 David Soave
David Soave Melisa Hayalioglu1
Melisa Hayalioglu1 Lei Sun
Lei Sun- 1Department of Mathematics, Wilfrid Laurier University, Waterloo, ON, Canada
- 2Ontario Institute for Cancer Research, Toronto, ON, Canada
- 3Department of Statistical Sciences, University of Toronto, Toronto, ON, Canada
- 4Division of Biostatistics, Dalla Lana School of Public Health, University of Toronto, Toronto, ON, Canada
For complex human traits, a large portion of genetic heritability remains unaccounted for beyond common genetic variants; therefore, estimating the contribution of rare variants (RVs) to the etiology of complex traits is of interest. Research in this domain has primarily focused on gene-based RV testing methods, in which information from multiple variants is combined to maximize statistical power in detecting genes associated with the trait of interest. However, after discovering an association, estimating individual effects becomes challenging due to sample size limitations. Hence, the focus may shift to estimating the average genetic effect (AGE) for the group of RVs analyzed. This study demonstrates that both AGEs and individual variant effects can be influenced by competing upward and downward biases, resulting from the winner’s curse and the heterogeneity of individual variant effects, respectively. Various bias-correction techniques, including bootstrap resampling and likelihood-based methods, have been proposed to address the winner’s curse bias. We conduct a simulation study to illustrate the ramifications of these competing biases on variant effect size estimation and how they complicate the precision of pooled estimates obtained from different bias-correction techniques. We then examine the individual effect estimates of the causal variants across the simulation replicates to show how they may contribute to the observed upward and downward biases when RVs are pooled.
1 Introduction
Common genetic variants generally account for only a portion of genetic heritability (Pritchard, 2001; Bodmer and Bonilla, 2008; Visscher et al., 2017). One of the proposed explanations for this missing heritability is that genetic architectures are highly polygenic, involving numerous variants with individually small effects that collectively contribute to complex traits (Park et al., 2010). Park et al. (2010) demonstrated that the distribution of effect sizes in genome-wide association studies (GWASs) follows an exponential-like decay, suggesting that many susceptibility loci remain undetected due to limited power in current study designs. Furthermore, linkage disequilibrium (LD) structure plays a key role in the proportion of heritability captured by GWASs as causal variants that are poorly tagged by genotyped markers lead to the underestimation of genetic contributions (Yang et al., 2012). These challenges are particularly relevant for rare variants (RVs), which, despite potentially larger effect sizes, remain difficult to detect due to their low minor allele frequency (MAF) and weaker LD with neighboring markers (Manolio et al., 2009; Wang et al., 2021).
To improve detection power, gene-based or pooled association tests have been developed to aggregate information across multiple RVs, allowing for the joint evaluation of their contribution to trait variation (Morgenthaler and Thilly, 2007; Li and Leal, 2008; Madsen and Browning, 2009; Price et al., 2010; Neale et al., 2011; Wu et al., 2011). Most of these gene-based RV tests belong to either the class of linear tests (e.g., the burden test) or the class of quadratic tests (e.g., variance components or SKAT methods) (Derkach et al., 2014). As each type of test can be more powerful under different scenarios, hybrid tests have also been proposed to combine the association evidence from two or more complementary approaches (Lee et al., 2012; Derkach et al., 2013; Liu et al., 2019).
Following association testing, it is of interest to estimate and understand the genetic effect sizes of the significant RVs. The gene-based approach, although effective at association testing, limits our ability to estimate the individual genetic effect size
For the analysis of individual common variants, several approaches have been proposed to correct for the winner’s curse, including bootstrap resampling (Sun and Bull, 2005; Faye et al., 2011), likelihood-based approaches (Zöllner and Pritchard, 2007; Ghosh et al., 2008; Zhong and Prentice, 2008; Xiao and Boehnke, 2011), and Bayesian approaches (Xu et al., 2011). More recently, additional work has explored the winner’s curse and proposed methods for correcting estimation in genome-wide association and polygenic risk score studies, along with eQTL and mediation analyses (Shi et al., 2016; Huang et al., 2018; Hu et al., 2019; Xie et al., 2021; Yang et al., 2021).
Focusing on the joint analysis of multiple rare variants using the combined multivariate and collapsing (CMC) method of Li and Leal (2008), Liu and Leal (2012) estimated
Our investigation builds upon the recent work by Grinde et al. (2017), with some notable distinctions. First, while Grinde et al. (2017) focused on case–control studies and used the squared difference of MAFs between cases and controls as the measure for an individual variant effect, we use a conventional effect size measure, which is the rate of change in outcome (either binary or continuous) per unit change in the genotype. Second, we consider both the individual variant effects and the average genetic effect. Finally, while Grinde et al. (2017) centered on the overestimation problem due to the winner’s curse, our study further considers the competing downward bias due to effect heterogeneity, providing a more nuanced understanding of the biases inherent in pooled analysis of multiple rare variants.
The remainder of this paper is organized as follows. Section 2 reviews existing methods, including association testing of rare variants, parameter estimation of the average genetic effect
2 Methods
2.1 Brief review of joint association testing of multiple rare variants
As association analyses between individual rare variants
There exist two classes of score tests that aggregate these scores in different ways (Derkach et al., 2013). The linear class
Depending on how the weights
In contrast, the quadratic class
where if
Hence, there exists another class of methods that can integrate the linear and quadratic statistics, such as the SKAT-O method (Lee et al., 2012), Fisher’s method (Derkach et al., 2013), or the more recent ACAT approach (Liu et al., 2019). Penalized regression approaches, which allow grouping of multiple regions at once, form yet another class of methods.
For the purpose of this study, we examine the linear and quadratic classes to better characterize the key factors contributing to the estimation bias. Within the two classes, without loss of generality, we then focus on the equal weighting methods, namely, CAST (Morgenthaler and Thilly, 2007) and C-Alpha (Neale et al., 2011), from the linear and quadratic classes, respectively. Both are unweighted methods from their respective classes, providing a straightforward interpretation of results. To facilitate the study of
2.2 Average genetic effect and CMC RV testing
It is usually desirable for investigators to estimate the genetic effect size after achieving a significant association testing result. This is commonly done for sample size estimation to conduct sufficiently powered replication studies. For the analysis of rare variants, Liu and Leal (2012) considered the following genetic model:
where
Unfortunately, due to the same logic that limits power in detecting causal SNPs individually, we are unable to estimate the individual
Possibly, the most organic choice of the parameter estimation method when association tests are based on weighting/collapsing variants is the AGE, defined as the change in the trait value per unit change in multivariate genotype coding
where
Under the CMC RV association method (Li and Leal, 2008), testing the null hypothesis
where
2.3 Winner’s curse and bias-corrected estimates
When analyzing individual common variants that have been filtered and selected based on statistically significant association, it is well established that the effect size estimate,
The bootstrap approach, in essence, splits the sample into two independent sub-samples, using one for hypothesis testing and the other for parameter estimation (Sun and Bull, 2005; Faye et al., 2011; Sun et al., 2011). In brief, for each bootstrap sample, a complete GWAS is conducted to determine the associated variants of interest. For each associated variant identified in the bootstrap sample, its effect size
For rare variants, Liu and Leal (2012) investigated the flexible bootstrap correction approach (using the median, instead of the mean, of
The following simulation study extends the investigation of Liu and Leal (2012) in several ways. First, we examine whether the original bootstrap procedure of using the mean of
To formalize the approach, we define the standardized effect size as follows:
Given an observed estimate
Since only values where
where
3 Simulation study
We conducted a simulation study using the Genetic Analysis Workshop 17 (GAW17) data (Almasy et al., 2011). The GAW17 “mini-exome” dataset consists of real human DNA sequence data from the 1000 Genomes Project (Consortium et al., 2010) and various qualitative and quantitative phenotype data simulated by the GAW17 data committee. For each phenotype, 200 replicate samples were generated by simulating different phenotype data based on the true genotype–phenotype model, conditional on the observed genotype data.
For this investigation, the sample for analysis consisted of the
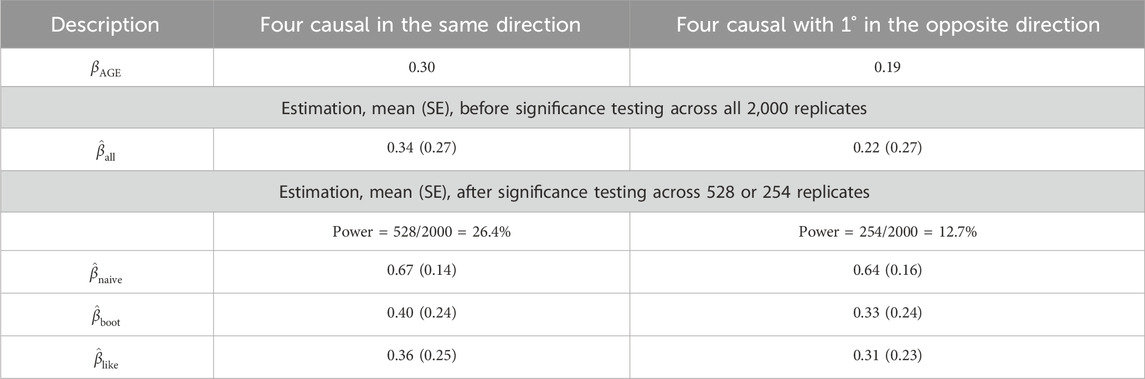
Table 1. GAW17 simulation summary statistics for
3.1 Setup
For each replicate sample, we conducted a linear regression analysis of the phenotype on the collapsed genotype parameter under the CMC pooling method (1), yielding an effect estimate
Next, we assessed the performance of the bootstrapping approach for effect size adjustment proposed by Liu and Leal (2012) (described in Section 2.3) and the approximate likelihood-based adjustment method proposed by Ghosh et al. (2008) by applying them to each of the statistically significant simulation replicates identified.
To identify the contribution of each of the four causal RVs to the pooled genotype testing procedures and the
We note that the effect sizes (
3.2 Performance of the bootstrapping and likelihood effect size adjustments
Summary statistics for the estimate of
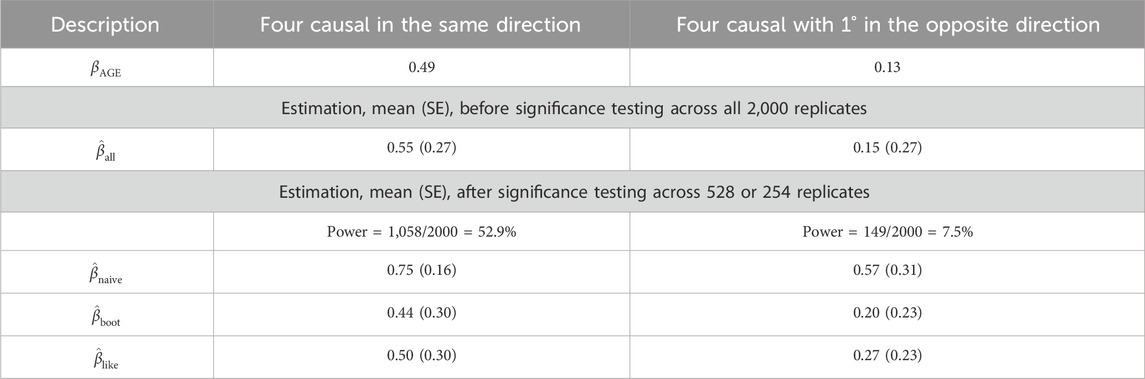
Table 2. GAW17 simulation summary statistics for
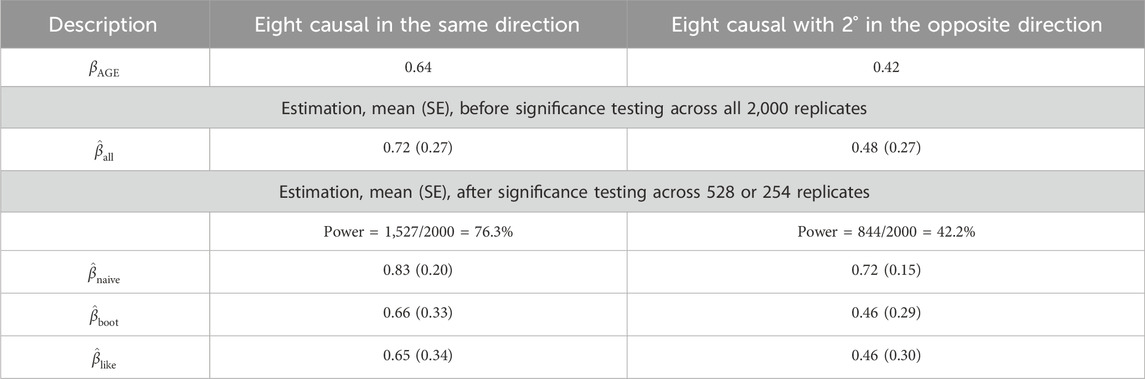
Table 3. GAW17 simulation summary statistics for
First, without conditioning on the association testing results, the parameter estimation is unbiased, as expected:
Second, when the average estimate was calculated among only the significant replicates, bias in
Third, similar to the results of Liu and Leal (2012), the bootstrap bias adjustment works well under the CMC RV association testing method. Using the bootstrap correction (Liu and Leal, 2012), the average absolute bias owing to the winner’s curse was greatly reduced,
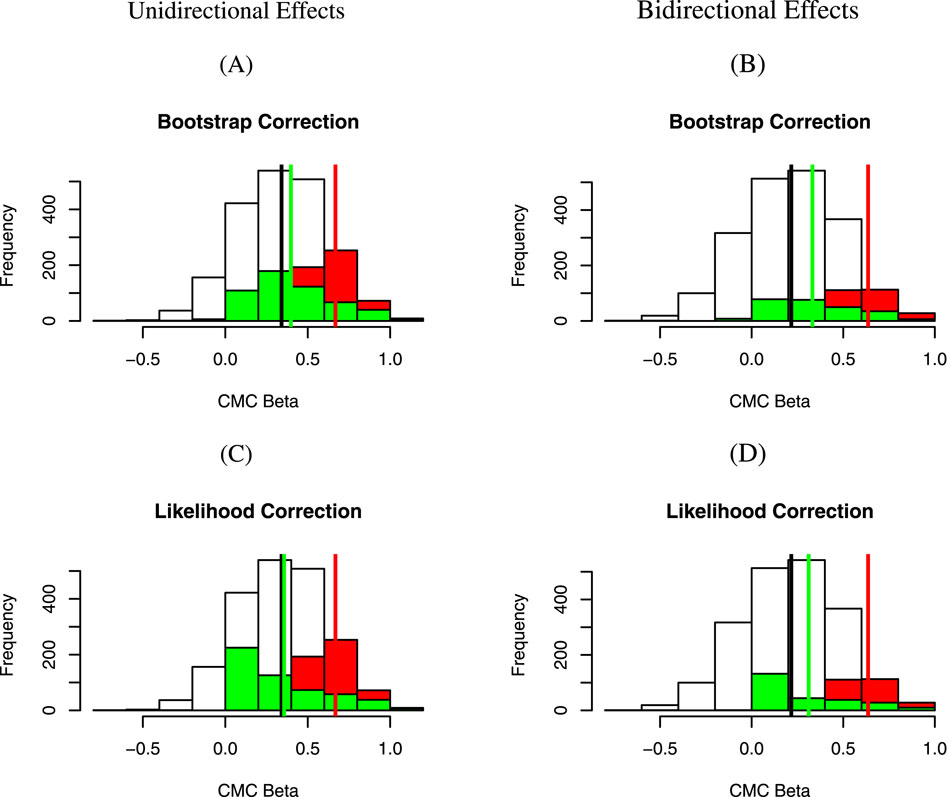
Figure 1. Distributions of
Fourth, the approximate likelihood approach proposed by Ghosh et al. (2008) appears to perform as well as, if not better than, the bootstrap approach, where
Finally, it is important to point out that
3.3 Bias–variance tradeoff in effect estimation
Estimates of effect sizes, such as the bias-corrected estimates investigated in this study, require standard error estimates for inference procedures. Ghosh et al. (2008) constructed confidence intervals with correct conditional coverage by applying the original Neymanian concept of confidence regions, where intervals are derived from the known conditional distribution of the test statistic after selection. Specifically, the acceptance region
Although Liu and Leal (2012) did not propose methods for calculating confidence intervals (CIs) or standard errors (SEs) for the bootstrap-corrected estimates of
Rather than investigating and reporting such standard error estimation methods, we report the empirical standard error estimates to investigate the bias–variance trade-off related to the bias-corrected estimate of
3.4 Individual variant effects
A limitation of the current state of the rare variant literature is a demonstration of the bias associated with the individual causal effects contributing to the winner’s curse bias. To this end, we examined the individual effect estimates of the causal variants over the GAW17 simulation replicates to shed light on some of the contributing factors that might play a role in the different scenarios of pooled effects. To this end, we first applied three RV association methods: CMC (Li and Leal, 2008), CAST (Morgenthaler and Thilly, 2007), and C-Alpha (Neale et al., 2011), jointly testing a group of variants. We then summarized the individual variant effect estimates, with or without conditioning on the testing results.
Results of the individual effect estimates (
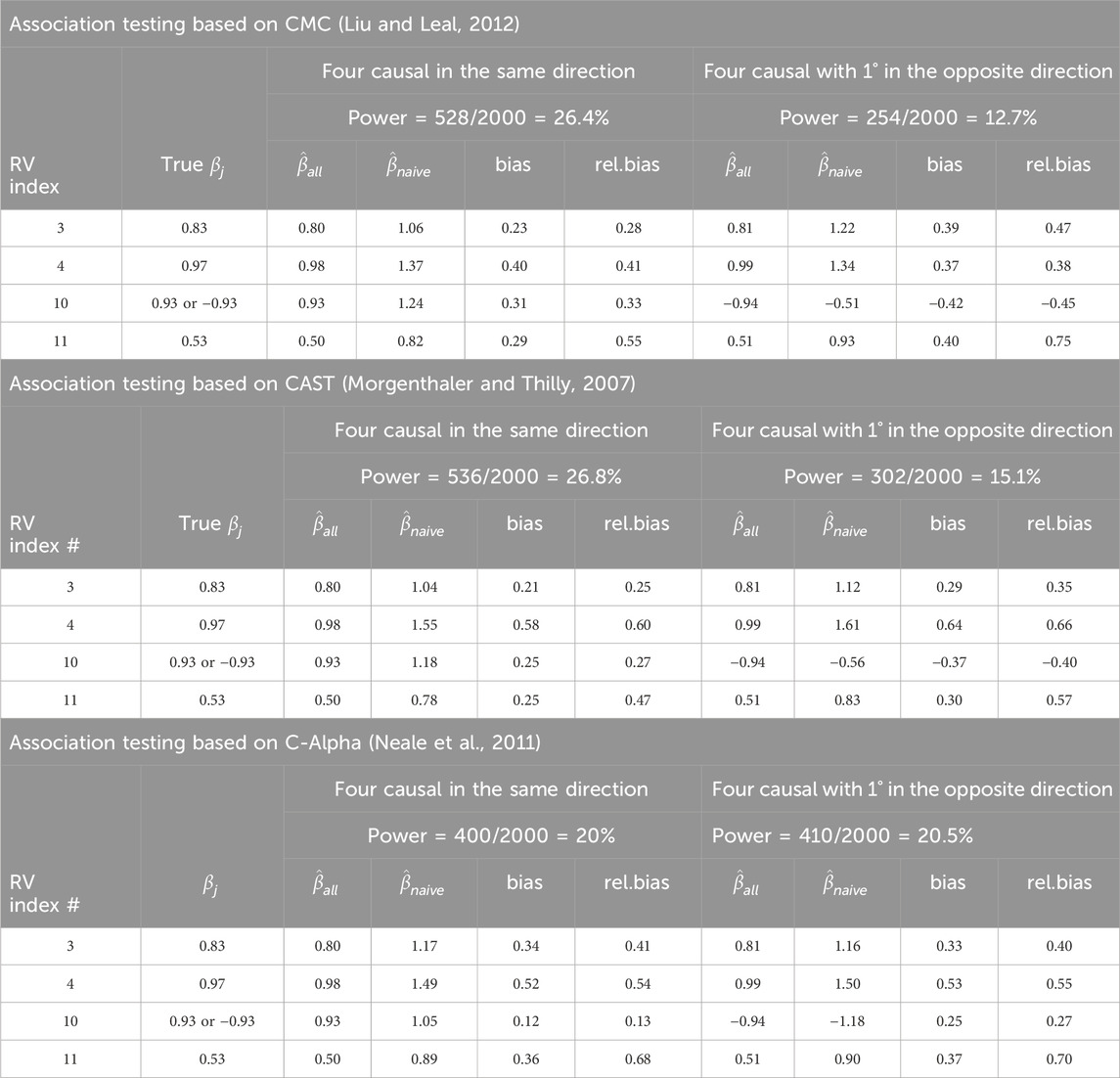
Table 4. Individual causal variant
Consistent with the previous literature on the power of RV association testing, when all causal SNPs have the same direction of the effect, the linear test has a slight advantage, but the power of the quadratic test is comparable (power of 20% for C-Alpha compared with 0.27 and 0.26 for CAST and CMC, respectively). On the other hand, when one of the four causal SNPs (25%) has an effect in the opposite direction to the others, the power is greater with the quadratic test statistic (0.21 for C-Alpha compared with 0.15 and 0.13 for CAST and CMC, respectively). Moreover, consistent with the previous literature on the winner’s curse for common variants, the relative bias increases as power decreases.
However, under the linear testing approaches (CMC and CAST), causal variants with effects in opposite directions (right side of Table 4) led to an increased upward bias for the individual
Unlike linear testing methods, quadratic testing (C-Alpha) involving causal variants with effects in opposite directions does not significantly alter the magnitudes of biases for the individually estimated
4 Discussion
The upward bias in effect size estimation due to selective inference conditional on significant association testing is well understood for common variants and has been demonstrated for rare variants in the context of
Starting with previously defined
We then examined how heterogeneous effect direction affects our interpretation of
First, as
Second, our simulation results showed that individual SNP effect estimate bias depends not only on the directionality of SNPs within the set but also on the choice of linear or quadratic association testing approach. For linear tests, individual estimates (
Although our simulation studies only focused on CAST (Morgenthaler and Thilly, 2007) from the linear class and C-Alpha (Neale et al., 2011) from the quadratic class, the general analytical form of these tests suggests that our observations can be generalized to other specific tests from each class, e.g., WSS (Madsen and Browning, 2009) from the linear class and SKAT (Wu et al., 2011) from the quadratic class. The behaviors of tests from the hybrid class, such as SKAT-O (Lee et al., 2012) and Fisher’s method (Derkach et al., 2013), however, are unknown and warrant future studies.
As rare variant studies scale to exome- and genome-wide datasets, computational efficiency becomes increasingly important. Bootstrap-based methods, while flexible, can be computationally expensive for large-scale analyses. Likelihood-based corrections, as evaluated in our study, offer a more scalable alternative, particularly when computational resources are limited. Additionally, for extremely rare variants (e.g., minor allele frequency
Existing bias-correction methods for common variants can be effectively applied to estimate the AGE for rare variants. However, pooling rare variants presents challenges for estimating individual effect sizes, particularly when causal variants have bidirectional effects. As rare variant studies expand with whole-genome sequencing and biobank-scale datasets, future work should focus on improving bias-correction methods for both pooled and individual effect estimates. Although likelihood-based methods are efficient, machine learning and Bayesian approaches could provide more flexible models for capturing complex effect architectures. Additionally, incorporating corrected rare variant effects into polygenic risk scores may enhance predictive accuracy, especially for diseases with strong rare variant contributions. Developing methods that optimally combine linear and variance-component tests, such as SKAT-O, could further improve estimation in the presence of effect heterogeneity. Advancing these techniques will strengthen our ability to quantify rare variant contributions to complex traits and improve genetic risk prediction.
Data availability statement
The “simulated” datasets presented in this study can be found at: https://github.com/dsoave/WinnersCurse_RareVariants_Public.
Author contributions
DS: Conceptualization, Formal Analysis, Methodology, Supervision, Writing – original draft, Writing – review and editing. MH: Formal Analysis, Writing – original draft, Writing – review and editing. LS: Conceptualization, Methodology, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Canadian Institutes of Health Research (CIHR, PJT-180460 to LS) and the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2018-04934 to LS, and RGPIN-2019-05912 to DS).
Acknowledgments
The authors would like to thank the Genetic Analysis Workshop 17 (GAW17) committee and the 1000 Genomes Project for providing the GAW17 application data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Almasy, L., Dyer, T. D., Peralta, J. M., Kent, J. W., Charlesworth, J. C., Curran, J. E., et al. (2011). Genetic analysis workshop 17 mini-exome simulation. BMC Proc. Biomed. Cent. 5, 1–9. doi:10.1186/1753-6561-5-S9-S2
Bodmer, W., and Bonilla, C. (2008). Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 40, 695–701. doi:10.1038/ng.f.136
Consortium, G. P., Abecasis, G. R., Altshuler, D., Auton, A., Brooks, L. D., Durbin, R. M., et al. (2010). A map of human genome variation from population scale sequencing. Nature 467, 1061–1073. doi:10.1038/nature09534
Decker, B., Allen, J., Luccarini, C., Pooley, K. A., Shah, M., Bolla, M. K., et al. (2017). Rare, protein-truncating variants in atm, chek2 and palb2, but not xrcc2, are associated with increased breast cancer risks. J. Med. Genet. 54, 732–741. doi:10.1136/jmedgenet-2017-104588
Derkach, A., Lawless, J. F., and Sun, L. (2013). Robust and powerful tests for rare variants using fisher’s method to combine evidence of association from two or more complementary tests. Genet. Epidemiol. 37, 110–121. doi:10.1002/gepi.21689
Derkach, A., Lawless, J. F., and Sun, L. (2014). Pooled association tests for rare genetic variants: a review and some new results. Stat. Sci. 29, 302–321. doi:10.1214/13-sts456
Efron, B. (2011). Tweedie’s formula and selection bias. J. Am. Stat. Assoc. 106, 1602–1614. doi:10.1198/jasa.2011.tm11181
Faye, L. L., Sun, L., Dimitromanolakis, A., and Bull, S. B. (2011). A flexible genome-wide bootstrap method that accounts for rankingand threshold-selection bias in gwas interpretation and replication study design. Statistics Med. 30, 1898–1912. doi:10.1002/sim.4228
Ghosh, A., Zou, F., and Wright, F. A. (2008). Estimating odds ratios in genome scans: an approximate conditional likelihood approach. Am. J. Hum. Genet. 82, 1064–1074. doi:10.1016/j.ajhg.2008.03.002
Göring, H. H., Terwilliger, J. D., and Blangero, J. (2001). Large upward bias in estimation of locus-specific effects from genomewide scans. Am. J. Hum. Genet. 69, 1357–1369. doi:10.1086/324471
Grinde, K. E., Arbet, J., Green, A., O’Connell, M., Valcarcel, A., Westra, J., et al. (2017). Illustrating, quantifying, and correcting for bias in post-hoc analysis of gene-based rare variant tests of association. Front. Genet. 8, 117. doi:10.3389/fgene.2017.00117
Hu, J., Zhang, W., Li, X., Pan, D., and Li, Q. (2019). Efficient estimation of disease odds ratios for follow-up genetic association studies. Stat. Methods Med. Res. 28, 1927–1941. doi:10.1177/0962280217741771
Huang, Q. Q., Ritchie, S. C., Brozynska, M., and Inouye, M. (2018). Power, false discovery rate and winner’s curse in eqtl studies. Nucleic acids Res. 46, e133. doi:10.1093/nar/gky780
Lee, S., Emond, M. J., Bamshad, M. J., Barnes, K. C., Rieder, M. J., Nickerson, D. A., et al. (2012). Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237. doi:10.1016/j.ajhg.2012.06.007
Li, B., and Leal, S. M. (2008). Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321. doi:10.1016/j.ajhg.2008.06.024
Liu, D. J., and Leal, S. M. (2012). Estimating genetic effects and quantifying missing heritability explained by identified rare-variant associations. Am. J. Hum. Genet. 91, 585–596. doi:10.1016/j.ajhg.2012.08.008
Liu, Y., Chen, S., Li, Z., Morrison, A. C., Boerwinkle, E., and Lin, X. (2019). Acat: a fast and powerful p value combination method for rare-variant analysis in sequencing studies. Am. J. Hum. Genet. 104, 410–421. doi:10.1016/j.ajhg.2019.01.002
Madsen, B. E., and Browning, S. R. (2009). A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 5, e1000384. doi:10.1371/journal.pgen.1000384
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi:10.1038/nature08494
Morgenthaler, S., and Thilly, W. G. (2007). A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (cast). Mutat. Research/Fundamental Mol. Mech. Mutagen. 615, 28–56. doi:10.1016/j.mrfmmm.2006.09.003
Neale, B. M., Rivas, M. A., Voight, B. F., Altshuler, D., Devlin, B., Orho-Melander, M., et al. (2011). Testing for an unusual distribution of rare variants. PLoS Genet. 7, e1001322. doi:10.1371/journal.pgen.1001322
Park, J.-H., Wacholder, S., Gail, M. H., Peters, U., Jacobs, K. B., Chanock, S. J., et al. (2010). Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. 42, 570–575. doi:10.1038/ng.610
Price, A. L., Kryukov, G. V., de Bakker, P. I., Purcell, S. M., Staples, J., Wei, L.-J., et al. (2010). Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 86, 832–838. doi:10.1016/j.ajhg.2010.04.005
Pritchard, J. K. (2001). Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 69, 124–137. doi:10.1086/321272
Shi, J., Park, J.-H., Duan, J., Berndt, S. T., Moy, W., Yu, K., et al. (2016). Winner’s curse correction and variable thresholding improve performance of polygenic risk modeling based on genome-wide association study summary-level data. PLoS Genet. 12, e1006493. doi:10.1371/journal.pgen.1006493
Sun, L., and Bull, S. B. (2005). Reduction of selection bias in genomewide studies by resampling. Genet. Epidemiol. Official Publ. Int. Genet. Epidemiol. Soc. 28, 352–367. doi:10.1002/gepi.20068
Sun, L., Dimitromanolakis, A., Faye, L. L., Paterson, A. D., Waggott, D., Bull, S. B., et al. (2011). Br-squared: a practical solution to the winner’s curse in genome-wide scans. Hum. Genet. 129, 545–552. doi:10.1007/s00439-011-0948-2
Taylor, J., and Tibshirani, R. J. (2015). Statistical learning and selective inference. Proc. Natl. Acad. Sci. 112, 7629–7634. doi:10.1073/pnas.1507583112
Visscher, P. M., Wray, N. R., Zhang, Q., Sklar, P., McCarthy, M. I., Brown, M. A., et al. (2017). 10 years of gwas discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. doi:10.1016/j.ajhg.2017.06.005
Wang, Q., Dhindsa, R. S., Carss, K., Harper, A. R., Nag, A., Tachmazidou, I., et al. (2021). Rare variant contribution to human disease in 281,104 UK biobank exomes. Nature 597, 527–532. doi:10.1038/s41586-021-03855-y
Wu, M. C., Lee, S., Cai, T., Li, Y., Boehnke, M., and Lin, X. (2011). Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93. doi:10.1016/j.ajhg.2011.05.029
Xiao, R., and Boehnke, M. (2011). Quantifying and correcting for the winner’s curse in quantitative-trait association studies. Genet. Epidemiol. 35, 133–138. doi:10.1002/gepi.20551
Xie, F., Wang, S., Beavis, W. D., and Xu, S. (2021). Estimation of genetic variance contributed by a quantitative trait locus: correcting the bias associated with significance tests. Genetics 219, iyab115. doi:10.1093/genetics/iyab115
Xu, L., Craiu, R. V., and Sun, L. (2011). Bayesian methods to overcome the winner’s curse in genetic studies. Ann. Appl. Statistics 5, 201–231. doi:10.1214/10-aoas373
Yang, J., Ferreira, T., Morris, A. P., Medland, S. E., Heath, A. C., Martin, N. G., et al. (2012). Conditional and joint multiple-snp analysis of gwas summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–S3. doi:10.1038/ng.2213
Yang, T., Niu, J., Chen, H., and Wei, P. (2021). Estimation of total mediation effect for high-dimensional omics mediators. BMC Bioinforma. 22, 414–417. doi:10.1186/s12859-021-04322-1
Zhong, H., and Prentice, R. L. (2008). Bias-reduced estimators and confidence intervals for odds ratios in genome-wide association studies. Biostatistics 9, 621–634. doi:10.1093/biostatistics/kxn001
Keywords: genome-wide association study, rare variants, joint analysis, estimation, selection bias, winner’s curse, effect heterogeneity
Citation: Soave D, Hayalioglu M and Sun L (2025) Winner’s curse in rare variant analysis: effect size estimation bias depends on effect direction and the association method used. Front. Genet. 16:1416673. doi: 10.3389/fgene.2025.1416673
Received: 12 April 2024; Accepted: 10 July 2025;
Published: 08 August 2025.
Edited by:
Stefan Böhringer, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Zhaoxia Yu, University of California, Irvine, United StatesRounak Dey, Insitro, Inc., United States
Copyright © 2025 Soave, Hayalioglu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Soave, ZHNvYXZlQHdsdS5jYQ==