 Jiao Xue
Jiao Xue Zhenfeng Song1
Zhenfeng Song1 Ying Zhang
Ying Zhang- 1 Department of Pediatric Neurology, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, China
- 2 Department of Anesthesiology, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, China
Background: Central nervous system-isolated hemophagocytic lymphohistiocytosis (CNS-HLH) is a rare disease caused by mutations in several genes.
Methods: Clinical information was obtained from medical records. Genetic analyses were performed using whole-exome sequencing (WES). NK cell function testing, Granzyme B staining, perforin staining, CD107a mobilization, and soluble CD25 levels were determined.
Results: We report the case of a 5-year-old girl who presented with involuntary movements, an unsteady gait, and a progressively irritable temper. Cranial MRI revealed bilateral multifocal white matter abnormalities. The patient harbored a homozygous missense mutation in the PRF1 gene (NM_001083116.3), c.1349C > T (p.Thr450Met), which is a maternal uniparental disomy. Based on the phenotype and absence of perforin expression, the patient was diagnosed with CNS-HLH.
Conclusion: We report a highly unusual case of CNS-HLH diagnosed by uniparental disomy of a PRF1 mutation. Exome sequencing should be considered in patients with chronic or recurrent brain inflammation who show partial or no response to conventional treatment regimens.
1 Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a multisystem inflammatory disorder that results from an unregulated cytokine storm and activation of cytotoxic T cells and antigen-presenting cells (Gupta et al., 2023). HLH typically affects multiple organ systems. HLH rarely involves the central nervous system, even in the absence of systemic inflammatory features (Benson et al., 2019). Here, we report a highly unusual case of a 5-year-old girl who presented with involuntary movements and an unsteady gait, and was diagnosed with central nervous system isolated HLH (CNS-HLH) by harboring uniparental disomy of a PRF1 mutation (c.1349C > T, p. Thr450Met).
2 Methods
The patient was admitted to our hospital in October 2024. Clinical information was obtained from medical records. Genetic analysis was performed using whole-exome sequencing (WES). NK cell functional testing, Granzyme B staining, perforin staining, CD107a mobilization, and soluble CD25 levels were determined at the Beijing Hightrust Diagnostic Medical Laboratory. Neural antibodies associated with autoimmune encephalitis, including NMDAR, LGI1, CASPR2, GABABR, AMPAR1, AMPAR2, IgLON5, DPPX, GAD65, mGluR5, GlyR, D2R, MOG, and GFAP in serum and cerebrospinal fluid (CSF) were tested using Cell-Based Assays with immunofluorescence double staining at the Jiangsu Simcere Diagnostic Laboratory (Jiangsu Simcere Diagnostics Co, Ltd., Nanjing 210002, China).
3 Results
A previously healthy 5-year-old girl presented to our hospital with a 1-week history of involuntary movements, unsteady gait, and a progressively irritable temper. Initially, twitching of her mouth, writhing trunk, and swinging limbs were uncontrolled, but her symptoms progressed to gait imbalance and frequent tripping.
Upon admission, the patient appeared restless. Her consciousness was clear and she had no other neurological symptoms, splenomegaly, hepatomegaly, lymph node swelling, or fever. Physical examination revealed dysmetria and intention tremors on finger-to-nose testing. The patient was unable to stand on both feet simultaneously druing Romberg test. Alternating movement and heel-to-shin tests was unnable to cooperate. The patient also exhibited a slight decrease in muscle strength and tone. No abnormality of the cranial nerves was found. The deep tendon reflexes were elicited symmetrically. Pathological reflex examination was negative. The family history was unremarkable.
Laboratory test results showed that red and white blood cell counts, and platelet counts were within normal ranges (Table 1). Blood chemistry analyses, including liver enzymes, renal function, and electrolytes, were normal. Triglycerides increased slightly (Table 1). The erythrocyte sedimentation rate, ferritin level, coagulation function, thyroid function, and antinuclear antibody levels were also normal (Table 1). Cranial MRI showed FLAIR hyperintensities in bilateral white matter, basal ganglia, corona radiata, brainstem, and anteroposterior horns of the lateral ventricle (Figures 1A–D). Cervical, thoracic, and lumbar MRI revealed no abnormalities. Video EEG showed a normal occipital background rhythm without epileptic discharges. CSF examination revealed pleocytosis (9 cells/mL) with normal glucose, protein and immunoglobulin levels. The CSF IL-6 and IL-8 levels were also elevated. The CSF bacterial smears and pathogenic sequencing results were negative. Comprehensive autoimmune encephalitis-associated antibodies in serum and CSF were normal. Blood amino acid and urine organic acid screening results were negative.
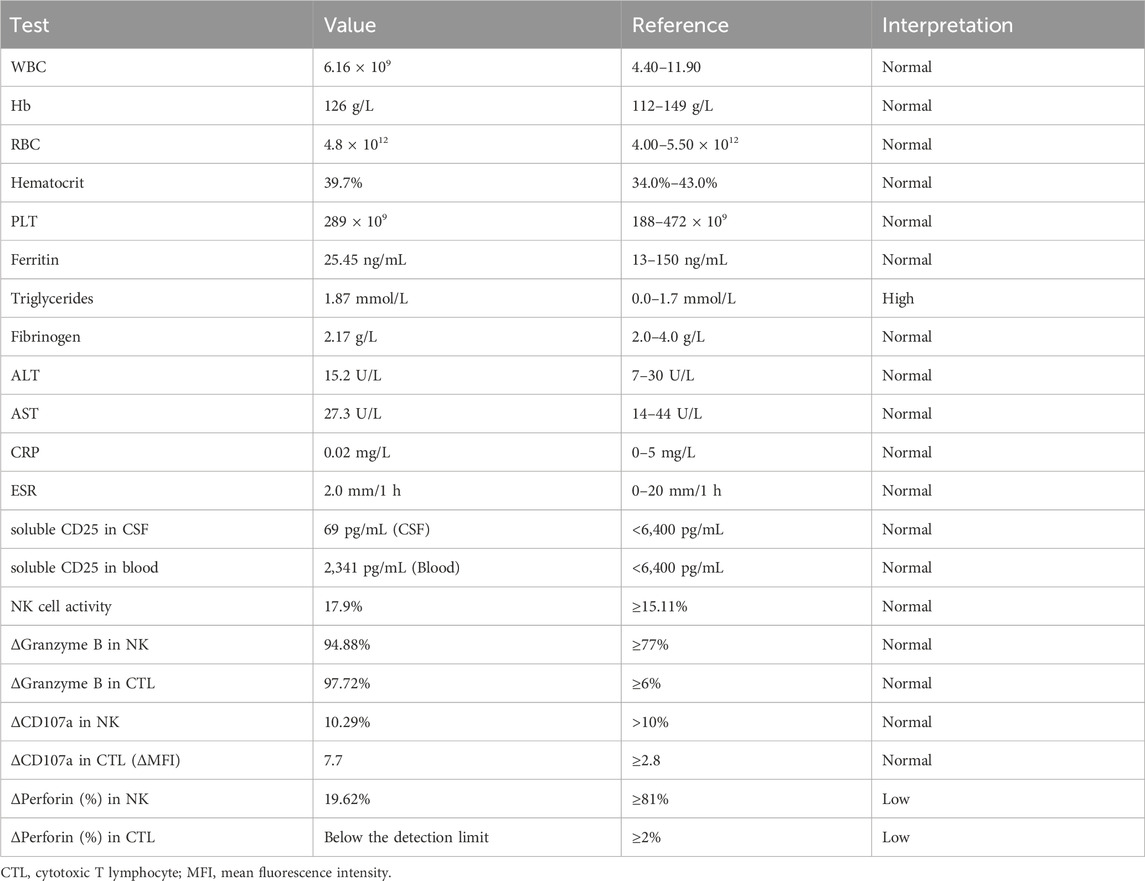
Table 1. Summary of Lab results.
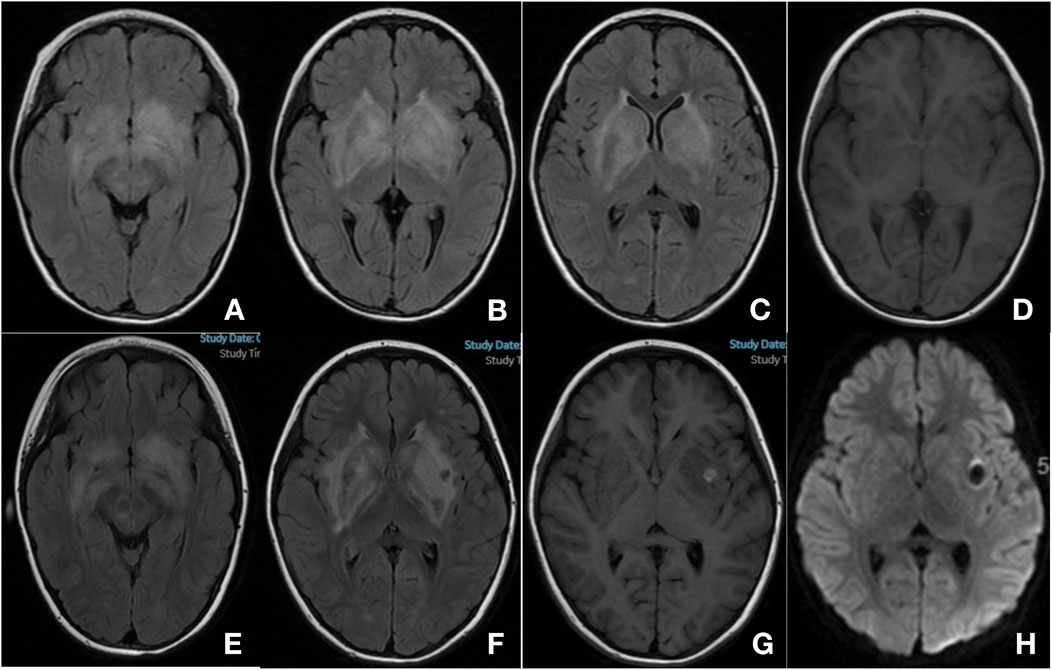
Figure 1. Cranial magnetic resonance imaging (MRI). MRI at 1 week of onset (A–D) bilateral basal ganglia, corona radiata, brainstem and anterior-posterior horns of lateral ventricle showed increased signal on T2-FLAIR (A–C) and decreased signal on T1 image (D) MRI at 1 month of onset (E–H) showed progression of the previously described lesions, and new-onset hemorrhage in the left basal ganglia [(E–F) T2-FLAIR; G: T1; H: DWI].
A diagnosis of an inflammatory disease of the central nervous system was presumed. Treatment with intravenous immunoglobulin (IVIG, 2 g/kg total) and intravenous methylprednisolone (IVMP, 4 mg/kg/d) resulted in clinical improvement. The patient’s involuntary movements were significantly reduced, and she could walk steadily, although she remained irritable. However, another brain MRI scan showed progression of the previously described lesions and a new-onset hemorrhage in the left basal ganglia (Figures 1E–H). A few days later, the result of WES returned and identified that she had a homozygous missense mutation in the PRF1 gene (NM_001083116.3): c.1349C > T (p.Thr450Met). The variant c.1349C > T was classified as pathogenic accoding to American College of Medical Genetics and Genomics (ACMG) guidelines for variant classification (PVS + PM + PP), and has been previously reported as pathogenic variant associated with familial HLH (PMIDs: 39434014, 26903364, 25297583, 25233452, etc.). Parental sample testing revealed that her mother was heterozygous and her father was wild-type. Furthermore, we selected multiple short tandem repeat (STR) sequences and used STR polymorphic linkage analysis to confirm her consanguinity with her parents and identify the maternal uniparental disomy of the patient.
Further workup of bone marrow aspiration revealed the phenomenon of cell phagocytosis. Repeat CSF examinations revealed no pleocytosis, oligoclonal bands, or intrathecal IgG synthesis. Perforin expression testing revealed that perforin is essentially absent (Table 1). NK cell activity, Granzyme B, and cd107a mobilization were normal (Table 1). The blood and CSF-soluble CD25 levels were normal (Table 1). Considering the homozygous pathogenic variant of PRF1 and supportive phenotype of the patient, a diagnosis of CNS-HLH was confirmed. The patient was transferred to the hematology department for chemotherapy. She was also a candidate for allogeneic bone marrow transplantation and HLA typing, and a search for a compatible stem cell donor was performed.
4 Discussion
HLH is a life-threatening hyperinflammatory syndrome that causes systemic inflammation and can lead to multiorgan failure and death (Griffin et al., 2020). It is characterized by edema, hepatosplenomegaly, and liver dysfunction (Griffin et al., 2020). Laboratory studies may show pancytopenia, coagulation abnormalities, hypofibrinogenemia, and hypertriglyceridemia (Griffin et al., 2020; Morimoto et al., 2016). The incidence of CNS involvement in HLH ranges from 10% to 73% (Solomon et al., 2018; Goo and Weon, 2007). HLH rarely presents with signs and symptoms that are isolated from the CNS (Benson et al., 2019). Patients with CNS-HLH are characterized by chronic inflammation restricted to the CNS that cannot be attributed to any other neuroinflammatory etiology, with no signs of systemic inflammation (Gupta et al., 2023; Benson et al., 2019). Here, we report the case of a 5-year-old girl diagnosed with CNS-HLH. Our case was unusual because the clinical presentation was restricted to the CNS and was alleviated by IVIG and steroid treatment. Uniparental disomy of the PRF1 mutation confirmed the diagnosis.
Common neurological symptoms of HLH include seizures, irritability, encephalopathy, gait ataxia, headache, hypotonia, cranial nerve palsy, and meningismus (Malik et al., 2021; Lehrer et al., 2021). Our patient presented with involuntary movement and an unsteady gait as the main manifestations, which are uncommon in CNS-HLH. Multifocal bilateral white matter abnormalities, mainly in basal ganglia and corona radiata, were seen in our case. The neuroradiologic features of CNS-HLH were heterogeneous (Benson et al., 2019; Li et al., 2019; Blincoe et al., 2020; Debinski et al., 2021). Nonspecific diffuse or multifocal white matter abnormalities were most commonly observed. The cerebellum was frequently involved, most often manifesting as white matter change, with lesions also observed anywhere in the brain (Benson et al., 2019; Li et al., 2019). MRI changes are more often bilateral in CNS-HLH than unilateral in autoimmune demyelinating disease (Lehrer et al., 2021). Hemorrhage was also observed; however, this was rare. Spinal cord involvement has also been reported but appears to be even rarer than hemorrhage (Li et al., 2019).
HLH is related to several genes, including PRF1, UNC13D, STX11, STXBP2, Rab27a, LYST, SH2D1A, BIRC4, ITK, AP3β1, MAGT1, and CD27. Mutations in PRF1, which affect perforin expression in T and NK cells, are most common in CNS-HLH (Benson et al., 2019; Blincoe et al., 2020). Our patient harbored a homozygous missense mutation, c.1349C > T (p.Thr450Met), of PRF1 gene, which was classified as pathogenic variant associated with familial HLH (Barmettler et al., 2016; Sun et al., 2014; Wang et al., 2014) and has been previously described in patients with CNS-HLH (Borda et al., 2024; Feng et al., 2020). For instance, Borda et al. (Borda et al., 2024) reported a 15-year-old male who was diagnosed as CNS-HLH and harbored compound heterozygous mutations of Thr450Met and Pro187Ser in PRF1 gene. Feng et al. (Feng et al., 2020) reported four Chinese pediatric patients who presented with neurologic manifestations as initial clinical presentation of HLH due to PRF1 mutation, and a heterozygous missense mutation Thr450Met that was present in two patients. In our patient, the absence of perforin expression in the blood also verified its pathogenicity. Maternal uniparental disomy is extremely rare, which indicates that suspicious genetic variants should not be dismissed. Timely recognition might be lifesaving, as hematopoietic stem cell transplantation might be well tolerated and effective for CNS-HLH (Gupta et al., 2023).
5 Conclusion
In conclusion, we report the case of a 5-year-old girl who presented with involuntary movements and an unsteady gait and was diagnosed with CNS-HLH due to uniparental disomy of a PRF1 mutation. Owing to its similarities with other CNS diseases, CNS-HLH remains difficult to diagnose. Exome sequencing should be considered in patients with chronic or recurrent brain inflammation who show partial or no response to conventional treatment regimens.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethical Committee of Affiliated Hospital of Qingdao University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JX: Funding acquisition, Writing – original draft. ZS: Conceptualization, Data curation, Writing – review and editing. HZ: Formal Analysis, Methodology, Writing – review and editing. CY: Investigation, Methodology, Writing – review and editing. FL: Data curation, Supervision, Writing – review and editing. ZY: Formal Analysis, Methodology, Writing – review and editing. KL: Investigation, Methodology, Writing – review and editing. YZ: Conceptualization, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Taishan Scholars Program of Shandong Province (NO. tsqn201909191) the Youth Fund of Shandong Natural Science Foundation (NO. ZR2021QH042) for the collection, analysis, and interpretation of data and in writing the manuscript.
Acknowledgments
We thank the patient and his family for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Barmettler, S., Nowak, R. J., Parker, T., and Price, C. (2016). Previously undiagnosed fatal familial haemophagocytic lymphohistiocytosis in a 24-year-old woman. BMJ Case Rep. 2016, bcr2015213698. doi:10.1136/bcr-2015-213698
Benson, L. A., Li, H., Henderson, L. A., Solomon, I. H., Soldatos, A., Murphy, J., et al. (2019). Pediatric CNS-Isolated hemophagocytic lymphohistiocytosis. Neurol. Neuroimmunol. Neuroinflamm 6 (3), e560. doi:10.1212/NXI.0000000000000560
Blincoe, A., Heeg, M., Campbell, P. K., Hines, M., Khojah, A., Klein-Gitelman, M., et al. (2020). Neuroinflammatory disease as an isolated manifestation of hemophagocytic lymphohistiocytosis. J. Clin. Immunol. 40 (6), 901–916. doi:10.1007/s10875-020-00814-6
Borda, M., Tian, H., Benitez, S., Bonheur, A., Dalvi, N., and Fraint, E. (2024). Pediatric CNS-Isolated hemophagocytic lymphohistiocytosis with brain hemorrhages: a case report. BMC Neurol. 24 (1), 404. doi:10.1186/s12883-024-03840-8
Debinski, C., Goergen, S., McLean, C., Buckland, M. E., Kumar, B., Tiller, G., et al. (2021). Exploring the intersection of Isolated-CNS hemophagocytic lymphohistiocytosis and pediatric chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. J. Child. Neurol. 36 (11), 935–942. doi:10.1177/08830738211009654
Feng, W. X., Yang, X. Y., Li, J. W., Gong, S., Wu, Y., Zhang, W. H., et al. (2020). Neurologic manifestations as initial clinical presentation of familial hemophagocytic lymphohistiocytosis Type2 due to PRF1 mutation in Chinese pediatric patients. Front. Genet. 11, 126. doi:10.3389/fgene.2020.00126
Goo, H. W., and Weon, Y. C. (2007). A spectrum of neuroradiological findings in children with haemophagocytic lymphohistiocytosis. Pediatr. Radiol. 37 (11), 1110–1117. doi:10.1007/s00247-007-0569-z
Griffin, G., Shenoi, S., and Hughes, G. C. (2020). Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best. Pract. Res. Clin. Rheumatol. 34 (4), 101515. doi:10.1016/j.berh.2020.101515
Gupta, J., Jauhari, P., Kumar, A., Gulati, S., Chakrabarty, B., Gupta, A. K., et al. (2023). Primary hemophagocytic lymphohistiocytosis with prolonged primary neurologic presentation. Pediatrics 151 (4), e2022057848. doi:10.1542/peds.2022-057848
Lehrer, H., Scigliano, E., and Chan, A. (2021). Central nervous system hemophagocytic lymphohistiocytosis (CNS-HLH) from leptomeningeal anaplastic large cell lymphoma: mild clinical neurologic syndrome with extensive multifocal white matter disease. Clin. Neuroradiol. 31 (3), 881–883. doi:10.1007/s00062-021-00998-3
Li, H., Benson, L. A., Henderson, L. A., Solomon, I. H., Kennedy, A. L., Soldatos, A., et al. (2019). Central nervous system-restricted familial hemophagocytic lymphohistiocytosis responds to hematopoietic cell transplantation. Blood Adv. 3 (4), 503–507. doi:10.1182/bloodadvances.2018027417
Malik, P., Antonini, L., Mannam, P., Aboobacker, F. N., Merve, A., Gilmour, K., et al. (2021). MRI patterns in pediatric CNS hemophagocytic lymphohistiocytosis. AJNR Am. J. Neuroradiol. 42 (11), 2077–2085. doi:10.3174/ajnr.A7292
Morimoto, A., Nakazawa, Y., and Ishii, E. (2016). Hemophagocytic lymphohistiocytosis: pathogenesis, diagnosis, and management. Pediatr. Int. 58 (9), 817–825. doi:10.1111/ped.13064
Solomon, I. H., Li, H., Benson, L. A., Henderson, L. A., Degar, B. A., Gorman, M. P., et al. (2018). Histopathologic correlates of familial hemophagocytic lymphohistiocytosis isolated to the central nervous system. J. Neuropathol. Exp. Neurol. 77 (12), 1079–1084. doi:10.1093/jnen/nly094
Sun, S., Guo, X., Zhu, Y., Yang, X., Li, Q., and Gao, J. (2014). Analysis of clinical phenotype and genetic mutations of a pedigree of familial hemophagocytic lymphohistiocytosis 31(5):570–573. doi:10.3760/cma.j.issn.1003-9406.2014.01.006
Keywords: central nervous system, hemophagocytic lympho-histiocytosis, PRF1 gene, uniparental disomy, pediatric
Citation: Xue J, Song Z, Zhao H, Yang C, Li F, Yi Z, Liu K and Zhang Y (2025) Case Report: Pediatric CNS-isolated hemophagocytic lymphohistiocytosis secondary to uniparental disomy of PRF1 mutation. Front. Genet. 16:1528844. doi: 10.3389/fgene.2025.1528844
Received: 12 December 2024; Accepted: 07 July 2025;
Published: 21 July 2025.
Edited by:
Randy Q. Cron, University of Alabama at Birmingham, United StatesReviewed by:
Magdeldin Elgizouli, University of Zurich, SwitzerlandLauren Henderson, Boston Children’s Hospital and Harvard Medical School, United States
Copyright © 2025 Xue, Song, Zhao, Yang, Li, Yi, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Zhang, emhhbmd5aW5nMDEyMjVAcWR1LmVkdS5jbg==