%), PMS2 (0.9%), ERCC2 (0.9%), CHEK2 (0.6%; 2/341), FAH (0.6%), NF1 (0.3%; 1/341), NF2 (0.3%), BRCA1 (0.3%), MEN1 (0.3%), PDE11A (0.3%), and RAD51C (0.3%).
Pathogenic and likely pathogenic variants in genes of uncertain cancer risk
We detected 50 participants with 20 unique p and LP variants distributed in 16 susceptibility genes with uncertain or non-well-established risk association for cancer (Table 3) but only nine of these participants had a personal history of cancer. Ten participants had more than one P or LP variant, of which nine had two P variants in two different genes, and one had two variants (one P in RTEL1 and one LP in ERCC2). The affected genes were RNASEL (3.5%, 12/341), BLM (2%; 7/341), FAH (2%), PDE11A (1.8; 6/341), ERCC2 (1.8%), ERCC3 (1.5%; 5/341), MC1R (0.9%; 3/341), RTEL1 (1.2%, 4/341), POLD1 (0.6%; 2/341), TMC6 (0.6); as well as in CYP21A2, FANCE, MITF, ERCC6, PTCH2, and SBDS genes (0.3%; 1/341). The frequent pathogenic monoallelic variant BLM p.Tyr736Leufs*5 found in seven participants is a well-known common founder allele in the Ashkenazi Jewish population and is associated with Bloom syndrome.
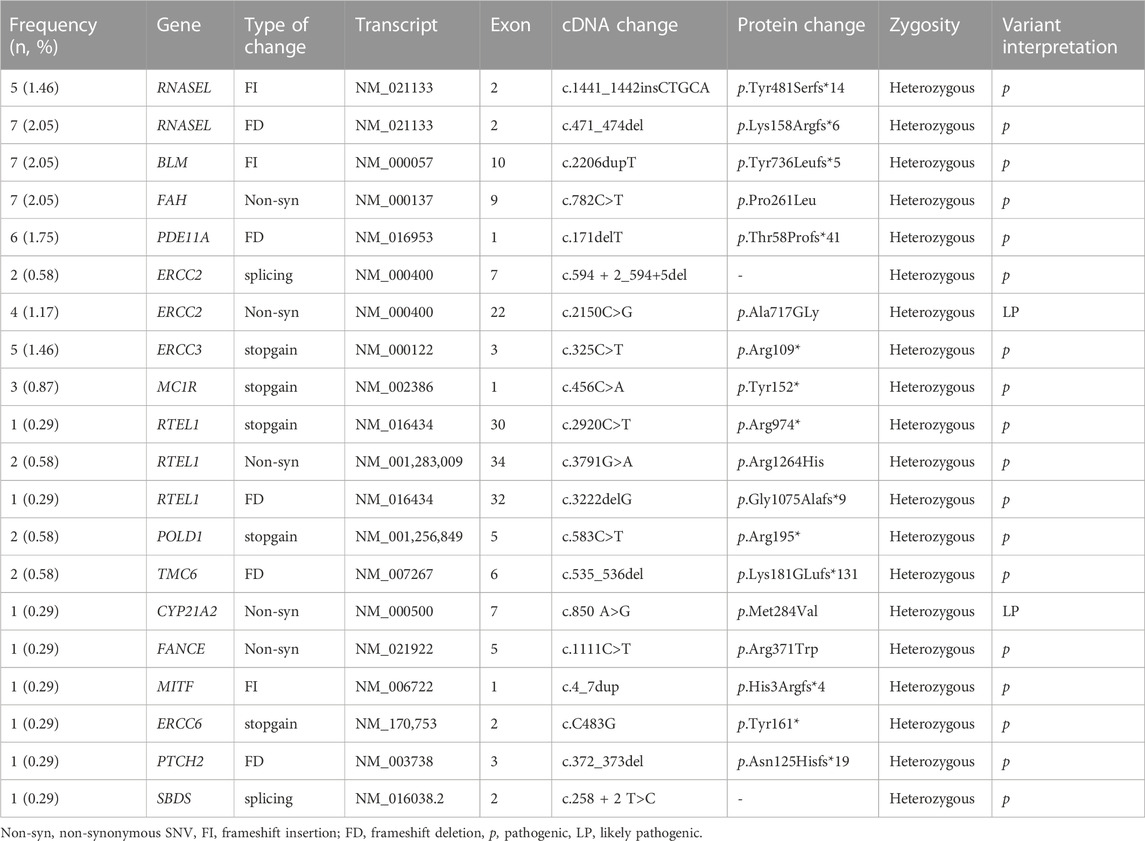
TABLE 3. Pathogenic and likely pathogenic alterations with unknown risk in 341 participants.
Discussion
Individuals of Ashkenazi Jewish ancestry have been identified as a population with a distinctive genetic background, characterized by a higher prevalence of certain pathogenic variants associated with susceptibility to both chronic and rare diseases, as evidenced by a relatively high frequency of alleles associated with breast and ovarian cancer (Ostrer, 2001; Xiao and Lauschke, 2021). We aimed to evaluate the germline cancer susceptibility mutation landscape in the AJ diaspora in Mexico. The landscape of germline pathogenic variants present in extensive panels of cancer associated genes have not been evaluated in recent generations of women of Ashkenazi Jewish origin, in particular in those populations after migration to Latin America. Moreover, most of the genetic studies on the AJ population are conducted in patients with a confirmed diagnosis of cancer and therefore, the reported frequency of founder pathogenic germline variants could be mainly influenced by the high proportion of inherited disease cases and may be underestimated in the unaffected AJ population. Population-based screening studies in AJ women without cancer diagnosis have reported BRCA1/2 mutation frequencies of 1.1%–2.9% (Metcalfe et al., 2010; Manchanda et al., 2015; 2020). Furthermore, the allelic composition of cancer susceptibility variants in genes distinct from BRCA1/2 is generally not evaluated in healthy populations at higher risk. As a result, it is possible that an important proportion of AJ germline carriers is not detected, especially in low- and middle-income countries where access to genetic counseling and germline testing is still restricted.
We showed that self-identification as AJ and the ancestry genetic composition agreed sufficiently to characterize AJ population in Mexico (Mathieson and Scally, 2020). As expected, admixture of genetic ancestries was present. However, a high proportion of participants (92%) shared ≥70% of the evaluated alleles with populations located in Europe. These combined results provide evidence that endogamy in the AJ population in Mexico is still persistent as confirmed by social studies showing that bicultural marriages were scarce among Jewish immigrants, as well as among the first generations born in Mexican territory (Goldsmit Brindis, 2010).
The proportion of AJ individuals carrying p or LP germline variants identified (26.7%) confirms the persistent elevated risk for chronic diseases, including cancer among AJ communities in Mexico as reported in a prior publication on this population (Morgenstern-Kaplan et al., 2022). Sixteen percent of the participants carried a p variant in a high-risk cancer gene. All p variants found correspond to founder mutations previously described in the AJ population, which have not been reported in the Mexican non-Jewish population (Ellis et al., 1998; Shaag et al., 2005) and were detected in individuals without personal history of cancer. Pathogenic and LP variants were significantly associated with younger age confirming the elevated prevalence of germline genetic variants in the AJ population and the need for timely genetic testing and counseling before disease onset. Interestingly, several reports have shown that almost 50% of Jewish participants with cancer that harbor a high-risk pathogenic variant have no family history (Walsh et al., 2017; Tennen et al., 2020).
In this work, we did not detect P or LP variants in BRCA1 or BRCA2 genes. However, CHEK2 LP variants were detected at a frequency of 2.9%. This frequency is not higher than those found in other previous reports for AJ breast cancer patients but slightly higher for cancer-free AJ women (Shaag et al., 2005). For cancer-free AJ women the probability of having a pathogenic mutation in CHEK2 is 1.4%–1.7% and the chance for a mutation in BRCA1 or BRCA2 is less than 2.5% (Walsh et al., 2017). This result highlights the importance of comprehensive genetic testing with a multigene panel to identify CHEK2 alterations, as well as private mutations in susceptibility genes for AJ women with no founder or p variant in BRCA1 or BRCA2. In addition, further evidence involving treatment decisions strongly reinforces consideration for CHEK2 testing. A recent study has shown poorer clinical outcomes for breast cancer patients diagnosed with invasive, early-onset breast cancer who carry a CHEK2 pathogenic variant (Greville-Heygate et al., 2020). These results also echo the recommendations regarding prudent use of consumer directed genetic tests targeting high-risk populations including exclusively highly prevalent founder pathogenic variants. For example, the FDA-Authorized, Direct-To-Consumer BRCA Test commercialized by 23andMeⓇ only identifies three BRCA1 p variants common in people of Ashkenazi Jewish ancestry. A negative result of this test must lead to complementary testing.
Among the recognized risk factors for breast cancer, the participants reported greater current alcohol consumption compared with the overall prevalence of alcohol consumption in Mexican women (64 percent versus 39 percent, respectively) (ENCODAT, 2017). In 2020, 1.3% (range 0.7%–2.1%) of all cancer cases diagnosed in women in Mexico were attributed to alcohol consumption (Rumgay et al., 2021). Considering the underlying elevated cancer risk AJ populations have, additional efforts promoting reduction of alcohol consumption should be envisioned in this community.
Interpretation of current results should consider some limitations in this study. Despite our stringent selection criteria to include a fair representation sample of the AJ community in Mexico, participants identified were born and lived in the central capital of the country and therefore do not fully represent AJ women from other regions of the country. Since in this study we investigated volunteers without medical indication, it is possible that there is a recruitment bias toward low-risk participants, as high-risk individuals may have already been analyzed by commercial laboratory testing. This may explain why common founder mutations in BRCA1/2 were not detected in this population. Despite the vast size of our gene panel, this strategy is not able to identify variants in promoters or remote regulatory regions, or those present in sites involved in mRNA processing such as 5′- and 3′-UTRs. Intronic variants that may affect splicing are also not covered. It is possible that these non-coding variants may contribute to the pathogenic allele pool of this population as has been shown in other reports (Hansmann et al., 2012; Burke et al., 2018; Evans et al., 2018; Montalban et al., 2019). Additional investigations involving whole genome sequencing and functional analysis are required to better understand the impact and composition of pathogenic genetic variation in this population.
This is the first study that estimates the prevalence of P and LP alleles in Mexican women who self-identify as AJ, showing an elevated frequency (26.7%) of participants with P or LP germline variants in cancer susceptibility genes, of which 14% were in high-risk genes. In the study sample of Ashkenazi Jewish individuals, we did not identify common pathogenic mutations in the BRCA1/2 genes but found high-penetrance mutations in other cancer associated genes such as MSH6, APC and MSH2. Pathogenic variants in HBOC moderate-risk genes such as CHEK2 were frequent in study participants without personal history of cancer. These findings need to be further explored as to better describe the cancer genetic risk of women who identify as AJ in Mexico and develop targeted cancer prevention programs focused both on genetic screening and modifiable risk factors such as reduction of alcohol consumption.
Data availability statement
The sequencing files can be accessed in the Sequence Reference Archive with the accession ID PRJNA914196. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA914196/.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Faculty of Health Sciences of the Anahuac University (201923). The patients/participants provided their written informed consent to participate in this study.
Author contributions
Conception of the study: RG and FVP; recruitment: AA, RG, and RKS; sequencing: CEDV; data collection: AA, RG, CEDV, JZ, and SP; data analysis: AA, CEDV, ADLCM, HMG, EARJ, RCCP, TWO, SP, and FVP; visualization: CEDV, HMG, EARJ, RCCP, and SP; drafting and revision: CEDV, RG, AA, RKS, ADLCM, HMG, EARJ, LIT, TWO, ELL, JZ, SP, and FVP; funding and resources: FVP, RG, and LIT. All authors reviewed and approved the final manuscript.
Funding
JZ (Translational Genomics Core, LSUHSC-New Orleans, LA): P20CA202922, P20 GM121288-01, P30GM114732. FP: UNAM PAPIIT IN225920, CONACYT Fondo Sectorial 272573, Fondo SEP CONACYT 285879. LT: CONACYT National Laboratories 2021 project 315817.
Acknowledgments
We are thankful with Laura Fejerman and Valentina Andrea Zavala for their useful comments about the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/World Health Organization.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1094260/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Principal component analysis showing ancestral composition of the reference populations from 1000 G using 57 pruned variants selected from a panel of 104 ancestry informative SNPs genotyped using the MassARRAY System (Agena Bioscience). POP, Populations; GBR, British; FIN, Finnish; IBS Iberian; CEU, Northern and Western European; TSI, Toscani; MXL, Mexican; PEL, Peruvian; CLM, Colombian; PUR, Puerto Rican; YRI, Yoruba.
SUPPLEMENTARY TABLE S1 | Ancestry components of the participants.
SUPPLEMENTARY TABLE S2 | Demographic variables for the 431 AJ participants according to presence of at least one pathogenic or likely pathogenic germline variant. For the Tobacco /Alcohol consumption variable, current smokers and current drinkers included those women who reported daily smoking or drinking at time of recruitment. Pregnancy variable included women who had at least one full term pregnancy. Contraceptive use refers to consecutively use during the last year.
SUPPLEMENTARY TABLE S3 | Variants with unknown clinical significance in genes associated with hereditary breast and ovarian cancer syndrome or other cancers.
References
Adaniel, C., Salinas, F., Donaire, J. M., Bravo, M. E., Peralta, O., Paredes, H., et al. (2019). Non-BRCA1/2 variants detected in a high-risk Chilean cohort with a history of breast and/or ovarian cancer. J. Glob. Oncol. 5, 1–14. doi:10.1200/JGO.18.00163
Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi:10.1038/nature15393
Avni, H., Liwerant, J. B., and Pergola, S. D. (2011). Pertenencia y alteridad: Judíos en/de América latina: Cuarenta años de cambios. America: Iberoamericana.
Behar, D. M., Hammer, M. F., Garrigan, D., Villems, R., Bonne-Tamir, B., Richards, M., et al. (2004). MtDNA evidence for a genetic bottleneck in the early history of the Ashkenazi Jewish population. Eur. J. Hum. Genet. EJHG 12, 355–364. doi:10.1038/sj.ejhg.5201156
Benson, A. B., Venook, A. P., Al-Hawary, M. M., Arain, M. A., Chen, Y.-J., Ciombor, K. K., et al. (2021). Colon cancer, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. JNCCN 19, 329–359. doi:10.6004/jnccn.2021.0012
Burke, L. J., Sevcik, J., Gambino, G., Tudini, E., Mucaki, E. J., Shirley, B. C., et al. (2018). BRCA1 and BRCA2 5’ noncoding region variants identified in breast cancer patients alter promoter activity and protein binding. Hum. Mutat. 39, 2025–2039. doi:10.1002/humu.23652
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 4, 7. doi:10.1186/s13742-015-0047-8
Cock-Rada, A. M., Ossa, C. A., Garcia, H. I., and Gomez, L. R. (2018). A multi-gene panel study in hereditary breast and ovarian cancer in Colombia. Fam. Cancer 17, 23–30. doi:10.1007/s10689-017-0004-z
Daly, M. B., Pal, T., Berry, M. P., Buys, S. S., Dickson, P., Domchek, S. M., et al. (2021). Genetic/Familial high-risk assessment: Breast, ovarian, and pancreatic, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 19, 77–102. doi:10.6004/jnccn.2021.0001
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., McGowan-Jordan, J., et al. (2016). HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 37, 564–569. doi:10.1002/humu.22981
Ellis, N. A., Ciocci, S., Proytcheva, M., Lennon, D., Groden, J., and German, J. (1998). The Ashkenazic Jewish Bloom syndrome mutation blmAsh is present in non-Jewish Americans of Spanish ancestry. Am. J. Hum. Genet. 63, 1685–1693. doi:10.1086/302167
ENCODAT (2017). Encuesta nacional de Consumo de Drogas, alcohol y tabaco, ENCODAT 2016-2017. Gob. Mx. Available at: http://www.gob.mx/salud%7Cconadic/acciones-y-programas/encuesta-nacional-de-consumo-de-drogas-alcohol-y-tabaco-encodat-2016-2017-136758 (Accessed August 30, 2022).
Evans, D. G. R., van Veen, E. M., Byers, H. J., Wallace, A. J., Ellingford, J. M., Beaman, G., et al. (2018). A dominantly inherited 5’ UTR variant causing methylation-associated silencing of BRCA1 as a cause of breast and ovarian cancer. Am. J. Hum. Genet. 103, 213–220. doi:10.1016/j.ajhg.2018.07.002
Fejerman, L., John, E. M., Huntsman, S., Beckman, K., Choudhry, S., Perez-Stable, E., et al. (2008). Genetic ancestry and risk of breast cancer among U.S. Latinas. Cancer Res. 68, 9723–9728. doi:10.1158/0008-5472.CAN-08-2039
Fokkema, I. F. A. C., Kroon, M., López Hernández, J. A., Asscheman, D., Lugtenburg, I., Hoogenboom, J., et al. (2021). The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. EJHG 29, 1796–1803. doi:10.1038/s41431-021-00959-x
Gallardo-Rincón, D., Álvarez-Gómez, R. M., Montes-Servín, E., Toledo-Leyva, A., Montes-Servín, E., Michel-Tello, D., et al. (2020). Clinical evaluation of BRCA1/2 mutation in Mexican ovarian cancer patients. Transl. Oncol. 13, 212–220. doi:10.1016/j.tranon.2019.11.003
Greville-Heygate, S. L., Maishman, T., Tapper, W. J., Cutress, R. I., Copson, E., Dunning, A. M., et al. (2020). Pathogenic variants in CHEK2 are associated with an adverse prognosis in symptomatic early-onset breast cancer. JCO Precis. Oncol. 4, 472–485. doi:10.1200/PO.19.00178
Gusev, A., Palamara, P. F., Aponte, G., Zhuang, Z., Darvasi, A., Gregersen, P., et al. (2012). The architecture of long-range haplotypes shared within and across populations. Mol. Biol. Evol. 29, 473–486. doi:10.1093/molbev/msr133
Hansmann, T., Pliushch, G., Leubner, M., Kroll, P., Endt, D., Gehrig, A., et al. (2012). Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum. Mol. Genet. 21, 4669–4679. doi:10.1093/hmg/dds308
Harris, P. A., Taylor, R., Minor, B. L., Elliott, V., Fernandez, M., O’Neal, L., et al. (2019). The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inf. 95, 103208. doi:10.1016/j.jbi.2019.103208
Harris, P. A., Taylor, R., Thielke, R., Payne, J., Gonzalez, N., and Conde, J. G. (2009). Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inf. 42, 377–381. doi:10.1016/j.jbi.2008.08.010
Inegi (2020). Inegi. Available at: https://www.inegi.org.mx (Accessed August 30, 2022).
Landrum, M. J., Chitipiralla, S., Brown, G. R., Chen, C., Gu, B., Hart, J., et al. (2020). ClinVar: Improvements to accessing data. Nucleic Acids Res. 48, D835–D844. doi:10.1093/nar/gkz972
Levy-Lahad, E., Catane, R., Eisenberg, S., Kaufman, B., Hornreich, G., Lishinsky, E., et al. (1997). Founder BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: Frequency and differential penetrance in ovarian cancer and in breast-ovarian cancer families. Am. J. Hum. Genet. 60, 1059–1067.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Li, Q., and Wang, K. (2017). InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 100, 267–280. doi:10.1016/j.ajhg.2017.01.004
Manchanda, R., Burnell, M., Gaba, F., Desai, R., Wardle, J., Gessler, S., et al. (2020). Randomised trial of population-based BRCA testing in Ashkenazi Jews: Long-term outcomes. BJOG Int. J. Obstet. Gynaecol. 127, 364–375. doi:10.1111/1471-0528.15905
Manchanda, R., Loggenberg, K., Sanderson, S., Burnell, M., Wardle, J., Gessler, S., et al. (2015). Population testing for cancer predisposing BRCA1/BRCA2 mutations in the ashkenazi-jewish community: A randomized controlled trial. J. Natl. Cancer Inst. 107, 379. doi:10.1093/jnci/dju379
Mathieson, I., and Scally, A. (2020). What is ancestry? PLOS Genet. 16, e1008624. doi:10.1371/journal.pgen.1008624
Metcalfe, K. A., Poll, A., Royer, R., Llacuachaqui, M., Tulman, A., Sun, P., et al. (2010). Screening for founder mutations in BRCA1 and BRCA2 in unselected Jewish women. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 387–391. doi:10.1200/JCO.2009.25.0712
Miller, D. T., Lee, K., Chung, W. K., Gordon, A. S., Herman, G. E., Klein, T. E., et al. (2021). ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of medical genetics and genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 23, 1381–1390. doi:10.1038/s41436-021-01172-3
Montalban, G., Bonache, S., Moles-Fernández, A., Gisbert-Beamud, A., Tenés, A., Bach, V., et al. (2019). Screening of BRCA1/2 deep intronic regions by targeted gene sequencing identifies the first germline BRCA1 variant causing pseudoexon activation in a patient with breast/ovarian cancer. J. Med. Genet. 56, 63–74. doi:10.1136/jmedgenet-2018-105606
Moreno-Ortiz, J. M., Ayala-Madrigal, M. de la L., Corona-Rivera, J. R., Centeno-Flores, M., Maciel-Gutiérrez, V., Franco-Topete, R. A., et al. (2016). Novel mutations in MLH1 and MSH2 genes in Mexican patients with lynch syndrome. Gastroenterol. Res. Pract. 2016, 5278024. doi:10.1155/2016/5278024
Morgenstern-Kaplan, D., Raijman-Policar, J., Majzner-Aronovich, S., Aradhya, S., Pineda-Alvarez, D. E., Aguinaga, M., et al. (2022). Carrier screening in the Mexican Jewish community using a pan-ethnic expanded carrier screening NGS panel. Genet. Med. Off. J. Am. Coll. Med. Genet. 24, 821–830. doi:10.1016/j.gim.2021.11.019
Oliver, J., Quezada Urban, R., Franco Cortés, C. A., Díaz Velásquez, C. E., Montealegre Paez, A. L., Pacheco-Orozco, R. A., et al. (2019). Latin American study of hereditary breast and ovarian cancer lacam: A genomic epidemiology approach. Front. Oncol. 9, 1429. doi:10.3389/fonc.2019.01429
Ostrer, H. (2001). A genetic profile of contemporary Jewish populations. Nat. Rev. Genet. 2, 891–898. doi:10.1038/35098506
Palamara, P. F., Lencz, T., Darvasi, A., and Pe’er, I. (2012). Length distributions of identity by descent reveal fine-scale demographic history. Am. J. Hum. Genet. 91, 809–822. doi:10.1016/j.ajhg.2012.08.030
Quezada Urban, R., Díaz Velásquez, C. E., Gitler, R., Rojo Castillo, M. P., Sirota Toporek, M., Figueroa Morales, A., et al. (2018). Comprehensive analysis of germline variants in Mexican patients with hereditary breast and ovarian cancer susceptibility. Cancers 10, E361. doi:10.3390/cancers10100361
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 17, 405–424. doi:10.1038/gim.2015.30
Rumgay, H., Shield, K., Charvat, H., Ferrari, P., Sornpaisarn, B., Obot, I., et al. (2021). Global burden of cancer in 2020 attributable to alcohol consumption: A population-based study. Lancet Oncol. 22, 1071–1080. doi:10.1016/S1470-2045(21)00279-5
Sandoval, R. L., Leite, A. C. R., Barbalho, D. M., Assad, D. X., Barroso, R., Polidorio, N., et al. (2021). Germline molecular data in hereditary breast cancer in Brazil: Lessons from a large single-center analysis. PloS One 16, e0247363. doi:10.1371/journal.pone.0247363
Serrano-Gomez, S. J., Sanabria-Salas, M. C., Hernández-Suarez, G., García, O., Silva, C., Romero, A., et al. (2016). High prevalence of luminal B breast cancer intrinsic subtype in Colombian women. Carcinogenesis 37, 669–676. doi:10.1093/carcin/bgw043
Shaag, A., Walsh, T., Renbaum, P., Kirchhoff, T., Nafa, K., Shiovitz, S., et al. (2005). Functional and genomic approaches reveal an ancient CHEK2 allele associated with breast cancer in the Ashkenazi Jewish population. Hum. Mol. Genet. 14, 555–563. doi:10.1093/hmg/ddi052
Solano, A. R., Mele, P. G., Jalil, F. S., Liria, N. C., Podesta, E. J., and Gutiérrez, L. G. (2021). Study of the genetic variants in BRCA1/2 and non-BRCA genes in a population-based cohort of 2155 breast/ovary cancer patients, including 443 triple-negative breast cancer patients, in Argentina. Cancers 13, 2711. doi:10.3390/cancers13112711
Tang, H., Peng, J., Wang, P., and Risch, N. J. (2005). Estimation of individual admixture: Analytical and study design considerations. Genet. Epidemiol. 28, 289–301. doi:10.1002/gepi.20064
Tennen, R. I., Laskey, S. B., Koelsch, B. L., McIntyre, M. H., and Tung, J. Y. (2020). Identifying Ashkenazi Jewish BRCA1/2 founder variants in individuals who do not self-report Jewish ancestry. Sci. Rep. 10, 7669. doi:10.1038/s41598-020-63466-x
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi:10.1093/bib/bbs017
Torres-Mejía, G., Royer, R., Llacuachaqui, M., Akbari, M. R., Giuliano, A. R., Martínez-Matsushita, L., et al. (2015). Recurrent BRCA1 and BRCA2 mutations in Mexican women with breast cancer. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 24, 498–505. doi:10.1158/1055-9965.EPI-13-0980
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.1–11.10.33. doi:10.1002/0471250953.bi1110s43
Villarreal-Garza, C., Alvarez-Gómez, R. M., Pérez-Plasencia, C., Herrera, L. A., Herzog, J., Castillo, D., et al. (2015). Significant clinical impact of recurrent BRCA1 and BRCA2 mutations in Mexico. Cancer 121, 372–378. doi:10.1002/cncr.29058
Walsh, T., Mandell, J. B., Norquist, B. M., Casadei, S., Gulsuner, S., Lee, M. K., et al. (2017). Genetic predisposition to breast cancer due to mutations other than BRCA1 and BRCA2 founder alleles among Ashkenazi jewish women. JAMA Oncol. 3, 1647–1653. doi:10.1001/jamaoncol.2017.1996
Wang, K., Li, M., and Hakonarson, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Weitzel, J. N., Lagos, V. I., Herzog, J. S., Judkins, T., Hendrickson, B., Ho, J. S., et al. (2007). Evidence for common ancestral origin of a recurring BRCA1 genomic rearrangement identified in high-risk Hispanic families. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 16, 1615–1620. doi:10.1158/1055-9965.EPI-07-0198
Keywords: Ashkenazi Jews, genetic screening, panel of genes, massive parallel sequencing, founder variant, APC
Citation: Díaz-Velásquez CE, Gitler R, Antoniano A, Kershenovich Sefchovich R, De La Cruz-Montoya AH, Martínez-Gregorio H, Rojas-Jiménez EA, Cortez Cardoso Penha R, Terrazas LI, Wegman-Ostrosky T, Levi-Lahad E, Zabaleta J, Perdomo S and Vaca-Paniagua F (2023) Evaluation of genetic alterations in hereditary cancer susceptibility genes in the Ashkenazi Jewish women community of Mexico. Front. Genet. 14:1094260. doi: 10.3389/fgene.2023.1094260
Received: 09 November 2022; Accepted: 24 January 2023;
Published: 10 February 2023.
Edited by:
Mehdi Pirooznia, Johnson & Johnson, United StatesReviewed by:
Clesson Turner, National Institutes of Health (NIH), United StatesLucas Delmonico, Oncoclinicas Group, Brazil
Miriam Bornhorst, Children’s National Hospital, United States
Copyright © 2023 Díaz-Velásquez, Gitler, Antoniano, Kershenovich Sefchovich, De La Cruz-Montoya, Martínez-Gregorio, Rojas-Jiménez, Cortez Cardoso Penha, Terrazas, Wegman-Ostrosky, Levi-Lahad, Zabaleta, Perdomo and Vaca-Paniagua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Perdomo, cGVyZG9tb3NAaWFyYy5mcg==; Felipe Vaca-Paniagua, ZmVsaXBlLnZhY2FAaXp0YWNhbGEudW5hbS5teA==
†These authors have contributed equally to this work and share first authorship