 Hua Liang1†
Hua Liang1† Liangping Zha
Liangping Zha- 1College of Pharmacy, Anhui University of Chinese Medicine, Hefei, China
- 2Institute of Conservation and Development of Traditional Chinese Medicine Resources, Anhui Academy of Chinese Medicine, Hefei, China
- 3MOE-Anhui Joint Collaborative Innovation Center for Quality Improvement of Anhui Genuine Chinese Medicinal Materials, Anhui University of Chinese Medicine, Hefei, China
Introduction: Atractylodes lancea (Thunb.) DC., a widely utilized herb in traditional Chinese medicine, contains sesquiterpenoids and polyacetylenes as its primary bioactive components. The WRKY gene family plays a critical role in regulating various biological processes in plants. However, the molecular mechanism underlying AlWRKY regulation of terpenoid synthesis unclear.
Methods: The AlWRKY gene family members were identified through bioinformatics approaches. Gene structures, motifs, and phylogenetic relationships were analyzed. Subsequently, their expression profiles across different geographical origins were investigated using transcriptome data. Furthermore, preliminary validation was performed via methyl jasmonate treatment and molecular docking, with a focus on the AlWRKY20 and AlWRKY37 genes.
Results: In this study, 65 AlWRKY genes with conserved domains were identified in A. lancea and classified into three groups: Group I (17 members), Group II (33 members), and Group III (15 members). Tissue-specific expression profiling revealed five rhizome-enriched AlWRKY genes (AlWRKY13, AlWRKY20, AlWRKY21, AlWRKY37, and AlWRKY49) were highly expressed in Hubei accessions compared to Jiangsu accessions, and co-expression analysis demonstrated their strong correlation with 16 AlTPS genes. Quantitative PCR (qPCR) validation confirmed the specific upregulation of AlWRKY20, AlWRKY21, AlWRKY37, and AlWRKY49 in Hubei rhizomes, consistent with the accumulation patterns of sesquiterpenes (hinesol, γ-eudesmol, and elemol). Methyl jasmonate (MeJA) induction experiments (12 h) revealed coordinated upregulation of AlWRKY20, AlWRKY37, AlTPS70, AlTPS71, concomitant with significantly increased cis-β-farnesene and α-curcumene content. Molecular docking analysis revealed strong binding affinities of AlWRKY20 to the AlTPS70/AlTPS71 promoter and of AlWRKY37 to the AlTPS70 promoter. Subcellular localization analysis demonstrated that both AlWRKY20 and AlWRKY37 are localized in the nucleus. These results suggest that AlWRKY20 and AlWRKY37 likely function as regulators of sesquiterpene biosynthesis, positively regulating cis-β-farnesene and α-curcumene production through AlTPS gene modulation.
Discussion: This study lays the groundwork for further exploration of the molecular mechanisms and functional validation of WRKY transcription factors in A. lancea.
1 Introduction
Atractylodes lancea (Thunb.) DC., a member of the Asteraceae family, constitutes a primary source of the traditional Chinese medicine Atractylodis, commonly referred to as “Cangzhu” in China. Dried rhizomes of this plant have been utilized for treating various diseases, such as spleen deficiency syndrome (SDS), across China, Japan, South Korea, and North Korea over an extended period (Peng et al., 2012; Xue et al., 2018). A. lancea contains volatile oils containing sesquiterpenes, terpenoids, polyacetylenes, monoterpenes, and steroids (Liu et al., 2016; Xu et al., 2016; Zhang et al., 2021). These components have garnered increasing scholarly interest in recent years. Prior research has identified hinesol, β-eudesmol, atractylon, and atractylodin as the main active components of the volatile oil components in A. lancea (Zhang et al., 2010; Tshering et al., 2021). Nevertheless, the composition of volatile oil in A. lancea varies across geographical regions. For example, the content of hinesol and β-eudesmol in Hubei significantly exceeds that in Jiangsu (Guo et al., 2008; Tsusaka et al., 2019; Xu et al., 2016; Zhang et al., 2023). This variation may correlate with gene regulation within terpenoid synthesis pathways (Zhang et al., 2023; Zhang et al., 2024).
Previous studies, including our own, have identified multiple genes associated with terpenoid synthesis in A. lancea. The AlAACT gene (Wu et al., 2022), along with AlDXS and AlDXR genes, were cloned in A. lancea and expressed in a prokaryotic system (Xu R. et al., 2023). Notably, AlSQS1 and AlSQS2 encode functional enzymes that catalyze the conversion of two farnesyl pyrophosphate molecules into squalene (Wu et al., 2021). Similarly, AlTPS1 and AlTPS2 utilize farnesyl pyrophosphate as a substrate to synthesize the sesquiterpenoids elemol and β-farnesene, respectively (Wu et al., 2023). Although significant progress has been made in identifying functional genes involved in terpenoid biosynthesis in A. lancea, limited research has been conducted on its transcription factors. In our preliminary transcriptome analysis of A. lancea rhizomes, AlWRKY genes exhibited co-expression patterns with AlTPS genes and correlated strongly with sesquiterpenoid content, prompting further investigation.
WRKY transcription factors (TFs) significantly contribute to the regulation of secondary metabolism in various medicinal plants. Recent studies have increasingly focused on the role of WRKY TFs in regulating terpenoid biosynthesis (Wang et al., 2021; Sun et al., 2018; Goyal et al., 2023). In Litsea cubeba, LcWRKY17 interacts with the W-box in the LcTPS42 promoter, and its overexpression markedly enhances monoterpene synthesis (Gao et al., 2023). Similarly, the PqWRKY1 transcription factor plays a pivotal role in regulating triterpene ginsenoside biosynthesis in Panax quinquefolius (Sun et al., 2013). In Artemisia annaua, WRKY1 (AaWRKY1) has been identified as a key regulator of amorpha-11-diene synthesis during terpene biosynthesis (Ma et al., 2009). Additionally, WRKY TFs involved in terpenoid biosynthesis have been identified in several medicinal plants, including Phyllostachys edulis (Zhang Z. J. et al., 2022), Carthamus tinctorius L (Li et al., 2020), Medicago sativa L (Li et al., 2023). A total of 86 HpWRKY and 63 AkWRKYS TFs have been identified in Hypericum perforatum and Akebia trifoliata (Zhou et al., 2022; Zhu et al., 2022). Similarly, 77 WRKY and 72 WRKY members have been identified in Scutellaria baicalensis Georgi and Taraxacum kok-saghyz Rodin genome (Zhang C. J. et al., 2022; Cheng et al., 2022). However, the whole-genome characterization of this gene family in A. lancea remains unexplored. Identifying WRKY genes in A. lancea will provide insights into the genetic mechanisms underlying its local adaptation and medicinal compound biosynthesis, thereby linking genomic variation with metabolomic diversity in this economically important medicinal species.
Although numerous WRKY genes have been functionally characterized in other species, a comprehensive analysis of the WRKY gene family in A. lancea is still lacking. In this study, 65 members of the WRKY gene family, designated AlWRKY, were identified in A. lancea. A systematic bioinformatics analysis was conducted, encompassing the phylogenetic relationships of AlWRKY proteins, conserved domain motifs, cis-elements and collinearity. Additionally, expression patterns of AlWRKY genes across various tissues and andmethyl jasmonate (MeJA) treatments were examined, followed by molecular docking analyses to assess AlWRKY binding potential with AlTPSs promoters. Collectively, this comprehensive study not only contribute to elucidating the mechanistic role of AlWRKY genes in modulating terpenoid biosynthesis pathways in A. lancea, but also opens avenues for metabolic engineering and sustainable harvesting strategies.
2 Materials and methods
2.1 Plant materials and treatment
Fresh plant tissues from 3-year-old wild A. lancea were collected from Yingshan (Hubei Province, China) and Nanjing (Jiangsu Province, China) for genome and transcriptome sequencing. Tissues were separated into roots, stems, and leaves, with three biological replicates per organ. These samples were immediately frozen in liquid nitrogen and stored at −80°C. Meanwhile, 3-month-old seedlings from Yingshan (Hubei, China) were treated with 200 μM methyl jasmonate (MeJA) for 0 h, 6 h, 12 h, and 24 h. Three replicates were performed for each treatment for quantitative real-time PCR (qPCR) and volatile chemical component analysis.
2.2 Identification and sequence analysis of WRKY genes in A. lancea
The hidden Markov model (HMM) file corresponding to the WRKY domain (PF03106) was downloaded from the Pfam protein family database (https://www.ebi.ac.uk/interpro/entry/pfam/#table, accessed 20 October 2023). HMMER 3.0 software (http://hmmer.org, accessed on 20 October 2023) was employed to search for WRKY genes in the A. lancea genome. Candidate WRKY protein sequences were submitted to the NCBI Conserved Domain Search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, accessed on 20 October 2023) for structural domain verification, and each sequence was manually inspected, resulting in the identification of 65 WRKY genes. Based on their chromosomal positions, these genes were designated “AlWRKYn,” where “Al” represents the Latin abbreviation for A. lancea, and “n” denotes their position on the chromosomes 1–12 from top to bottom. Physicochemical properties of the AlWRKY proteins, including isoelectric point, molecular weight, and instability index, were predicted using the ExPASy website.
2.3 Phylogenetic and sequence feature analysis
Multiple sequence alignments of AlWRKY protein sequences were conducted using MAFFT v.7 (Katoh and Standley, 2013), with manual refinement performed in BioEdit 7.0.9 (Hall, 1999). Then the AlWRKYs were then divided into different groups based on the classification of the Arabidopsis thaliana WRKY proteins sequences. Phylogenetic trees based on sequence alignment were constructed from the sequence alignments using IQ-TREE software with the maximum likelihood-based method, and the VT + R5 model was identified as the most appropriate (Nguyen et al., 2015). Finally, the phylogenetic tree was visualized and identified using the iTOL software (https://itol.embl.de/, accessed on 12 January 2024).
2.4 Conserved motifs and gene structure analysis
Conserved motifs of A. lancea WRKY protein sequences were identified using the MEME online tool (https://meme-suite.org/meme/tools/meme, accessed on 10 November 2023) (He et al., 2012), and the predicted results were visualized with TBtools. According to the gene annotation GFF files, the exon-intron structure was analyzed using the gene structure shower tool.
2.5 Analysis of cis-acting elements in promoters
The 2 kb upstream region sequences of AlWRKY genes were extracted and submitted to the PlantCARE website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 16 January 2024) for cis-element analysis. Cis-acting elements were subsequently visualized using TBtools (Lescot et al., 2002).
2.6 Chromosomal location, collinearity analysis, gene replication, and Ka/Ks analysis
Chromosomal positions of the AlWRKY genes were determined from the genomic structure annotation file and displayed using TBtools software (Wang et al., 2012). Subsequently, the Simple Ka/Ks Calculator tool was used to calculate the Ka/Ks ratios of the AlWRKY genes. Collinearity analysis was performed using two representative species—Helianthus annuus (a closely related Asteraceae member to A. lancea) to identify conserved syntenic blocks and A. thaliana (a distantly related eudicot model) to detect ancestral WRKY arrangements. Genomic data for A. thaliana and Helianthus were sourced from NCBI (Wang et al., 2010).
2.7 Gene co-expression and protein-protein interaction network analysis
Differentially expressed genes (DEGs) across various A. lancea tissues were identified using the DESeq2 v1.4.5 software, with a Q-value (Adjusted P-value) ≤ 0.05. Fragments per kilobase per million mapped reads (FPKM) of the genes were calculated using RSEM v1.3.1, and an expression atlas and gene co-expression network were generated using TBtools (Love et al., 2014). STRING (htKIN://string-db.org/) was used to construct the functional interaction network of the proteins.
2.8 Gene expression analysis using quantitative real-time PCR (qRT-qPCR)
Expression levels of terpene synthesis-related genes in A. lancea roots were assessed via qRT-PCR. The PCR primer sets are listed in Supplementary Table S1, with β-actin serving as the internal reference. Analysis was performed on an Agilent Mx3000P system (Agilent Technologies) using a 2x Realab Green PCR Fast Mixture Kit (LabLead Biotechnology, Beijing, China). Relative expression of the genes was calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
2.9 Determination of volatile chemical composition using GC–MS
Dried rhizome samples of A. lancea were pulverized and sieved through a 50-mesh screen. Precisely, 0.1 g of powder was ultrasonically extracted with 3 mL of n-hexane for 30 min, cooled to room temperature, and replenished with fresh n-hexane. Following centrifugation, the supernatant was filtered through a 0.22 μm nylon membrane and analyzed via GC–MS using a DB-5 capillary column (30 m × 0.25 mm, 0.25 μm). GC–MS parameters aligned with those used in our previous study (Yu et al., 2019).
2.10 Molecular docking analysis of AlWRKY TFs with AlTPS promoters
Potential interactions between AlWRKY TFs (AlWRKY20 and AlWRKY37) and the promoter regions of AlTPS genes (AlTPS70 and AlTPS71) were investigated using molecular docking analysis on the HDOCK server (Yan et al., 2017) with default parameters. Prior to docking simulations, the tertiary structures of AlWRKY20 and AlWRKY37 were predicted via homology modeling using the SWISS-MODEL web server (https://swissmodel.expasy.org/). Based on established criteria (Yan et al., 2017; Castillo-Zeledón et al., 2023), docking poses with scores <−200 and confidence scores >0.7 were considered to represent high-affinity binding interactions. Results were analyzed and visualized using PyMOL (version 3.1.3.1).
2.11 Cloning of AlWRKYs and subcellular localization assay
To analyze the subcellular localization of AlWRKY20 and AlWRKY37 proteins, we first predicted their localization using two widely used online tools, WoLF PSORT (https://www.genscript.com/wolf-psort.html) and CELLO (https://cello.life.nctu.edu.tw/). Subsequently, the open reading frames (ORFs) of these AlWRKYs were fused to the pBI121-GFP vector. Fusion plasmids and an empty vector were then transferred into GV3101 Agrobacterium tumefaciens in Nicotiana benthamiana leaves via Agrobacterium-mediated transformation. GFP signals were observed 2–3 days post-infection using a laser scanning microscope (LSM 900, ZEISS, Germany) and nuclei were stained with DAPI.
3 Results
3.1 Identification of the WRKY genes in A. lancea
A total of 65 WRKY genes were identified in A. lancea and designated AlWRKY01 through AlWRKY65 based on their chromosomal locations. Comprehensive data for these genes were presented, including gene ID, gene name, amino acid numbers, molecular weight (MW), isoelectric point (PI), and instability index, were compiled for these genes. The fundamental physical and chemical properties of the samples are listed in Table 1. The WRKY proteins ranged in length from 100 to 1,631 amino acids (AAs), with molecular weights spanning from 11284.44 to 183204.79 Da, and isoelectric points ranging from 5.00 to 10.02. Protein structure analyses confirmed that these selected proteins contained a complete WRKY domain or zinc finger structure.
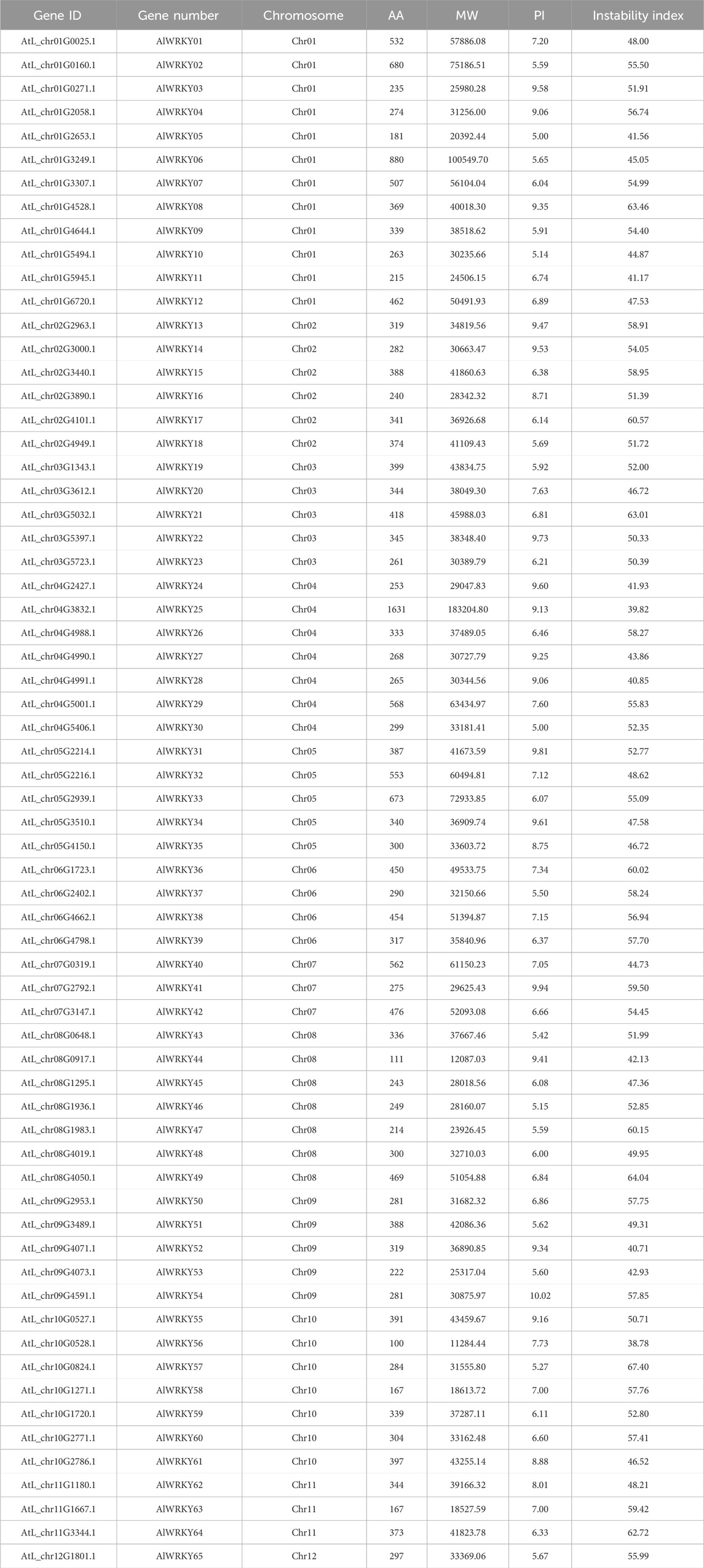
Table 1. Information on the WRKY transcription factor family of A. lancea.
3.2 Phylogenetic analysis and classification of AlWRKYs
A circular phylogenetic tree comprising 65 A. lancea WRKY genes was constructed using the maximum-likelihood (ML) method to classify and elucidate the evolutionary relationships among the AlWRKY genes. Seventy-one AtWRKY genes from A. thaliana, representing distinct classification groups, served as references (Li et al., 2019). The AlWRKY proteins were categorized into three groups: Group I (17 members), Group II (33 members), and Group III (15 members) (Figure 1; Supplementary Table S2). Group II was further divided into five subgroups—IIa (four members), IIb (seven members), IIc (seven members), IId (five members), and IIe (ten members). Consequently, all 65 AlWRKY genes were systematically classified into three primary groups. Of the 17 AlWRKY proteins in Group I, only six possessed two WRKY domains. Phylogenetic analysis of full-length WRKY proteins revealed that 11 AlWRKY genes (AlWRKY02, AlWRKY14, AlWRKY15, AlWRKY19, AlWRKY21, AlWRKY33, AlWRKY37, AlWRKY49, AlWRKY51, AlWRKY61, and AlWRKY64) with incomplete or absent C-terminal WRKY structures were classified into Group I. In contrast, Group II included 33 WRKY proteins, each harboring either a single WRKY domain or a zinc finger structure. This group was further divided into five subgroups, distinguished by variations in their zinc finger structures. Subgroup IIa contains the CX5CPV(T/A)KKKVQ motif; subgroup IIb contains the CX5CPVRKQ(H)VQ; subgroup IIc has CX4C; subgroup IId features CX5CP(K)ARKH(R)VE(Q); and subgroup IIe includes CX5CXAR(K)K(R)VE. Group III comprised 15 proteins, each with a single WRKY domain and a C2HC zinc finger structure (C-X7-CX23–31-H-X1-C) (Cheng et al., 2023). Notably, there were a few exceptions: AlWRKY02, AlWRKY14, AlWRKY15, AlWRKY37, and AlWRKY51 proteins, although structurally aligned with Group II, clustered with Group I members in the ML analysis. We hypothesized that these WRKY proteins lost their C-terminal WRKY domain during evolution; alternatively, the C-terminal region may have been inaccurately annotated.
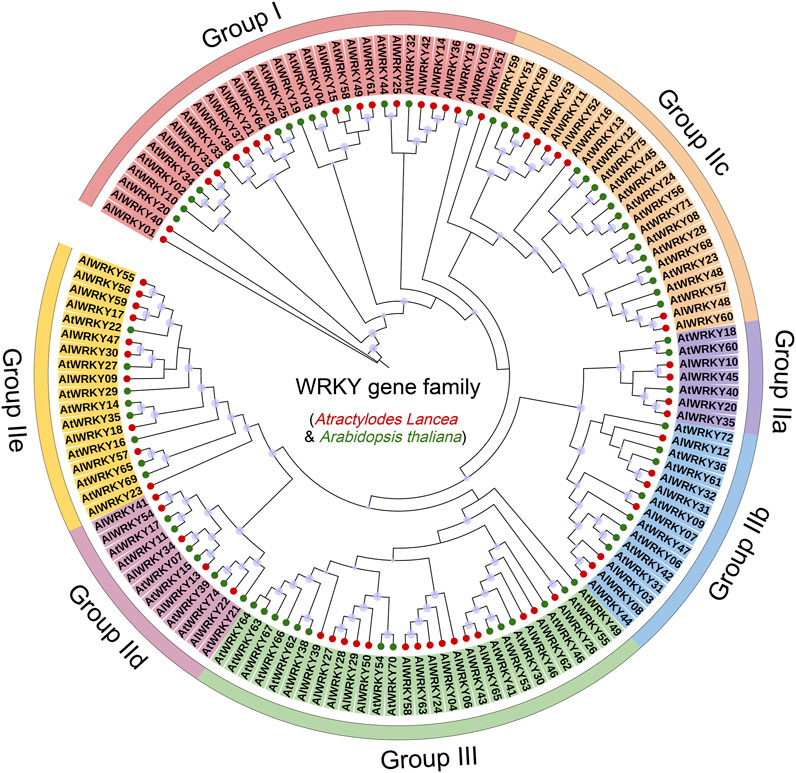
Figure 1. The maximum likelihood phylogenetic tree of WRKY protein of Arabidopsis and A. lancea. Red circles, A. lancea WRKY proteins; green circles, Arabidopsis WRKY proteins; light purple dot, a bootstrap value of ≥70, the larger the dot, the higher the value. The WRKY protein sequences are shown in Supplementary Table S2.
Through multiple sequence alignment of WRKY domains from A. lancea, the structure of the highly conserved WRKY domain heptapeptide was identified as WRKYGQK (Supplementary Figure S1). Variants including WRKYGKK, WKKYGEK, WEKYGQT, and WKKYGTK were observed in subgroups IIc, IId, and III (Song X. M. et al., 2023; Liu Q. G. et al., 2023).
3.3 AlWRKY gene chromosomal locations, duplications, and synteny analyses
The genomic distribution of AlWRKY genes was examined by mapping the approximate positions of the 65 AlWRKYs on the twelve chromosomes of A. lancea. As illustrated in Supplementary Figure S2, the distribution of AlWRKY genes across chromosomes was uneven. Specifically, chromosomes 3, 5, and 9 each harbored six genes, while chromosomes 4, 8, and 10 contained seven AlWRKY genes each. In contrast, chromosome 12 contained only a single gene, AlWRKY65. Segmental and tandem duplications are recognized as significant contributors to the expansion of plant gene families. To investigate the evolutionary regulation of the A. lancea WRKY gene family, segmental and tandem duplications of the 65 AlWRKY genes were analyzed using TBtools and MCScanX. The analysis identified four genes involved in tandem duplications: AlWRKY27 and AlWRKY28, as well as AlWRKY55 and AlWRKY56 (Figure 2A). Furthermore, the parameters Ks (synonymous substitution rate) and Ka (nonsynonymous substitution rate) for duplication events were computed using the Simple Ka/Ks Calculator available in TBtools. The Ka/Ks ratio could not be determined for only one pair of AlWRKY segmental duplications (AlWRKY15 and AlWRKY60), while the Ka/Ks ratios for the remaining 15 pairs of AlWRKY tandem duplications were found to be less than 1. This suggests that these AlWRKY gene pairs have undergone purifying selection.
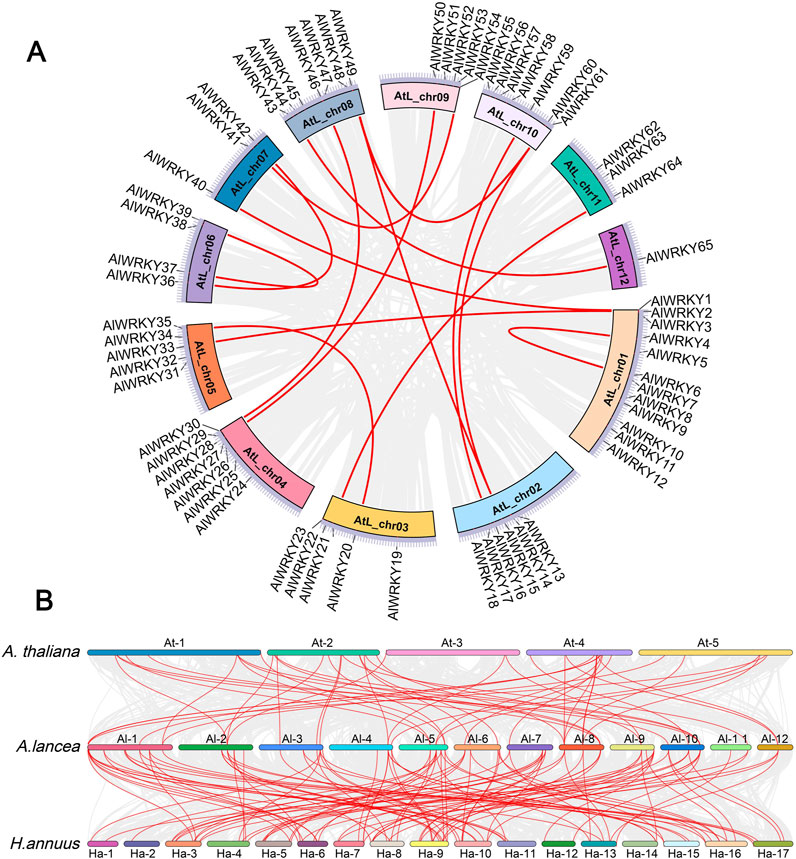
Figure 2. Collinearity of AlWRKYs. The collinearity relationships are marked with the red line. (A) The collinearity relationships of AlWRKYs within A. lancea. (B) The collinearity relationships of WRKYs between A. lancea, A. thaliana, and H. annuus.
To further explore the molecular evolutionary relationships between species, H. annuus and A. thaliana were used to perform an interspecies collinearity analysis of the A. lancea WRKY family (Figure 2B). The results showed that 46 and 107 pairs of AlWRKY genes exhibited collinear relationships with WRKY genes in A. thaliana and H. annuus, respectively. These results indicated that the AlWRKY genes exhibited higher homology with Asteraceae (H. annuus), potentially attributable to a close genetic relationship.
3.4 Gene structure and motif composition of AlWRKYs
To gain deeper insights into the critical role of exon-intron structural features in the evolution of A. lancea gene families, the structure of AlWRKY genes was obtained through Visualize Gene Structure analysis (Figure 3B). All 65 WRKY genes were interrupted by introns, with the number of exons ranging from two to eight. Specifically, subgroups Ⅱa—IIe contained two to five exons, Group I had two to eight, and Group III had three to seven.
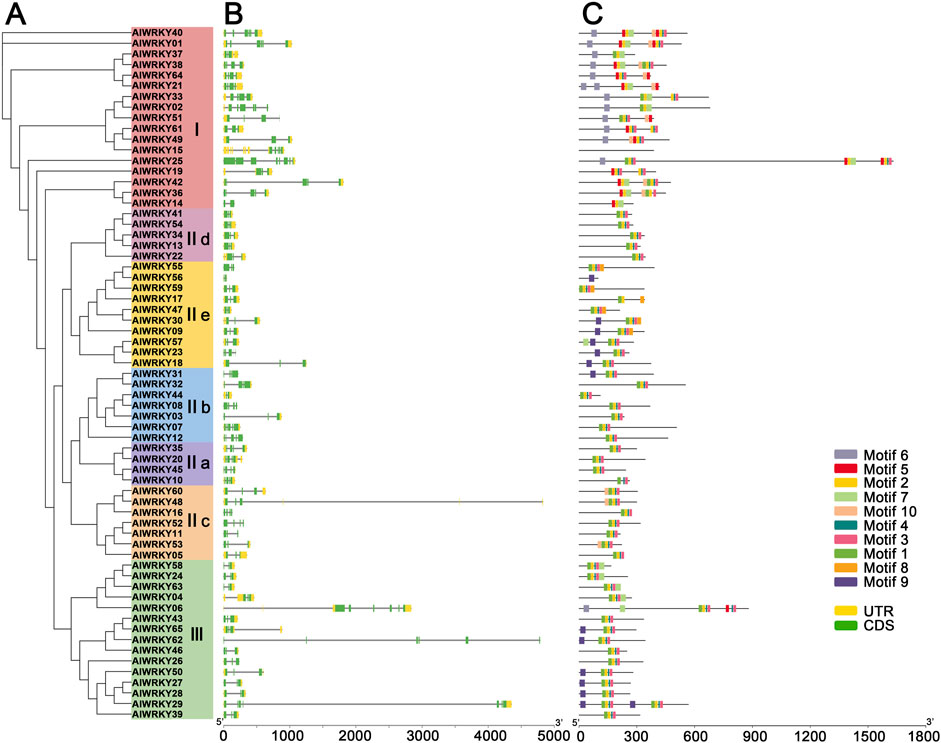
Figure 3. The grouping, gene structure and motif analyses of the AlWRKY family. (A) The maximum likelihood phylogenetic includes 65 AlWRKY proteins from A. lancea, which are clustered into 7 subgroups, sequentially designated as Ⅰ, Ⅱa, Ⅱb, Ⅱc, Ⅱd, Ⅱe and Ⅲ. (B) Exon-intron structure of A. lancea WRKY genes. Yellow boxes indicate untranslated 5′- and 3′-regions; green boxes indicate exons; black lines indicate introns. (C) The motif composition of A. lancea WRKY proteins. 10 different motifs are displayed in different colored boxes. The length of protein can be estimated using the scale at the bottom.
To further investigate the gene structure of AlWRKY, conserved motifs in AlWRKY proteins were predicted using MEME to assess functional regions (Figure 3C; Supplementary Table S3). Ten conserved motifs were identified and designated as motifs 1–10. Notably, motifs 1 and 5 included the heptapeptide sequence WRKYGQK, with most AlWRKY proteins possessing one or two WRKYGQK motifs. Genes within the same group or subgroup exhibited similar motif composition, suggesting functional conservation. For instance, motif 9 was predominantly found in subgroups IIe and III, whereas motif 10 was primarily observed in subgroups IIc and I. Additionally, motifs 5 and 6 were mainly distributed within group I.
3.5 Cis elements analysis of AlWRKY genes
Most biological processes are predicted to involve various metabolic pathways and to respond to stressful conditions. To further explore the evolution and potential functions of AlWRKY genes under abiotic stress, an analysis of the upstream 2.0 kb promoter regions of AlWRKY genes was conducted using the PlantCARE database (Figure 4; Supplementary Table S4). The promoter regions of AlWRKY genes contain seven stress-responsive elements, including the TC-rich repeats (a cis-acting element involved in defense and stress responsiveness), the LTR (cis-acting element associated with low-temperature responsiveness), the ARE (cis-acting regulatory element essential for the anaerobic induction), the GC-motif (an enhancer-like element involved in anoxic specific inducibility), the DRE element (cis-acting element related to dehydration, low-temperature and salt stresses), the MBS element (a MYB binding site associated with drought inducibility), and the WUN-motif (a wound-responsive element), among others. ARE elements were the most abundant in the promoter regions of the AlWRKY genes, accounting for 57% of the total. LTR elements were identified in 30 promoters, whereas WUN-motif elements were found in four AlWRKY genes. Notably, AlWRKY18 contains a single DRE motif. Furthermore, five hormone-responsive cis-elements were identified: those involved in MeJA, abscisic acid, gibberellin, auxin, and salicylic acid responsiveness. Among these, the CGTCA-motif elements were present in 65 AlWRKY genes, representing 46% of the abiotic stress-related elements, followed by abscisic acid responsive elements (ABREs), which accounted for 27%.
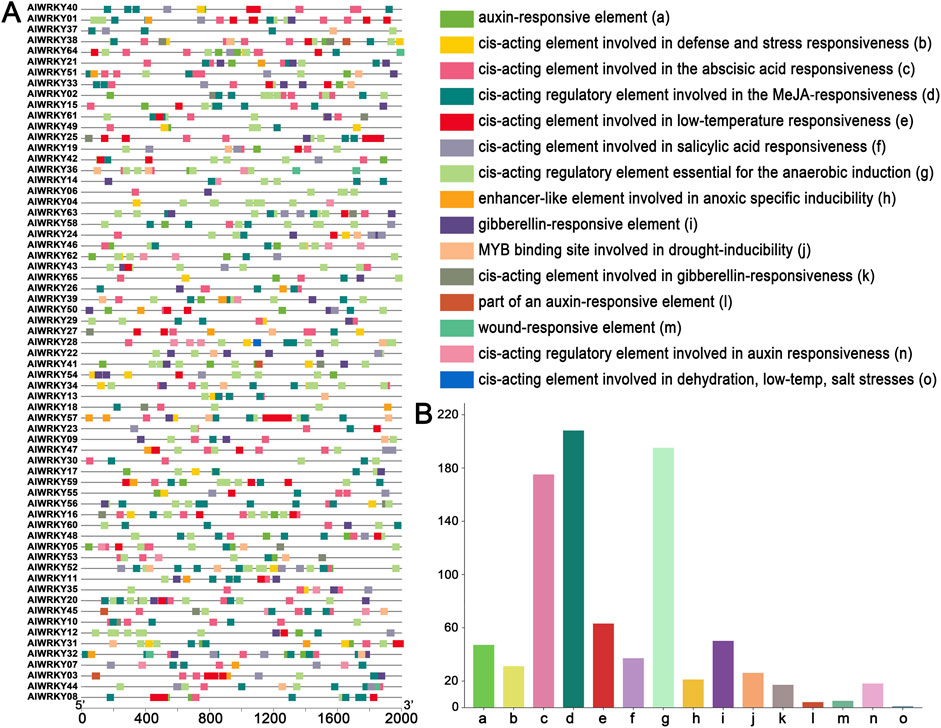
Figure 4. Cis-acting elements analysis of the promoters of A. lancea WRKY genes. (A) cis-acting element distribution. (B) Number of cis-acting element genes.
3.6 Interaction analysis of specific AlWRKY proteins
To gain a deeper insight into the biological roles and the intricate regulatory networks associated with AlWRKYs, the potential protein-protein interactions (PPIs) among these proteins were forecasted employing an orthology-based approach. The outcomes revealed that 20 of the AlWRKY proteins shared orthologous connections with their counterparts in Arabidopsis (Supplementary Figure S3). We identified five interacting proteins with high-confidence interactions (score >0.7), including VQ proteins (such as Meckel syndrome, type 1 [MKS1] and sigma factor binding protein 1 [SIB1]), which are implicated in the regulation of plant defense responses, probable NADH dehydrogenase kinase F28J15.12 and T17B22.21, and TIFY6A proteins involved in repress transcription of jasmonate-responsive genes. The AlWRKY21 protein is highly homologous to Arabidopsis WRKY33, suggesting that it may have stronger interactions with most plant defense proteins (MKS1 and SIB1). Moreover, AlWRKY48 showed a close relationship with TIFY6A, a known repressor of jasmonate responses.
3.7 Expression patterns of AlWRKY genes in different organs of two chemotypes of A. lancea
WRKY TFs play a critical role in plant growth and development, often exhibiting tissue-specific expression patterns (Song H. et al., 2023). To identify WRKY TFs associated with biosynthesis of sesquiterpenoids, particularly hinesol and β-eudesmol, the expression profiles of the 65 AlWRKY genes were characterized in the rhizome, leaves, and stem tissues of A. lancea from the Hubei and Jiangsu regions (Figure 5; Supplementary Table S5). As sesquiterpenoids are predominantly found in rhizomes, and the concentrations of hinesol and β-eudesmol are significantly higher in Hubei than in Jiangsu (Xu et al., 2016), two chemical types of A. lancea rhizomes were used for comparative analysis. Differential gene expression analysis identified 11 DEGs: AlWRKY06, AlWRKY10, AlWRK13, AlWRKY18, AlWRKY20, AlWRKY21, AlWRKY32, AlWRKY36, AlWRKY37, AlWRKY40, and AlWRKY49. Among these, AlWRKY13, AlWRKY20, AlWRKY21, AlWRKY37 and AlWRKY49 were highly expressed in rhizomes of A. lancea from Hubei, consistent with the distribution of sesquiterpenes. Therefore, these TFs may activate genes involved in terpenoid biosynthesis, thereby regulating the synthesis of related terpenoid metabolites in A. lancea.
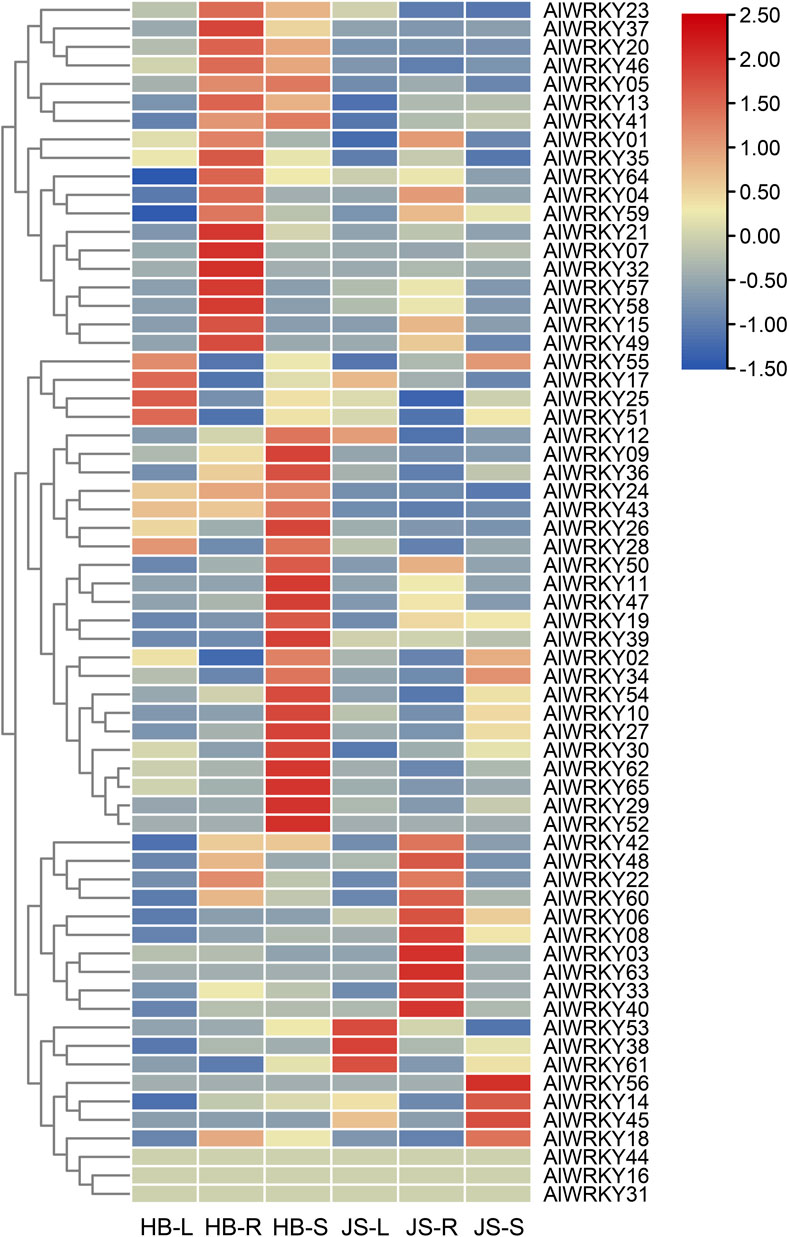
Figure 5. Heatmap of AlWRKY genes expression in leaves (L), rhizomes (R) and stem (S) of two chemotypes A. lancea. Hubei (HB), Jiangsu (JS).
3.8 Co-expression analysis of candidate AlWRKY and AlTPS genes
Co-expression network analysis was conducted to identify genes exhibiting coordinated expression patterns across various samples. A co-expression network was constructed using AlWRKY and AlTPS genes from A. lancea. The AlWRKY unigene set was combined with the expression of candidate AlTPS genes to assume that WRKY unigenes may be involved in terpenoid biosynthesis. The transcription levels of the two co-expressed gene sets displayed similar expression profiles throughout the samples. Pearson’s correlation analysis was conducted between the five differentially expressed AlWRKY genes and all 74 AlTPS genes, revealing 16 AlTPS genes that showed significant correlations (|r| > 0.8, p < 0.05) with these AlWRKY TFs. Subsequently, the 16 AlTPS genes exhibiting positive correlations with differentially expressed AlWRKY genes were selected for co-expression network analysis, as illustrated in Figure 6A. The gene family members AlWRKY21 and AlWRKY49 in A. lancea were highly correlated with AlTPS2, AlTPS32, AlTPS70 and AlTPS71 expression, whereas AlWRKY20, AlWRKY13 and AlWRKY37 were positively correlated with AlTPS1 expression. AlWRKY49 was negatively correlated with AlTPS18, AlTPS41, AlTPS54, and AlTPS57.
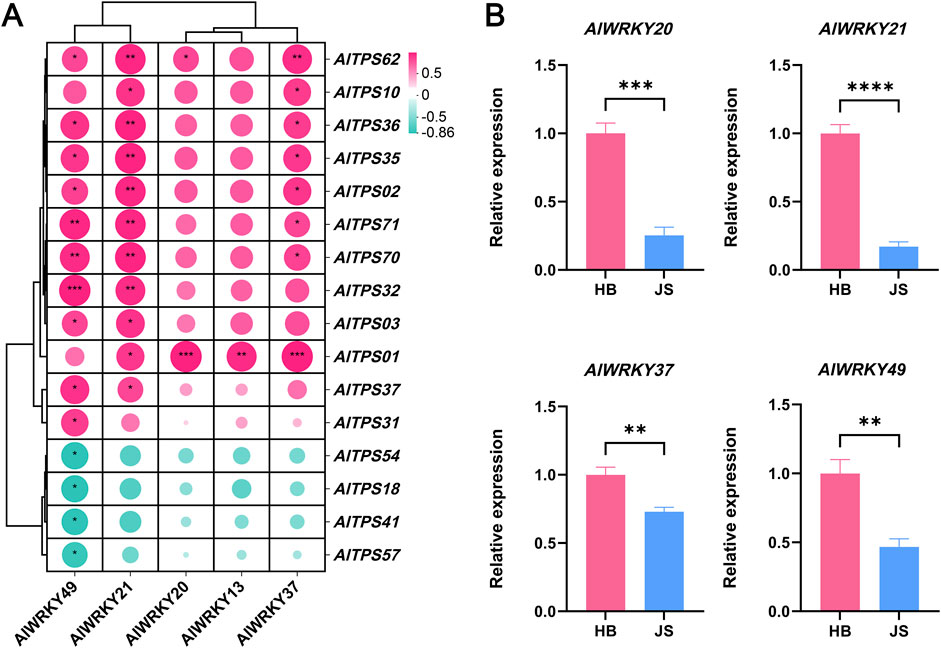
Figure 6. Correlation analysis of AlWRKYs and AlTPSs and expression profiling of candidate AlWRKYs in A. lancea. (A) AlWRKYs of A. lancea correlation with candidated AlTPS genes. (B) RT-qPCR validation of AlWRKY gene expression in Hubei (HB) and Jiangsu (JS). Statistical significance was assessed by Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001).
3.9 Comparative analysis of volatile components and AlWRKY gene expression between two chemotypes of A. lancea
GC–MS analysis of two A. lancea chemotypes identified 18 major volatile compounds, predominantly sesquiterpenoids, by comparison with the NIST mass spectral library. As summarized in Table 2, both chemotypes shared several common compounds including berkheyaradulene, β-caryophyllene, γ-elemene, humulene, β-sesquiphellandrene, cubenol, γ-eudesmol, hinesol, β-eudesmol, and atractylodin. However, distinct chemotypic differences were observed: β-elemene, β-himachalene, β-selinene, selina-3,7(11)-diene, and atractylone were absent in Yingshan populations, while zingiberene, elemol, and aristolene were undetectable in Nanjing specimens. Notably, hinesol and β-eudesmol collectively constituted more than 60% of volatile oils in Yingshan chemotypes, compared to only 3%–4% in Nanjing samples. Conversely, atractylon and atractylodin dominated the Nanjing chemotypes (exceeding 50% of total volatiles) but were nearly negligible (0%–1%) in the Yingshan populations.
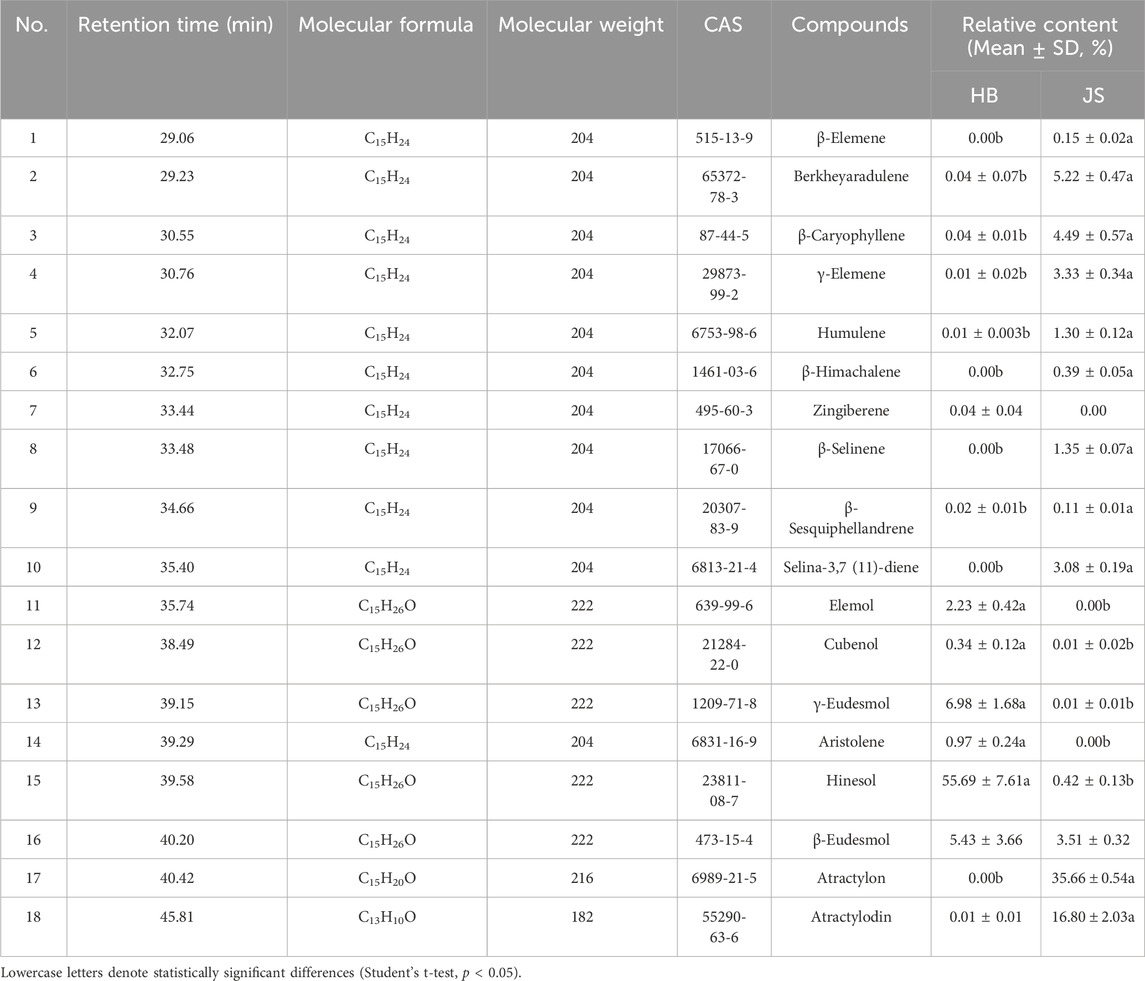
Table 2. Contents of the main components obtained from the rhizome of A. lancea.
Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed to verify the hub WRKY genes in A. lancea: AlWRKY13, AlWRKY20, AlWRKY21, AlWRKY37, and AlWRKY49. Rhizomes from two A. lancea chemotypes, sourced from Hubei and Jiangsu, were used for the qPCR validation. As shown in Figure 6B, expression levels of AlWRKY20, AlWRKY21, AlWRKY37 and AlWRKY49 genes were higher in Hubei rhizomes than in Jiangsu rhizomes, consistent with the distribution of sesquiterpenes (hinesol, γ-eudesmol, and elemol). No differential expression of AlWRKY13 was observed between the two regions (Supplementary Figure S4).
3.10 Comparative analysis of volatile components and AlWRKY/AlTPS gene expression between MeJA-treated samples
To further validate the functions of these four genes, methyl jasmonate (MeJA) treatment was applied to A. lancea plants. The correlation between their differential expression patterns and corresponding chemical composition changes was analyzed to predict their putative biological roles. GC–MS analysis of A. lancea chemotypes treated with MeJA at different time points (0 h, 6 h, 12 h, and 24 h) identified 12 major volatile compounds, predominantly sesquiterpenoids (Figure 7A), by comparison with the NIST mass spectral database. The relative content of α-grujunene and zingiberene showed a “decrease-increase-decrease” trend, whereas cis-β-farnesene, β-himachalene, α-curcumene, and β-sesquiphellandrene exhibited an “initial increase followed by a decrease” pattern. In contrast, γ-maaliene, elixene, atractylon, and atractylodin displayed an “initial decrease followed by an increase” trend. In addition, γ-elemene and humulene demonstrated a “fluctuating (increase-decrease-increase)” pattern. Significant increases in cis-β-farnesene and α-curcumene were observed at 12 h compared with the control (CK) (P < 0.05).
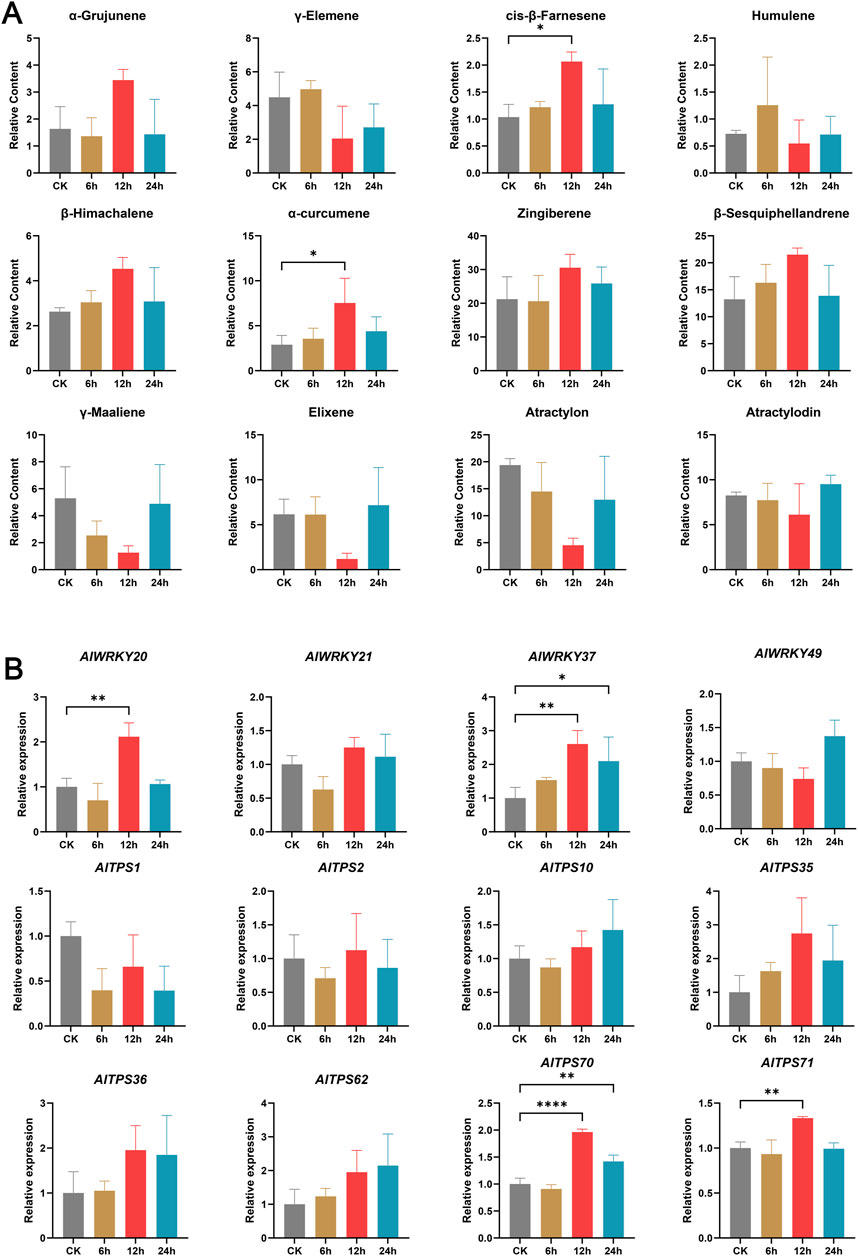
Figure 7. Dynamic changes of major components and expression patterns of AlWRKYs and AlTPSs in A. lancea rhizomes under MeJA treatment. (A) Dynamic changes of major components. (B) Expression patterns of AlWRKYs and AlTPSs. Columns and bars separately represent the means and standard deviation (n = 3), and the data was determined by One-way ANOVA (*p < 0.05, **p < 0.01, ****p < 0.0001).
RT-PCR analysis of MeJA-treated A. lancea samples revealed that expression levels of AlWRKY21 and AlWRKY49 remained unchanged across treatment durations, whereas AlWRKY20 exhibited significantly higher expression at 12 h, and AlWRKY37 showed elevated expression at both 12 h and 24 h compared with the CK (Figure 9B) (p < 0.05). Subsequently, eight TPS genes (AlTPS1, AlTPS2, AlTPS10, AlTPS35, AlTPS36, AlTPS62, AlTPS70, and AlTPS71) predicted to interact with AlWRKY20 and AlWRKY37 (Figure 6A), were examined. The results showed that the expression level of AlTPS1 at 12 h was lower than that of the CK, whereas AlTPS2, AlTPS10, AlTPS35, AlTPS36, and AlTPS62 exhibited higher expression levels than the CK at 12 h. In addition, AlTPS70 showed significantly higher expression levels than CK at both 12 and 24 h, and AlTPS71 was significantly upregulated compared to CK at 12 h (p < 0.05) (Figure 7B). Notably, the coordinated upregulation of AlWRKY20, AlWRKY37, AlTPS70, and AlTPS71 at 12 h positively correlated with cis-β-farnesene and α-curcumene accumulation, suggesting that AlWRKY20 and AlWRKY37 likely promote the biosynthesis of cis-β-farnesene and α-curcumene through their regulatory effects on AlTPS gene expression.
3.11 Molecular docking analysis of AlWRKY TFs with AlTPS promoters
Homology modeling was performed for AlWRKY20 using A0A2J6JT24.1. A as the template, yielding good model quality with 87.04% sequence identity and a GMQE score of 0.63. Similarly, AlWRKY37 was modeled using A0A118K6T8.1. A as the template, achieving 83.97% sequence identity ansd a GMQE score of 0.54. Molecular docking analysis conducted with HDOCK revealed the following: (1) The AlWRKY37-AlTPS71 promoter complex exhibited poor binding potential with a docking score greater than −200 and confidence score less than 0.7; (2) in contrast, AlWRKY20 exhibited strong binding to both the AlTPS70 and AlTPS71 promoters, while AlWRKY37 showed good binding to the AlTPS70 promoter, all with docking scores less than −200 and confidence scores exceeding 0.7, indicating high reliability of these complex models (Supplementary Table S6). Structural analysis (Figure 8) demonstrated that: (1) Both the AlWRKY20-AlTPS70 and AlWRKY20-AlTPS71 promoter complexes formed 11 hydrogen bonds, respectively, with binding interfaces predominantly involving Lys, Gln, and Ser residues; (2) the AlWRKY37-AlTPS70 promoter interaction formed 15 hydrogen bonds, with the binding interface primarily composed of Thr, Lys, and Arg residues.
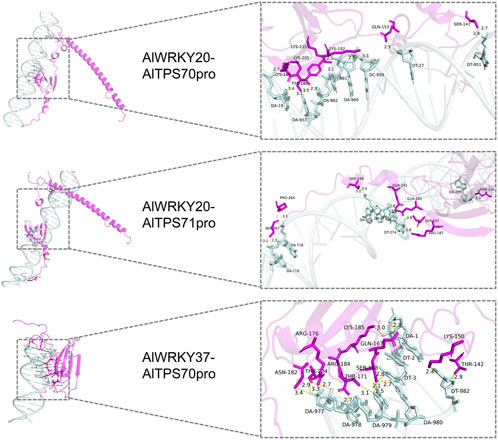
Figure 8. Molecular docking of AlWRKY transcription factors with AlTPS promoters. The interacting amino acid residues forming hydrogen bonds are depicted in magenta, nucleotide bases are shown in pale cyan, and hydrogen bond interactions are highlighted in yellow.
3.12 Cloning, bioinformatics and subcellular localization analysis of AlWRKY20 and AlWRKY37
Two candidate genes (AlWRKY20 and AlWRKY37) were successfully cloned for subsequent functional studies. The ORFs of AlWRKY20 and AlWRKY37 were 1,035 bp and 873 bp, encoding proteins of 344 and 290 AAs, respectively (Figure 9A). Each of AlWRKY20 and AlWRKY37 possesses one WRKYGQK motif, the signature sequence of WRKY transcription factors (Figure 9A). The protein tertiary structures were constructed using the structures of A. thaliana WRKY proteins as models (Figure 9B).
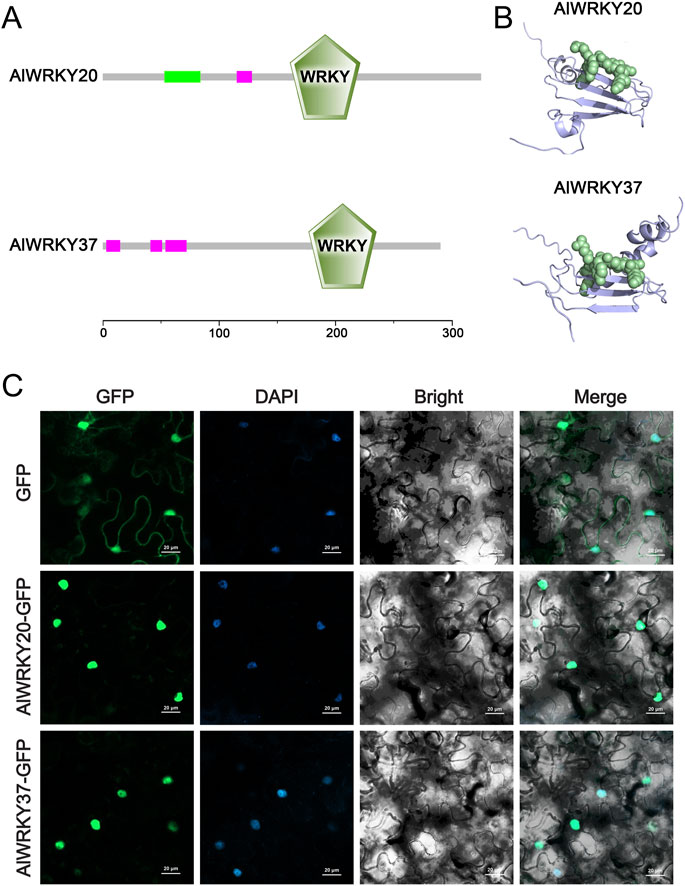
Figure 9. Conserved motif, protein tertiary structure analyses and subcellular localisation of AlWRKY20 and AlWRKY37 genes. (A) Conserved motif of these two genes. (B) Protein tertiary structure of these two genes. (C) Subcellular localisation of these two genes. GFP was used as the negative control. The green fluorescence indicates the location of fusion proteins. Scale bars = 20 µm.
Subcellular localization prediction analyses using WoLF PSORT and CELLO strongly suggested nuclear localization of AlWRKY20 and AlWRKY37 (Supplementary Table S7). To further study the subcellular localization of these two AlWRKY proteins, recombinant plasmids were constructed and transiently expressed in tobacco leaves with the empty GFP vector as a control. The results showed that the fluorescent signals of AlWRKY20-GFP and AlWRKY37-GFP fusion proteins were predominantly localized in the nucleus, consistent with previous predictions. The signal of 35S-GFP was detected in the nucleus and cytoplasm. Notably, the green fluorescence emitted from AlWRKY20-GFP and AlWRKY37-GFP fusion protein matched the blue fluorescence produced by DAPI staining of nuclei (Figure 9C), suggesting that AlWRKY20 and AlWRKY37 is a nucleus-localized protein.
4 Discussion
Recent years have seen a deepening understanding of the pharmacological effects of the major active components of A. lancea (Sun et al., 2022; Na-Bangchang et al., 2017), establishing it as one of the best-selling traditional Chinese medicines. WRKY TFs rank among the largest TF families and serve as key regulators of numerous plant processes. This family has been characterized across a wide range of model plants and medicinal plant species, including 71 AtWRKY genes in A. thaliana (Abdullah-Zawawi et al., 2021), 122 AaWRKY genes in Artemisia annua (Paolis et al., 2020), 63 DoWRKY genes in Dendrobium officinale (Wang et al., 2018), 64 CeqWRKY genes in Casuarina equisetifolia (Zhao et al., 2024) and 79 WfWRKY genes in weeping forsythia (Yang et al., 2023). In the present study, 65 AlWRKY genes were identified and the first genome-wide analysis of the WRKY gene family in A. lancea was performed. The WRKY gene family in A. lancea is relatively small compared with that of other medicinal plants. This contraction parallels observations in Salvia miltiorrhiza (Li et al., 2015), where metabolic specialization was associated with a reduction in regulatory genes.
Members of the WRKY protein family are defined by a conserved structural feature comprising the WRKYGQK motif and a zinc finger structure. Based on these attributes, the 65 AlWRKY proteins can be classified into three primary groups (I–III) and five subgroups (IIa, IIb, IIc, IId, and IIe). Group II contains the largest proportion of AlWRKY proteins. This classification is consistent with the findings for Prunus sibirica and Neolamarckia cadamba (Yu et al., 2024; Xu ZW. et al., 2023). Analysis of the core domain of AlWRKY proteins and the structure of AlWRKY genes revealed a strong correlation between motif structure and phylogenetic relationships, further supporting the classification observed in the HuWRKY gene family (Chen et al., 2022). In parallel, multiple sequence alignments of the conserved domains of 65 AlWRKY proteins identified four variants of the AlWRKY domain: WRKYGKK, WKKYGEK, WEKYGQT, and WRKFGQK. Notably, the WRKY domains of four AlWRKY proteins (AlWRKY05, AlWRKY11, AlWRKY52, and AlWRKY53) in Group IIc contained the heptapeptide variant WRKYGKK, a variation consistent with forms commonly observed in Caragana korshinskii and Asteranae (Liu J. H. et al., 2023; Guo et al., 2019). This suggests potential variability in the DNA-binding affinity associated with these variants (Chen et al., 2018). Variations in exon number, gene structure, and coding sequence (CDS) length among AlWRKY genes across different classifications, combined with the uneven distribution of gene numbers among various groups and subgroups and their irregular chromosomal localization, indicate that distinct WRKY genes may have undergone diverse evolutionary processes (Qiao et al., 2018).
In plant genomes, differentiation of WRKY genes has resulted in disparities in genes numbers within groups, with gene family expansion driven significantly by copy number expansion and tandem or local duplications (Cannon et al., 2004). Collinearity analysis identified two pairs of tandem duplications (AlWRKY27 and AlWRKY28, AlWRKY55, and AlWRKY56) and 16 pairs of segmental duplications in the A. lancea WRKY gene family, a phenomenon likely contributing substantially to the expansion of this gene family, which is consistent with the situation in Platycodon grandiflorus, Zea mays, and Cucumis sativus (Yu et al., 2024; Hu et al., 2021; Chen et al., 2020). Comparative transcriptomics revealed divergent expression patterns between tandem duplicates and non-duplicated WRKYs. AlWRKY27 displays stem-specific expression, whereas AlWRKY28 exhibits preferential expression in both leaves and stems, suggesting subfunctionalization between these paralogs. Phylogenetic similarities were observed between AlWRKY27 and AlWRKY28 as well as between AlWRKY55 and AlWRKY56. Subsequent examination of cis-acting elements in their promoters revealed involvement in defense (TC-rich repeats), low-temperature responsiveness (LTR), and anaerobic induction (ARE). Collinearity analysis with other plants. demonstrated the existence of conserved WRKY genes in A. lancea that are evolutionarily related to those in other plants, such as A. thaliana, known as orthologous genes. Therefore, the functional analysis and validation of AlWRKYs can be guided by the functions of WRKYs in other plants.
WRKY proteins act as critical regulators of secondary metabolite production in various biological processes (Eulgem et al., 1999; Ulker and Somssich, 2004). Evidence suggests that specific WRKY proteins, either independently or in synergy with other TFs, play pivotal roles in the biosynthesis of valuable natural products (Hsin et al., 2022). Terpene synthase is a fundamental enzyme in terpene biosynthesis, with transcriptional levels of TPS genes involved in terpenoid biosynthesis modulated by WRKY TFs (Wei et al., 2023). Research on A. annua has indicated that AaWRKY1 activates the expression of AaADS and AaCYP71AV1 to control the production of artemisinin (Ma et al., 2009). In L. cubeba, LcWRKY17 transactivates the promoters of the monoterpene synthase genes LcTPS42, contributing to monoterpene synthesis (Gao et al., 2023). A strong correlation has been observed between six AvWRKY unigenes and eight deduced AvTPS unigenes in Amomum villosum, indicating that these WRKY genes may play crucial roles in regulating terpene biosynthesis (He et al., 2018). In this study, MeJA induction (12 h) triggered coordinated expression of AlWRKY20/AlWRKY37 and AlTPS70/AlTPS71, correlating with elevated cis-β-farnesene and α-curcumene accumulation alongside. Molecular docking confirmed binding of both AlWRKY20 and AlWRKY37 to AlTPS promoters. TcWRKY47 from Taxus chinensis significantly upregulates taxol-biosynthesis-related genes (Zhang et al., 2018), and both AlWRKY20 and TcWRKY47 belong to Group IIa. Similarly, SmWRKY2 in S. miltiorrhiza primarily enhances tanshinone biosynthesis by activating SmCPS (Deng et al., 2019), while AlWRKY37 and SmWRKY2 are classified under Group I. These findings indicated that AlWRKY20 and AlWRKY37 may be involved in the generation of sesquiterpenes through AlTPS gene modulation. However, further validation of the specific functions of AlWRKY20 and AlWRKY37 in terpenoid metabolism in A. lancea is needed to comprehensively understand their mechanisms of action.
5 Conclusion
This study provides the first comprehensive genome-wide analysis of WRKY transcription factors in Atractylodes lancea, identifying 65 AlWRKY genes with conserved domains. Phylogenetic classification revealed three major groups: Group I (17 members), Group II (33 members), and Group III (15 members). Tissue-specific expression profiling identified five rhizome-enriched AlWRKY genes that showed chemotype-dependent expression patterns in Hubei and Jiangsu populations. Multiple lines of evidence supporting that AlWRKY20 and AlWRKY37 play the potential regulatory roles in sesquiterpene biosynthesis regulation, as evidenced by their nuclear localization, co-expression with terpene synthase genes (AlTPSs), molecular docking, and response to MeJA treatment. These results suggest that AlWRKY20 and AlWRKY37 likely function as regulators of sesquiterpene biosynthesis, positively regulating cis-β-farnesene and α-curcumene production through AlTPS gene modulation. These findings not only contribute to elucidating the molecular mechanisms underlying WRKY-mediated regulation of terpenoid biosynthesis in A. lancea but also provide valuable genetic resources for future metabolic engineering efforts aimed at improving medicinal compound production in this important traditional herb. Further studies should focus on validating these regulatory networks through genetic transformation and detailed functional analyses.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
HL: Data curation, Writing – original draft, Writing – review and editing. WL: Data curation, Writing – original draft, Writing – review and editing. ZZ: Writing – review and editing. YL: Writing – review and editing. LZ: Data curation, Funding acquisition, Methodology, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant number 82073957), Excellent Young Scholars Project of Natural Science Foundation of Anhui Province in China (grant number 2208085Y30); Science Research Project at the Universities of Anhui Province for Distinguished Young Scholars (grant number 2023AH020036); Young Elite Scientists Sponsorship Program by CACM (grant number CACM-2023-QNRC2-B23); the Key Project Foundation of Support Program for the Excellent Young Faculties in Universities of Anhui Province in China (grant number gxyqZD2022051); research Funds of Joint Research Center for Chinese Herbal Medicine of Anhui of IHM (grant number yjzx2023002) and Traditional Chinese Medicine high-level key discipline construction project of National Administration of Traditional Chinese Medicine-Science of Chinese medicinal material resources (pharmaceutical botany) (zyyzdxk-2023095); Scientific Research Team Program of Anhui Colleges and Universities (Grant no. 2022AH010036).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1551991/full#supplementary-material
References
Abdullah-Zawawi, M. R., Ahmad-Nizammuddin, N. F., Govender, N., Harun, S., Mohd-Assaad, N., and Mohamed-Hussein, Z. A. (2021). Comparative genome-wide analysis of WRKY, MADS-box and MYB transcription factor families in Arabidopsis and rice. Sci. Rep. 11 (1), 19678. doi:10.1038/s41598-021-99206-y
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., and May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4, 10. doi:10.1186/1471-2229-4-10
Castillo-Zeledón, A., Rivas-Solano, O., Villalta-Romero, F., Villalta-Romero, F., Gómez-Espinoza, O., Moreno, E., et al. (2023). The Brucella abortus two-component system response regulator BvrR binds to three DNA regulatory boxes in the upstream region of omp25. Front. Microbiol. 14, 1241143. doi:10.3389/fmicb.2023.1241143
Chen, C. B., Xie, F. F., Shah, K. R., Hua, Q. Z., Chen, J. Y., Zhang, Z. K., et al. (2022). Genome-wide identification of WRKY gene family in pitaya reveals the involvement of HmoWRKY42 in betalain biosynthesis. Int. J. Mol. Sci. 23 (18), 10568. doi:10.3390/ijms231810568
Chen, C. H., Chen, X. Q., Han, J., Lu, W. L., and Ren, Z. H. (2020). Genome-wide analysis of the WRKY gene family in the cucumber genome and transcriptome-wide identification of WRKY transcription factors that respond to biotic and abiotic stresses. BMC Plant Biol. 20 (1), 443. doi:10.1186/s12870-020-02625-8
Chen, F., Hu, Y., Vannozzi, A., Wu, K. C., Cai, H. Y., Qin, Y., et al. (2018). The WRKY transcription factor family in model plants and crops. Crit. Rev. Plant Sci. 36 (5), 311–335. doi:10.1080/07352689.2018.1441103
Cheng, L., Yu, J. J., Zhang, L. C., Yao, Y. Y., Sun, Z., Han, M., et al. (2023). Identification of SbWRKY transcription factors in Scutellaria baicalensis Georgi under drought stress and their relationship with baicalin. Agronomy 13 (10), 2564. doi:10.3390/agronomy13102564
Cheng, Y. F., Luo, J. X., Li, H., Wei, F., Zhang, Y. Q., Jiang, H. Y., et al. (2022). Identification of the WRKY Gene family and characterization of stress-responsive genes in Taraxacum kok-saghyz Rodin. Int. J. Mol. Sci. 23 (18), 10270. doi:10.3390/ijms231810270
Deng, C. P., Hao, X. H., Shi, M., Fu, R., Wang, Y., Zhang, Y., et al. (2019). Tanshinone production could be increased by the expression of SmWRKY2 in Salvia miltiorrhiza hairy roots. Plant Sci. 284, 1–8. doi:10.1016/j.plantsci.2019.03.007
Eulgem, T., Rushton, P. J., Schmelzer, E., Hahlbrock, K., and Somssich, I. E. (1999). Early nuclear events in plant defence signalling: rapid gene activation by WRKY transcription factors. EMBO J. 18 (17), 4689–4699. doi:10.1093/emboj/18.17.4689
Gao, J., Chen, Y. C., Gao, M., Wu, L. W., Zhao, Y. X., and Wang, Y. D. (2023). LcWRKY17, a WRKY transcription factor from litsea cubeba, effectively promotes monoterpene synthesis. Int. J. Mol. Sci. 24 (8), 7210. doi:10.3390/ijms24087210
Goyal, P., Devi, R., Verma, B., Hussain, S., Arora, P., Tabassum, R., et al. (2023). WRKY transcription factors: evolution, regulation, and functional diversity in plants. Protoplasma 260 (2), 331–348. doi:10.1007/s00709-022-01794-7
Guo, H. Y., Zhang, Y. T., Wang, Z., Lin, L. M., Cui, M. H., Long, Y. H., et al. (2019). Genome-wide identification of WRKY transcription factors in the Asteranae. Plants (Basel) 8 (10), 393. doi:10.3390/plants8100393
Guo, L. P., Huang, L. Q., Hu, J., and Shao, A. J. (2008). Variation rules and chemotype classification of Atractylodes Lancea essential oil based on bio-information science. Resour. Sci. 30, 770–777. doi:10.3724/SP.J.1006.2008.01484
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98. doi:10.1021/bk-1999-0734.ch008
He, H. S., Dong, Q., Shao, Y. H., Jiang, H. Y., Zhu, S. W., Cheng, B. J., et al. (2012). Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 31 (7), 1199–1217. doi:10.1007/s00299-012-1241-0
He, X. Y., Wang, H., Yang, J. F., Deng, K., and Wang, T. (2018). RNA sequencing on Amomum villosum Lour. induced by MeJA identifies the genes of WRKY and terpene synthases involved in terpene biosynthesis. Genome 61 (2), 91–102. doi:10.1139/gen-2017-0142
Hsin, K. T., Hsieh, M. C., Lee, Y. H., Lin, K. C., and Cheng, Y. S. (2022). Insight into the phylogeny and binding ability of WRKY transcription factors. Int. J. Mol. Sci. 23 (5), 2895. doi:10.3390/ijms23052895
Hu, W. J., Ren, Q. Y., Che, Y. L., Xu, G. L., and Qian, Y. X. (2021). Genome-wide identification and analysis of WRKY gene family in maize provide insights into regulatory network in response to abiotic stresses. BMC Plant Biol. 21 (1), 427. doi:10.1186/s12870-021-03206-z
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi:10.1093/molbev/mst010
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Peer, Y. Y. D., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic. Acids. Res. 30 (1), 325–327. doi:10.1093/nar/30.1.325
Li, C. L., Li, D. Q., Shao, F. J., and Lu, S. F. (2015). Molecular cloning and expression analysis of WRKY transcription factor genes in Salvia miltiorrhiza. BMC Genomics 16 (1), 200. doi:10.1186/s12864-015-1411-x
Li, H. Y., Li, L. X., ShangGuan, G. D., Jia, C., Deng, S. N., Noman, M., et al. (2020). Genome-wide identification and expression analysis of bZIP gene family in Carthamus tinctorius L. Sci. Rep. 10 (1), 15521. doi:10.1038/s41598-020-72390-z
Li, X. Y., He, F., Zhao, G. Q., Li, M. N., Long, R. C., Kang, J. M., et al. (2023). Genome-wide identification and phylogenetic and expression analyses of the PLATZ gene family in Medicago sativa L. Int. J. Mol. Sci. 24 (3), 2388. doi:10.3390/ijms24032388
Li, Y. X., Zhang, L., Zhu, P. P., Cao, Q. H., Sun, J., Li, Z. Y., et al. (2019). Genome-wide identification, characterisation and functional evaluation of WRKY genes in the sweet potato wild ancestor Ipomoea trifida (H.B.K.) G. Don. under abiotic stresses. BMC Genet. 20 (1), 90. doi:10.1186/s12863-019-0789-x
Liu, J. H., Li, G. J., Wang, R. G., Wang, G. X., and Wan, Y. Q. (2023a). Genome-wide analysis of WRKY transcription factors involved in abiotic stress and ABA response in Caragana korshinskii. Int. J. Mol. Sci. 24 (11), 9519. doi:10.3390/ijms24119519
Liu, Q., Kong, D., Luo, J., Kong, W., Guo, W., and Yang, M. (2016). Quantitative and fingerprinting analysis of Atractylodes rhizome based on gas chromatography with flame ionization detection combined with chemometrics. J. Sep. Sci. 39 (13), 2517–2526. doi:10.1002/jssc.201501275
Liu, Q. G., Wang, S. P., Wen, J. X., Chen, J. H., Sun, Y. Q., and Dong, S. J. (2023b). Genome-wide identification and analysis of the WRKY gene family and low-temperature stress response in Prunus sibirica. BMC Genomics 24 (1), 358. doi:10.1186/s12864-023-09469-0
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25 (4), 402–408. doi:10.1006/meth.2001.1262
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi:10.1186/s13059-014-0550-8
Ma, D. M., Pu, G. B., Lei, C. Y., Ma, L. Q., Wang, H. H., Guo, Y. W., et al. (2009). Isolation and characterization of AaWRKY1, an Artemisia annua transcription factor that regulates the amorpha-4,11-diene synthase gene, a key gene of artemisinin biosynthesis. Plant Cell Physiol. 50 (12), 2146–2161. doi:10.1093/pcp/pcp149
Na-Bangchang, K., Plengsuriyakarn, T., and Karbwang, J. (2017). Research and development of Atractylodes lancea (Thunb) DC. as a promising candidate for cholangiocarcinoma chemotherapeutics. Evid. Based Complement. Altern. Med. 2017, 5929234. doi:10.1155/2017/5929234
Nguyen, L. T., Schmidt, H. A., Haeseler, A. V., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi:10.1093/molbev/msu300
Paolis, D. A., Caretto, S., Quarta, A., Di Sansebastiano, G. P., Sbrocca, I., Mita, G., et al. (2020). Genome-wide identification of WRKY genes in Artemisia annua: characterization of a putative ortholog of AtWRKY40. Plants (Basel) 9 (12), 1669. doi:10.3390/plants9121669
Peng, H. S., Yuan, Q. J., Li, Q. Q., and Huang, L. Q. (2012). Molecular systematics of genus Atractylodes (Compositae, Cardueae): evidence from internal transcribed spacer (ITS) and trnL-F sequences. Int. J. Mol. Sci. 13 (11), 14623–14633. doi:10.3390/ijms131114623
Qiao, X., Yin, H., Li, L. T., Wang, R. Z., Wu, J. Y., Wu, J., et al. (2018). Different modes of gene duplication show divergent evolutionary patterns and contribute differently to the expansion of gene families involved in important fruit traits in pear (Pyrus bretschneideri). Front. Plant Sci. 9, 161. doi:10.3389/fpls.2018.00161
Song, H., Cao, Y. P., Zhao, L. G., Zhang, J. C., and Li, S. (2023a). Review: WRKY transcription factors: understanding the functional divergence. Plant Sci. 334, 111770. doi:10.1016/j.plantsci.2023.111770
Song, X. M., Hou, X. F., Zeng, Y. L., Jia, D. H., Li, Q., Gu, Y. G., et al. (2023b). Genome-wide identification and comprehensive analysis of WRKY transcription factor family in safflower during drought stress. Sci. Rep. 13 (1), 16955. doi:10.1038/s41598-023-44340-y
Sun, W. J., Zhan, J. Y., Zheng, T. R., Sun, R., Wang, T., Tang, Z. Z., et al. (2018). The jasmonate-responsive transcription factor CbWRKY24 regulates terpenoid biosynthetic genes to promote saponin biosynthesis in Conyza blinii H. Lév. J. Genet. 97 (5), 1379–1388. doi:10.1007/s12041-018-1026-52041-018-1026-5
Sun, Y. Z., Niu, Y. Y., Xu, J., Li, Y., Luo, H. M., Zhu, Y. J., et al. (2013). Discovery of WRKY transcription factors through transcriptome analysis and characterization of a novel methyl jasmonate-inducible PqWRKY1 gene from Panax quinquefolius. Plant Cell Tiss. Organ Cult. 114, 269–277. doi:10.1007/s11240-013-0323-1
Sun, Z. J., Zhang, Y. T., Peng, X., Huang, S. J., Zhou, H. H., Xu, J., et al. (2022). Diverse sesquiterpenoids and polyacetylenes from Atractylodes lancea and their anti-osteoclastogenesis activity. J. Nat. Prod. 85 (4), 866–877. doi:10.1021/acs.jnatprod.1c00997
Tshering, G., Plengsuriyakarn, T., Na-Bangchang, K., and Pimtong, W. (2021). Embryotoxicity evaluation of atractylodin and β-eudesmol using the zebrafish model. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 239, 108869. doi:10.1016/j.cbpc.2020.108869
Tsusaka, T., Makino, B., Ohsawa, R., and Ezura, H. (2019). Genetic and environmental factors influencing the contents of essential oil compounds in Atractylodes lancea. PLoS One 14 (5), e0217522. doi:10.1371/journal.pone.0217522
Ulker, B., and Somssich, I. E. (2004). WRKY transcription factors: from DNA binding towards biological function. Curr. Opin. Plant Biol. 7 (5), 491–498. doi:10.1016/j.pbi.2004.07.012
Wang, D. P., Zhang, Y. B., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinforma. 8 (1), 77–80. doi:10.1016/S1672-0229(10)60008-3
Wang, M. Z., Qiu, X. X., Pan, X., and Li, C. L. (2021). Transcriptional factor-mediated regulation of active component biosynthesis in medicinal plants. Curr. Pharm. Biotechnol. 22 (6), 848–866. doi:10.2174/1389201021666200622121809
Wang, T., Song, Z., Wei, L., and Li, L. (2018). Molecular characterization and expression analysis of WRKY family genes in Dendrobium officinale. Genes Genomics 40 (3), 265–279. doi:10.1007/s13258-017-0602-z
Wang, Y. P., Tang, H. B., Debarry, J. D., Tan, X., Li, J. P., Wang, X. Y., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic. Acids. Res. 40 (7), e49. doi:10.1093/nar/gkr1293
Wei, J. C., Yang, Y., Peng, Y., Wang, S. Y., Zhang, J., Liu, X. B., et al. (2023). Biosynthesis and the transcriptional regulation of terpenoids in tea plants (Camellia sinensis). Int. J. Mol. Sci. 24 (8), 6937. doi:10.3390/ijms24086937
Wu, J. X., Hu, J. P., Yu, H. W., Lu, J. M., Jiang, L., Liu, W. W., et al. (2023). Full-length transcriptome analysis of two chemotype and functional characterization of genes related to sesquiterpene biosynthesis in Atractylodes lancea. Int. J. Biol. Macromol. 225, 1543–1554. doi:10.1016/j.ijbiomac.2022.11.210
Wu, J. X., Liu, W. W., Lu, J. M., Xu, R., Xie, J., and Zha, L. P. (2022). Cloning, prokaryotic expression, and purification of acetyl-CoA C-acetyltransferase from Atractylodes lancea. Protein Pept. Lett. 29 (2), 156–165. doi:10.2174/0929866528666211126162838
Wu, J. X., Xu, R., Lu, J. M., Liu, W. W., Yu, H. W., Liu, M. L., et al. (2021). Molecular cloning and functional characterization of two squalene synthase genes in Atractylodes lancea. Planta. Planta 255 (1), 8. doi:10.1007/s00425-021-03797-9
Xu, K., Jiang, J. S., Feng, Z. M., Yang, Y. N., Li, L., Zang, C. X., et al. (2016). Bioactive sesquiterpenoid and polyacetylene glycosides from Atractylodes lancea. J. Nat. Prod. 79 (6), 1567–1575. doi:10.1021/acs.jnatprod.6b00066
Xu, R., Wu, J. X., Zhang, Y. Z., Lu, J., Yao, J. C., Zha, L. P., et al. (2023a). Isolation, characterisation, and expression profiling of DXS and DXR genes in Atractylodes lancea. Genome 66 (6), 150–164. doi:10.1139/gen-2022-0084
Xu, Z. W., Liu, Y. T., Fang, H. T., Wen, Y. Q., Wang, Y., Zhang, J. X., et al. (2023b). Genome-wide identification and expression analysis of WRKY gene family in Neolamarckia cadamba. Int. J. Mol. Sci. 24 (8), 7537. doi:10.3390/ijms24087537
Xue, D. H., Liu, Y. Q., Cai, Q., Liang, K., Zheng, B. Y., Li, F. X., et al. (2018). Comparison of bran-processed and crude Atractylodes Lancea effects on spleen deficiency syndrome in rats. Pharmacogn. Mag. 14 (54), 214–219. doi:10.4103/pm.pm_126_17
Yan, Y., Zhang, D., Zhou, P., Li, B. T., and Sheng-You Huang, S. Y. (2017). HDOCK: a web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 45 (W1), W365–W373. doi:10.1093/nar/gkx407
Yang, Y. L., Cushman, S. A., Wang, S. C., Wang, F., Li, Q., Liu, H. L., et al. (2023). Genome-wide investigation of the WRKY transcription factor gene family in weeping Forsythia: expression profile and cold and drought stress responses. Genetica 151 (2), 153–165. doi:10.1007/s10709-023-00184-y
Yu, D. Q., Han, X. J., Shan, T. Y., Xu, R., Hu, J., Cheng, W. X., et al. (2019). Microscopic characteristic and chemical composition analysis of three medicinal plants and surface frosts. Molecules 24 (24), 4548. doi:10.3390/molecules24244548
Yu, H. W., Li, J., Chang, X. W., Dong, N., Chen, B. W., Wang, J. T., et al. (2024). Genome-wide identification and expression profiling of the WRKY gene family reveals abiotic stress response mechanisms in Platycodon grandiflorus. Int. J. Biol. Macromol. 257 (Pt 1), 128617. doi:10.1016/j.ijbiomac.2023.128617
Zhang, C. C., Wang, H. Y., Lyu, C., Wang, Y. H., Sun, J. H., Zhang, Y., et al. (2023). Authenticating the geographic origins of Atractylodes lancea rhizome chemotypes in China through metabolite marker identification. Front. Plant Sci. 14, 1237800. doi:10.3389/fpls.2023.1237800
Zhang, C. C., Wang, S., Sun, J. H., Li, X. K., Wang, H. Y., Guo, X. Z., et al. (2024). Genome resequencing reveals the genetic basis of population evolution, local adaptation, and rewiring of the rhizome metabolome in Atractylodes lancea. Hortic. Res. 11 (8), uhae167. doi:10.1093/hr/uhae167
Zhang, C. J., Wang, W. T., Wang, D. H., Hu, S. Y., Zhang, Q., Wang, Z. Z., et al. (2022a). Genome-wide identification and characterization of the WRKY gene family in Scutellaria baicalensis Georgi under diverse abiotic stress. Int. J. Mol. Sci. 23 (8), 4225. doi:10.3390/ijms23084225
Zhang, L., Ouyang, Z., Zhao, M., Wang, P. X., and Fang, J. (2010). Simultaneous determination of atractylone, hinesol, beta-eudesmol, atrctylodin in Atractylodes lancea and hierarchical cluster analysis. Zhongguo Zhong Yao Za Zhi 35 (6), 725–728. doi:10.4268/cjcmm20100615
Zhang, M., Chen, Y., Nie, L., Jin, X. F., Liao, W. F., Zhao, S. Y., et al. (2018). Transcriptome-wide identification and screening of WRKY factors involved in the regulation of taxol biosynthesis in Taxus chinensis. Sci. Rep. 8 (1), 5197. doi:10.1038/s41598-018-23558-1
Zhang, W. J., Zhao, Z. Y., Chang, L. K., Cao, Y., Wang, S., Kang, C. Z., et al. (2021). Atractylodis rhizoma: a review of its traditional uses, phytochemistry, pharmacology, toxicology and quality control. J. Ethnopharmacol. 266, 113415. doi:10.1016/j.jep.2020.113415
Zhang, Z. J., Yu, P. Y., Hang, B., Ma, R. F., Vinod, K. K., and Ramakrishnan, M. (2022b). Genome-wide identification and expression characterization of the DoG gene family of moso bamboo (Phyllostachys edulis). BMC Genomics 23 (1), 357. doi:10.1186/s12864-022-08551-3
Zhao, X. H., Qi, G. N., Liu, J. H., Chen, K., Miao, X. X., Hussain, J. S., et al. (2024). Genome-wide identification of WRKY transcription factors in Casuarina equisetifolia and the function analysis of CeqWRKY11 in response to NaCl/NaHCO3 stresses. BMC Plant Biol. 24 (1), 376. doi:10.1186/s12870-024-04889-w
Zhou, W., Yang, S., Yang, L., Xiao, R. Y., Chen, S. Y., Wang, D. H., et al. (2022). Genome-wide identification of the Hypericum perforatum WRKY gene family implicates HpWRKY85 in drought resistance. Int. J. Mol. Sci. 24 (1), 352. doi:10.3390/ijms24010352
Keywords: Atractylodes lancea, WRKY transcription factors, genome-wide analysis, expression patterns, sesquiterpenes
Citation: Liang H, Liu W, Zhao Z, Li Y and Zha L (2025) Genome-wide identification and expression analysis of the WRKY transcription factors related to sesquiterpenes biosynthesis in Atractylodes lancea. Front. Genet. 16:1551991. doi: 10.3389/fgene.2025.1551991
Received: 27 December 2024; Accepted: 25 April 2025;
Published: 15 May 2025.
Edited by:
Zhengyi Wei, Guangxi Academy of Agricultural Sciences, ChinaReviewed by:
Yimian Ma, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaGang Zhang, Shaanxi University of Chinese Medicine, China
Zishan Ahmad, Nanjing Forestry University, China
Copyright © 2025 Liang, Liu, Zhao, Li and Zha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangping Zha, emxwX2FodGNtQDEyNi5jb20=
†These authors have contributed equally to this work