 Rongbo Lin1,2†Wenhui Zhang3†
Rongbo Lin1,2†Wenhui Zhang3† Mingwei Huang4†Yansheng Shen4Jianxiang Liao1Ping Song2Ying Qi2Jie He4Yuanxiang Xia4
Mingwei Huang4†Yansheng Shen4Jianxiang Liao1Ping Song2Ying Qi2Jie He4Yuanxiang Xia4 Jing Duan1
Jing Duan1 Yuanzhen Ye1
Yuanzhen Ye1 Qiuwei Yi5Pei Lan1Lingyu Kong1*
Qiuwei Yi5Pei Lan1Lingyu Kong1* Zhanqi Hu3*
Zhanqi Hu3*- 1Department of Neurology, Shenzhen Children’s Hospital, Shenzhen, China
- 2Department of Emergency, Shenzhen Children’s Hospital, Shenzhen, China
- 3Department of Pediatrics, Shenzhen Guangming District People’s Hospital, Shenzhen, China
- 4Aegicare (Shenzhen) Technology Co., Ltd., Shenzhen, China
- 5Department of Respiratory Medicine, Shenzhen Children’s Hospital, Shenzhen, China
Chromosomal abnormality is a significant cause of neurodevelopmental delay and congenital malformation. Only a few cases of chromosome 7 imbalances with both duplication of the distal long arm (7q) and deletion of the distal short arm (7p) have been reported without a systematic analysis of the genotype-phenotype relationship. We identify a new case of chromosome 7 imbalance with dup 7q36.3-qter and del 7pter-p22.3 and thoroughly characterize the chromosomal abnormality in the patient and related family members using a variety of genetic tests. More importantly, similar cases of 7q duplication and 7p deletion arising from parental pericentric inversion are reviewed to clarify the genotype-phenotype correlation of the disease. In summary, in cases of normal prenatal and early postnatal growth, progressive neurodevelopmental delay, intellectual disability, limited speech, and mild facial dysmorphism, the rare combination of duplication and deletion of distal ends of chromosome 7 may be suspected. Parental pericentric chromosomal inversion is likely a genetic contributor to the duplication-deletion imbalance in the offspring despite normal phenotypes in the inversion carrier, so genetic testing and counseling are recommended for better disease management and prevention.
1 Introduction
Only a few cases of chromosome 7 imbalances with both duplication of the distal long arm (7q) and deletion of the distal short arm (7p) have been reported previously, thus making the diagnosis and disease management of those patients and future ones challenging. The first two documented patients in 1978 were from a single big family and carried the exact duplication of 7q32-qter and deletion of 7pter-p22. By traditional chromosomal banding and meiotic analysis, the abnormalities in chromosome 7 were revealed as a result of meiotic crossing-over and recombination of a parental pericentric inversion, inv(7)(p22q32) (Winsor et al., 1978). Subsequent occasional reports on different cases of chromosome 7 duplication-deletion described similar distal short-arm deletion of 7pter-p22 but varying lengths of long-arm duplication (from 7q11.22-qter to 7q35-qter), which possibly explain the broad spectrum of clinical phenotypes (Ishii et al., 1997; Goodman et al., 1999; Lukusa et al., 2002; Tchirikov et al., 2010). The most recent case study identified a stillborn boy with a very large duplication that included almost the entire long arm of chromosome 7, who showed severe prenatal growth retardation, micrognathia, ventricular septal defect, aortic coarctation, bradyarrhythmia, pericardial effusion, bilateral hydronephrosis, infravesical obstruction, and cerebellar hypoplasia (Tchirikov et al., 2010). To date, no systematic analysis has been conducted on those patients to clarify the potential genotype-phenotype relationship.
In this study, we report on a new case of chromosome 7 imbalance with a small duplication of distal 7q36.3 and deletion of distal 7p22.3. Notable clinical features of our proband include progressive neurodevelopmental delay, mild facial dysmorphism, and drug-controlled seizures. A variety of genetic tests are carried out to thoroughly characterize the chromosomal abnormality in the patient and related family members. We also review similar cases of 7q duplication and 7p deletion arising from parental pericentric inversion to clarify the genotype-phenotype correlation of the disease.
2 Materials and methods
2.1 Whole genome sequencing (WGS) and whole exome sequencing (WES)
Genomic DNA was isolated from peripheral blood samples using a previously published method (Duan et al., 2022). WGS and WES were performed on the BGI DNBSEQ-T7RS sequencer platform on MGIEasy universal DNA library for the proband and his parents, respectively.
2.2 The SNP array
The CNVs were detected by Affymetrix CytoScan750K_Array (Affymetrix, Santa Clara, CA). The SNP array was performed on DNA isolated from the proband’s EDTA-treated blood (Zhang et al., 2021). Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, GmbH, Germany), then quantified, digested, ligated, fragmented, labeled, hybridized, stained, and scanned following the Affymetrix protocol.
2.3 The optical genome mapping (OGM)
Fresh peripheral blood from the patient was collected and stored. Subsequently, ultra-high molecular weight DNA was isolated, labeled, uploaded, and scanned. Labeled DNA was especially uploaded to nanochannel chips and scanned using a Saphyr instrument (Bionano Genomics). Finally, image analysis was performed using the Bionano de novo genome assembly pipeline. The genome maps obtained were aligned and assembled with Human Genome Reference Consortium GRCh37/hg19 for structural variants (SVs) detection.
2.4 The translocation breakpoints analysis
Genomic DNA was isolated from peripheral blood from the patient and two healthy boys as controls, purified, amplified, and quantified. Subsequently, the polymerase chain reaction (PCR) products were displayed with 2% agarose gel electrophoresis and analyzed using Sanger sequencing. The PCR and Sanger sequencing primers, spanning the translocation breakpoints, were CGGCGGCTTCCCCTCCTTCC (forward) and GCTGCCGCTGTCCCTCCACC (reverse).
3 Result
3.1 Clinical presentation
We present the case of a 3-year-old boy with no notable family history of disease. He was born full-term via cesarean section to a healthy mother. Typical developmental milestones were achieved before the age of 5 months. However, following the first episode of seizure, progressive neurodevelopmental delay was observed, characterized by intellectual disability, limited speech, distinctive facial features, and a middle cranial fossa meningioma (Figure 1). His facial dysmorphism included a high and prominent forehead, large fontanels with wide cranial sutures, a round face, a high-arched palate, bitemporal narrowing, an arched mouth, a short and prominent nose, and small, low-set ears (Figures 1A–C). No abnormalities were detected in the trunk or limbs (Figures 1E,F). Systematic laboratory tests ruled out ocular or congenital heart diseases. His seizures have been successfully controlled with sodium valproate oral solution (Sanofi, 3.5 mL orally, twice daily).
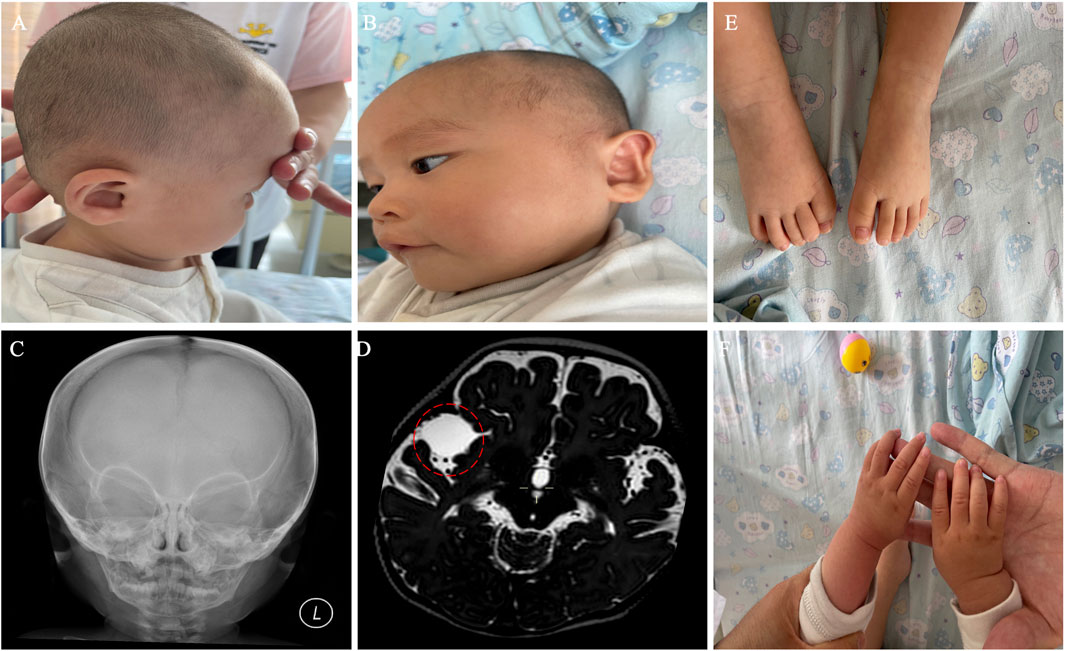
Figure 1. Clinical phenotypes of the proband with 7p22.3 deletion and 7q36.3 duplication at 10 months of age. (A,B) Dysmorphic facial features of the proband. (C) X-ray of the face. (D) Brain MRI showed a middle cranial fossa meningioma. The dashed red circle marks the approximate boundaries of the region associated with a middle cranial fossa meningioma. (E,F) normal trunk and limbs.
3.2 Chromosomal abnormalities determined through multiple tests
To identify potential genetic causes of the disease, WGS and WES were performed on the index patient and his unaffected parents, respectively, with a mean sequencing depth of 35% and 98.98% of target coding regions being sequenced 15 times or more. No single nucleotide variants or small insertions/deletions were found to match the clinical presentations described above. However, we identified two copy number variants (CNVs) in the patient, a 7p22.3 deletion (1.66 Mb) and a 7q36.3 duplication (3.87 Mb), both of which were absent in his parents (Figures 2A,B). Subsequently, both CNVs were further confirmed by single nucleotide polymorphism (SNP) array analysis using CytoScan 750K Array (Figure 2C). Due to the relatively small sizes of our identified CNVs, we did not carry out traditional karyotyping such as GTG banding, which typically has a low resolution (5–10 Mb) (Mantere et al., 2021).
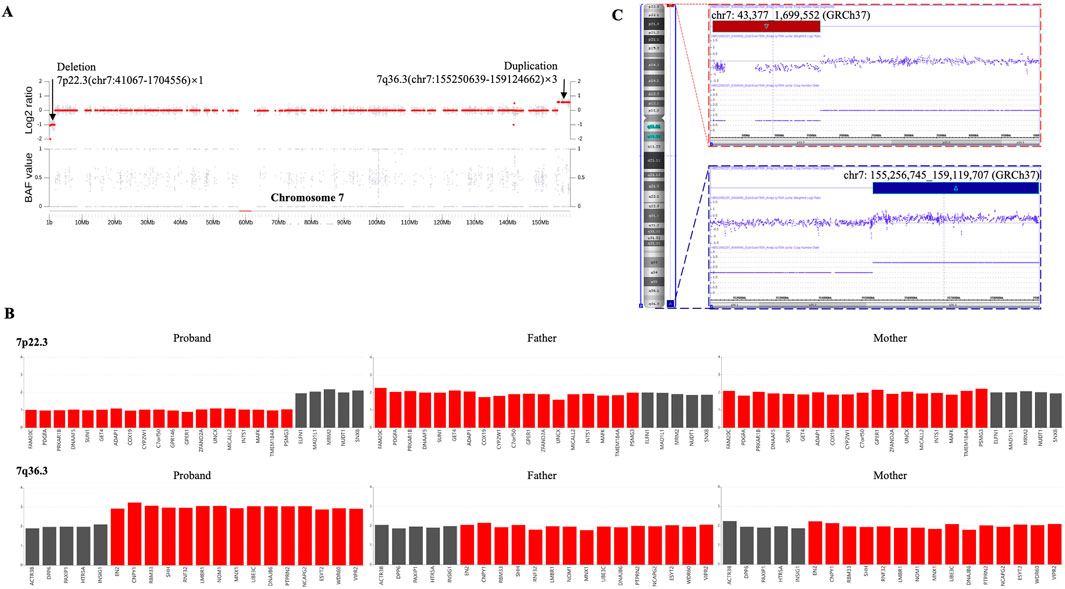
Figure 2. The heterozygous 7p22.3 deletion and 7q36.3 duplication were identified via CNV analysis utilizing next-generation sequencing (NGS) and SNP array in a male patient. (A) Scatter diagram of the CNV analysis with NGS data. This diagram shows the log2 ratio of sample/batch median (upper) and BAF value (lower) for chromosome 7, where deleted 7p22.3 and duplicated 7q36.3 are marked with chromosome coordinates (GRCh37). (B) The CNV variants from the proband are absent in both of his parents. This diagram shows the copy numbers for genes within deleted 7p22.3 (upper) and duplicated 7q36.3 (lower). Individual genes within each diagram are arranged according to their sequential locations on the chromosome. Red bars indicate genes affected by the CNV deletion/duplication in the patient, whereas black bars indicate genes unaffected. (C) CNVs were confirmed by SNP array. This diagram shows the log2 ratio of sample/batch median for chromosome 7, where deleted 7p22.3 (red) and duplicated 7q36.3 (blue) are marked with chromosome coordinates (GRCh37).
Despite the identification of both CNVs, the localization and orientation of the duplicated 7q36.3 region could not be determined by WGS or SNP array. Therefore, optical genome mapping (OGM), which images very long linear DNA libraries (median size larger than 250 kb) and allows comprehensive identification of structural variants (Mantere et al., 2021), was performed on the patient blood sample. As illustrated in Figure 3A, OGM identified an intrachromosomal fusion of two distal chromosome 7 fragments, which matches the duplicated 7q36.3 and a region next to the deleted 7p22.3 (fus(7; 7)(p22.3; q36.3)), suggesting the duplicated 7q36.3 is translocated to where the deleted 7p22.3 normally locates.
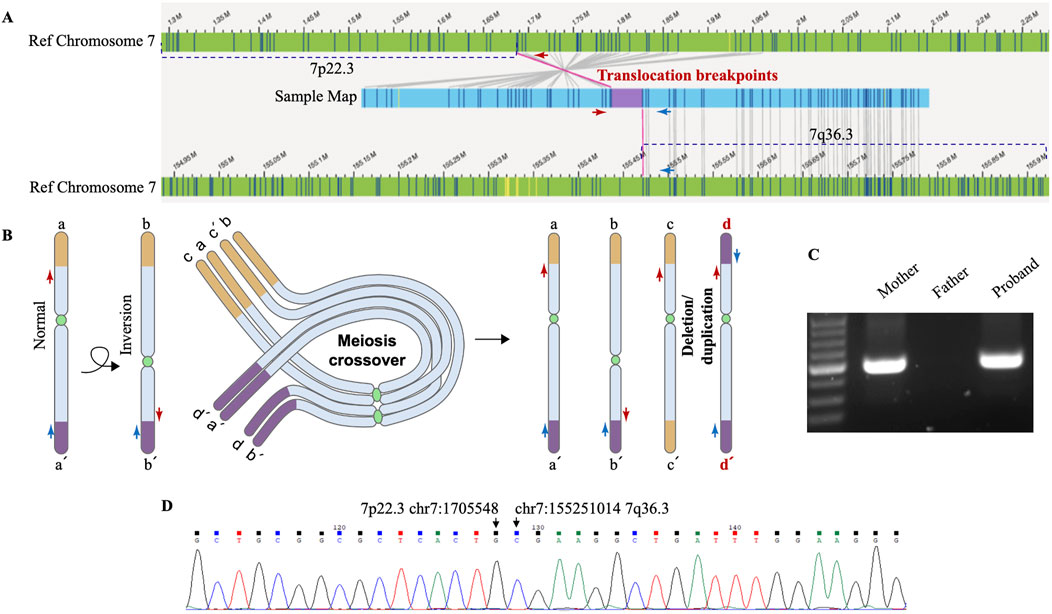
Figure 3. Genomic rearrangements disclosed by OGM and verified by Sanger sequencing. (A) OGM detects an intrachromosomal fusion of two distal chromosome 7 fragments (Sample map, blue ribbon), which matches a region next to the deleted 7p22.3 (left) and the duplicated 7q36.3 (right). The upper reference OGM map of normal Chromosome 7 (green ribbons) shows the deleted 7p22.3 (marked with a dashed line) and the neighboring region, and the lower reference map (green ribbons) shows the duplicated 7q36.3 (marked with a dashed line) and the neighboring region. Bands within the blue and green ribbons indicate specific labeling patterns of different chromosome regions via OGM. The matching of bands (gray connecting lines) suggests identical sequence compositions between compared regions. OGM does not specify the exact translocation breakpoints within the purple region. PCR primers designed to span the assumed translocation breakpoints are shown as red and blue arrows at corresponding chromosome regions. (B) Schematic representation of a proposed mechanism for how deletion/duplication imbalances of Chromosome 7 in the proband result from a balanced chromosome inversion carried by a parent. Normal chromosome (a-a’) may undergo pericentric inversion to form a balanced product (b-b’). In a heterozygous carrier, a-a’ and b-b’ can pair up in a loop configuration during meiosis, when a single crossover may lead to the production of four gametes, including a-a’ and b-b’, as well as recombinant c-c’ and d-d’ with deletion/duplication imbalances. The distal ends of the short and long arms of chromosome 7 are marked as yellow and purple, respectively. d-d’ with deleted 7p22.3 and duplicated 7q36.3 is present in the proband. Red and blue arrows indicate PCR primers for breakpoint verification and sequencing. (C) PCR results verify chromosome 7 translocation in the proband and inversion in his mother. PCR amplification was carried out using primers indicated above and DNA isolated from the proband’s and his parents’ blood samples. The results were analyzed by 2% agarose gel electrophoresis, and the size of the expected amplification product is 291 bp. (D) The exact breakpoints are identified by Sanger sequencing. DNA was gel purified from the bands shown above and then subjected to Sanger sequencing. The breakpoints in hg19 are chr7: (G)1705548 (p22.3) and chr7: (G)155251014 (q36.3), as indicated by arrows.
The translocation breakpoints are located within a region marked as purple inside the sample map of the fused fragment (Figure 3A), but OGM does not reveal a nucleotide-resolution sequence of the region due to its technological limitation. As such, PCR primers were designed to span the breakpoints so that Sanger sequencing of the PCR products could reveal the precise breakpoints. Besides, previous cases of intrachromosomal deletion-duplication imbalances of chromosome 7 all originated from a parent with chromosome 7 pericentric inversion. We proposed a mechanism for how deletion/duplication imbalances of Chromosome 7 in the proband results from a balanced chromosome inversion carried by a parent (Figure 3B). PCR results verified chromosome 7 translocation in the proband and inversion in his mother rather than his father (Figure 3C), supporting maternal balanced chromosomal changes as a source of chromosome imbalances in the proband. More importantly, Sanger sequencing of the PCR products identified the exact sequence of the breakpoints (Figure 3D), which are chr7: g.1705548 (p22.3) and chr7: g.155251014 (q36.3).
4 Discussion
To the best of our knowledge, our patient is the only documented case of chromosome 7 imbalances with distal 7p22.3 deletion and 7q36.3 duplication as a result of parental chromosome pericentric inversion and subsequent meiotic crossing-over and recombination. Table 1 compiles similar cases of chromosome 7 imbalances with both 7p deletion and 7q duplication, including those previously reported in the literature and our proband. To illustrate the potential genotype-phenotype relationship, these cases are arranged according to the size of the unbalanced regions, from large to small.
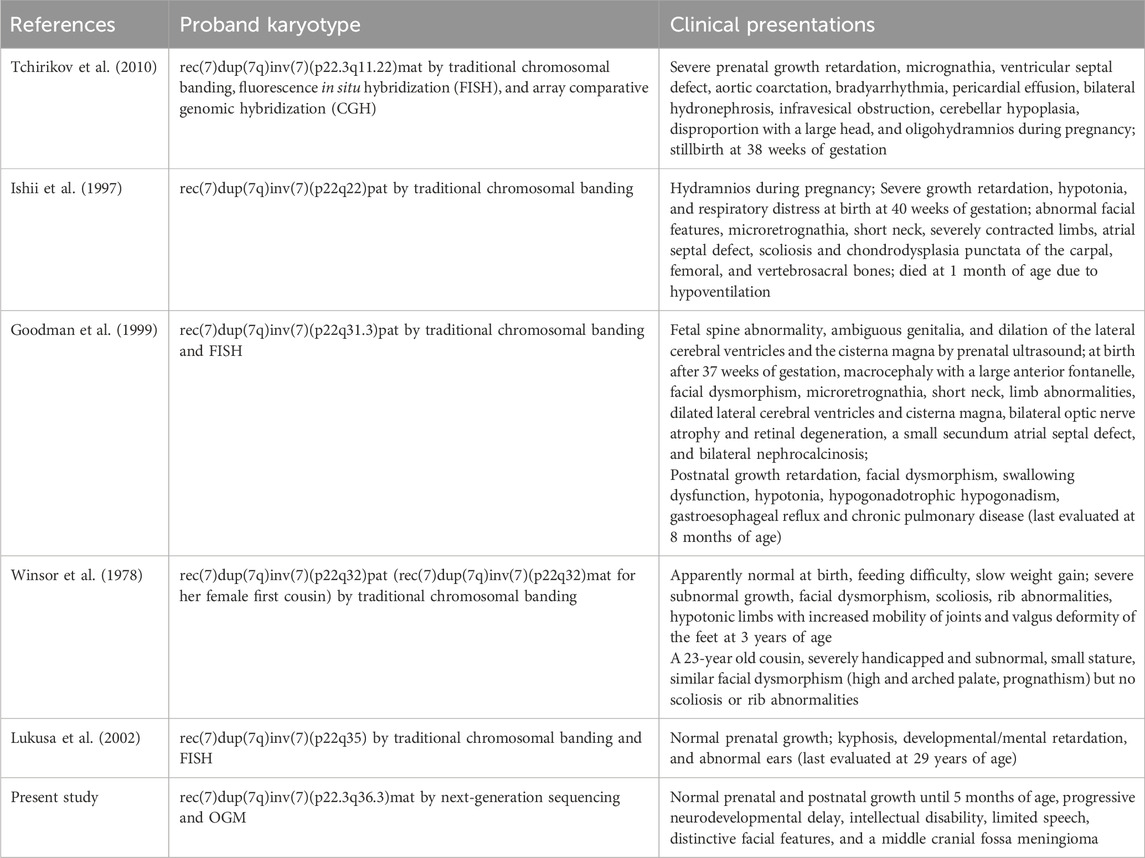
Table 1. Cases of chromosome 7 imbalances with both 7p deletion and 7q duplication.
It is interesting to note that all surveyed cases present with distal 7p22 deletion despite the fact that the patients are unrelated to each other. Since the deletion-duplication imbalances presumably arise from parental chromosome pericentric inversion, which has been confirmed in nearly all parents, we postulate that 7p22 may be a hotspot for chromosome inversion. Our study identifies the exact sequence of the breakpoints with the help of OGM and Sanger sequencing. In contrast, previous reports only utilized more traditional chromosomal banding and/or complementary methods to map the approximate locations (Table 1). Applying high-precision tools such as OGM in future cases will enable better characterization and verification of the 7p22 inversion breakpoints.
Because the compiled cases have similar distal 7p22 deletions, their different clinical presentations could mainly be attributed to differences in distal 7q duplications. Despite interpersonal variability and various genetic backgrounds, a marked proportionality between the size of 7q duplication and the survival and prenatal/postnatal growth is observed, consistent with an earlier study on patients with partial trisomy 7q (Forabosco et al., 1988). The most severe case with the largest 7q duplication, 7q11.22-qter, that included almost the entire long arm of chromosome 7, showed dramatic prenatal growth retardation, cerebellar hypoplasia, micrognathia, aortic coarctation, ventricular septal defect, hydronephrosis, and stillbirth at 38 weeks of gestation (Tchirikov et al., 2010). Slightly shorter duplication of 7q22-qter correlated with severe growth retardation at birth, abnormal facial features, microretrognathia, short neck, severely contracted limbs, atrial septal defect, scoliosis, and premature death at 1 month of age (Ishii et al., 1997). A case with decreased duplication of 7q31.3-qter presented with significant prenatal and postnatal growth retardation, facial dysmorphism, and abnormalities in multiple body systems but survived to at least 8 months of age by the last evaluation (Goodman et al., 1999). With even shorter duplication of 7q (7q32-qter and 7q35-qter), the patients were apparently normal at birth, showed seemingly fewer clinical symptoms, and survived to older ages (Winsor et al., 1978; Lukusa et al., 2002). In our case of 7q36.3 duplication, the shortest one identified so far, normal prenatal and postnatal growth was observed until 5 months of age, when progressive neurodevelopment delay and intellectual disability started to manifest. Even though the proband is only 3 years of age currently, his relatively mild symptoms suggest that he could survive to an old age. Follow-ups of the cases compiled in Table 1 and future investigation of new cases will significantly enrich the genetic and phenotypic spectrums of chromosome 7 imbalances with both 7p deletion and 7q duplication, promoting a better understanding of the genotype-phenotype correlation of the disease.
Of the two breakpoints identified in our study, chr7: g.1705548 (p22.3) maps to 145 bp upstream of the ELFN1 gene, whereas chr7: g.155251014 (q36.3) maps to the first exon of EN2. Considering the mother’s overall healthy condition, we reason that these breakpoints in a heterozygous state do not lead to detrimental disruption in gene regulation or expression. Similarly, other parental carriers of balanced chromosome 7 pericentric inversions presented with no notable clinical symptoms (Winsor et al., 1978; Ishii et al., 1997; Goodman et al., 1999; Lukusa et al., 2002; Tchirikov et al., 2010). However, under extremely rare circumstances, homozygosity in pericentric inversion of chromosome 7 may lead to significant disruption in a HOXA13 enhancer sequence and contribute to disease in a patient with hand-foot-genital syndrome (Watson et al., 2016). More importantly, such pericentric inversions carry well-known risks for duplication and/or deletion of chromosome fragments in the offspring through crossing-over and recombination during meiosis (Liehr et al., 2019; Ishii et al., 1997; Goodman et al., 1999). Therefore, mapping out the inversion breakpoints as precisely as possible in the carriers would be of critical value for genetic counseling and disease prevention. Even though the telomeric repeat sequences can render such nuanced genomic characterization challenging in our case, an integrated approach with complementary tools such as NGS, OGM, SNP array, and PCR-based Sanger sequencing would meet the need.
Notable genes within the 1.66 Mb CNV deletion of 7p22.3 include FAM20C, PDGFA, PRKAR1B, DNAAF5, SUN1, GET4, ADAP1, COX19, CYP2W1, C7orf50, GPR146, GPER1, ZFAND2A, UNCX, MICALL2, INTS1, MAFK, TMEM184A, and PSMG3 (Figure 2B), but none of them is haploinsufficient individually according to the Clinical Genome Resource (ClinGen) (Wright et al., 2024). Chromosome 7p22.3 deletions alone have been associated with neurodevelopmental delays and cardiac defects (Skvortsova et al., 2024; Mastromoro et al., 2020). More specifically, a minimal deleted region of less than 200 kb, which spans MAD1L1, FTSJ2, NUDT1, and SNX8, was delineated for cardiac anomalies (Richards et al., 2011), but a later case study on a smaller deletion involving the SNX8 gene supported SNX8 haploinsufficiency in neurodevelopment rather than cardiac development (Mastromoro et al., 2020). In our case of neurodevelopmental delay without apparent cardiac defects, the region from MAD1L1 to SNX8 was unaffected by the deletion (Figure 2B). In contrast, a recent case of 7p22.3 deletion, which includes this region from MAD1L1 to SNX8, reported both neurodevelopmental delay and heart anomalies (Skvortsova et al., 2024), supporting the likely involvement of the MAD1L1-SNX8 region in cardiac development (but cannot exclude the possible role of SNX8 in neurodevelopment). Among the genes affected by our 7p22.3 deletion, PRKAR1B is of particular interest due to its established autosomal dominant inheritance pattern in Marbach-Schaaf neurodevelopmental syndrome (Marbach et al., 2021). It is suggested that PRKAR1B haploinsufficiency is the primary mechanism underlying the intellectual disability phenotype for 7p22.3 deletion (Skvortsova et al., 2024). However, the identification of only missense variants for Marbach-Schaaf neurodevelopmental syndrome and the abundant presence of loss-of-function variants (nonsense, frameshift, and splice variants) in the population database gnomAD (https://gnomad.broadinstitute.org/gene/ENSG00000188191) indicate that PRKAR1B is not haploinsufficient, consistent with its low statistical values for haploinsufficiency index and probability of loss-of-function intolerance (https://search.clinicalgenome.org/kb/genes/HGNC:9390). Other genes with an established autosomal recessive inheritance pattern, such as FAM20C and INTS1, could not be the primary cause of disease in our case of 7p22.3 deletion either.
Similarly, the 3.87 Mb CNV duplication of 7q36.3 include EN2, CNPY1, RBM33, SHH, RNF32, LMBR1, NOM1, MNX1, UBE3C, DNAJB6, PTPRN2, NCAPG2, ESYT2, WDR60, and VIPR2 (Figure 2B), but there is no sufficient evidence for triplosensitivity for any of the individual genes by ClinGen (Wright et al., 2024). Therefore, we reason that no deletion or duplication of single genes is responsible for the disease presentation in our case, where the disturbed expression and regulation of multiple genes ultimately lead to disease. With this reasoning, our case of chromosome 7 abnormalities could be syndromic, because several body systems would be affected by this broad range of genetic disruptions, even though only minor symptoms have manifested at the current age. Future enrichment of the genetic and phenotypic spectrums of related cases, together with better molecular tools to define the chromosomal abnormalities, may contribute to identifying candidate genes for genotype-phenotype correlation.
5 Conclusion
In summary, in cases of normal prenatal and early postnatal growth, progressive neurodevelopmental delay, intellectual disability, limited speech, and mild facial dysmorphism, the rare combination of duplication and deletion of distal ends of chromosome 7 may be suspected. Parental pericentric chromosomal inversion is likely a genetic contributor to the duplication-deletion imbalance in the offspring despite normal phenotypes in the inversion carrier, so genetic testing and counseling are recommended for better disease management and prevention.
Data availability statement
The data presented in the study are deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences that are publicly accessible at https://bigd.big.ac.cn/gsa-human/browse/HRA012222.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Shenzhen Children’s Hospital (Shenzhen, China). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
RL: Conceptualization, Data curation, Writing – original draft, Writing – review and editing. WZ: Conceptualization, Writing – original draft, Writing – review and editing, Data curation. MH: Conceptualization, Writing – original draft, Writing – review and editing, Data curation. YS: Methodology, Writing – review and editing. JL: Data curation, Writing – review and editing. PS: Data curation, Writing – review and editing. YQ: Data curation, Writing – review and editing. JH: Methodology, Writing – review and editing. YX: Methodology, Writing – review and editing. JD: Data curation, Project administration, Writing – review and editing. YY: Data curation, Project administration, Writing – review and editing. QY: Data curation, Project administration, Writing – review and editing. PL: Data curation, Project administration, Writing – review and editing. LK: Conceptualization, Funding acquisition, Writing – review and editing. ZH: Conceptualization, Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Guangdong High-level Hospital Construction Fund, Shenzhen Fund for Guangdong Provincial High-Level Clinical Key Specialties (No. SZGSP012); the National Natural Science Foundation of Shenzhen Children’s Hospital (ynkt2021-zz11); the Shenzhen Science and Technology Program (JCYJ20220530160005012); Shenzhen Science and Technology Innovation Committee (JCYJ20190809112205541).
Acknowledgments
We sincerely thank the proband and his parents for participating in our study.
Conflict of interest
Authors MH, YS, JH, and YX are employed by Aegicare Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Duan, J., Ye, Y., Cao, D., Zou, D., Lu, X., Chen, L., et al. (2022). Clinical and genetic spectrum of 355 Chinese children with epilepsy: a trio-sequencing-based study. Brain 145, e43–e46. doi:10.1093/brain/awac053
Forabosco, A., Baroncini, A., Dalpra, L., Chessa, L., Giannotti, A., Maccagnani, F., et al. (1988). The phenotype of partial dup(7q) reconsidered: a report of five new cases. Clin. Genet. 34, 48–59. doi:10.1111/j.1399-0004.1988.tb02615.x
Goodman, B. K., Stone, K., Coddett, J. M., Cargile, C. B., Gurewitsch, E. D., Blakemore, K. J., et al. (1999). Molecular cytogenetic analysis and clinical findings in a newborn with prenatally diagnosed rec(7)dup(7q)inv(7)(p22q31.3)pat. Prenat. Diagn 19, 1150–1156. doi:10.1002/(sici)1097-0223(199912)19:12<1150::aid-pd733>3.0.co;2-0
Ishii, F., Fujita, H., Nagai, A., Ogihara, T., Kim, H. S., Okamoto, R., et al. (1997). Case report of rec(7)dup(7q)inv(7)(p22q22) and a review of the recombinants resulting from parental pericentric inversions on any chromosomes. Am. J. Med. Genet. 73, 290–295. doi:10.1002/(sici)1096-8628(19971219)73:3<290::aid-ajmg12>3.0.co;2-e
Liehr, T., Weise, A., Mrasek, K., Ziegler, M., Padutsch, N., Wilhelm, K., et al. (2019). Recombinant chromosomes resulting from parental pericentric inversions-two new cases and a review of the literature. Front. Genet. 10, 1165. doi:10.3389/fgene.2019.01165
Lukusa, T., Van Buggenhout, G., Devriendt, K., and Fryns, J. P. (2002). Pericentric inversion with partial 7(q35--qter) duplication and 7pter deletion: diagnosis by cytogenetic and fish analysis in a 29-year-old male patient. Genet. Couns. 13, 1–10.
Mantere, T., Neveling, K., Pebrel-Richard, C., Benoist, M., van der Zande, G., Kater-Baats, E., et al. (2021). Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 108, 1409–1422. doi:10.1016/j.ajhg.2021.05.012
Marbach, F., Stoyanov, G., Erger, F., Stratakis, C. A., Settas, N., London, E., et al. (2021). Variants in PRKAR1B cause a neurodevelopmental disorder with autism spectrum disorder, apraxia, and insensitivity to pain. Genet. Med. 23, 1465–1473. doi:10.1038/s41436-021-01152-7
Mastromoro, G., Capalbo, A., Guido, C. A., Torres, B., Fabbretti, M., Traversa, A., et al. (2020). Small 7p22.3 microdeletion: case report of Snx8 haploinsufficiency and neurological findings. Eur. J. Med. Genet. 63, 103772. doi:10.1016/j.ejmg.2019.103772
Richards, E. G., Zaveri, H. P., Wolf, V. L., Kang, S. H., and Scott, D. A. (2011). Delineation of a less than 200 kb minimal deleted region for cardiac malformations on chromosome 7p22. Am. J. Med. Genet. A 155A, 1729–1734. doi:10.1002/ajmg.a.34041
Skvortsova, L., Perfilyeva, A., Bespalova, K., Kuzovleva, Y., Kabysheva, N., and Khamdiyeva, O. (2024). 7p22.3 microdeletion: a case study of a patient with congenital heart defect, neurodevelopmental delay and epilepsy. Orphanet J. Rare Dis. 19, 301. doi:10.1186/s13023-024-03321-8
Tchirikov, M., Merinsky, A., Strohner, M., Bonin, M., Beyer, V., Haaf, T., et al. (2010). Prenatal diagnosis of a recombinant chromosome 7 resulting in trisomy 7q11.22 --qter. Am. J. Med. Genet. A 152A, 721–725. doi:10.1002/ajmg.a.33238
Watson, C. M., Crinnion, L. A., Harrison, S. M., Lascelles, C., Antanaviciute, A., Carr, I. M., et al. (2016). A chromosome 7 pericentric inversion defined at single-nucleotide resolution using diagnostic whole genome sequencing in a patient with hand-foot-genital syndrome. PLoS One 11, e0157075. doi:10.1371/journal.pone.0157075
Winsor, E. J., Palmer, C. G., Ellis, P. M., Hunter, J. L., and Ferguson-Smith, M. A. (1978). Meiotic analysis of a pericentric inversion, inv(7) (p22q32), in the father of a child with a duplication-deletion of chromosome 7. Cytogenet Cell Genet. 20, 169–184. doi:10.1159/000130849
Wright, M. W., Thaxton, C. L., Nelson, T., DiStefano, M. T., Savatt, J. M., Brush, M. H., et al. (2024). Generating clinical-grade gene-disease validity classifications through the ClinGen data platforms. Annu. Rev. Biomed. Data Sci. 7, 31–50. doi:10.1146/annurev-biodatasci-102423-112456
Keywords: neurodevelopmental delay, facial dysmorphism, chromosome 7 imbalance, dup 7q36.3-qter, del 7pter-p22.3, parental pericentric inversion, genotype-phenotype correlation, genetic counseling
Citation: Lin R, Zhang W, Huang M, Shen Y, Liao J, Song P, Qi Y, He J, Xia Y, Duan J, Ye Y, Yi Q, Lan P, Kong L and Hu Z (2025) Case Report: A rare chromosomal imbalance with dup 7q36.3-qter and del 7pter-p22.3 arising from parental pericentric inversion. Front. Genet. 16:1564711. doi: 10.3389/fgene.2025.1564711
Received: 22 January 2025; Accepted: 27 June 2025;
Published: 17 July 2025.
Edited by:
Jordi Pérez-Tur, Spanish National Research Council (CSIC), SpainReviewed by:
Thomas Liehr, Friedrich Schiller University Jena, GermanyHajkhelil Amel, University of Monastir, Tunisia
Copyright © 2025 Lin, Zhang, Huang, Shen, Liao, Song, Qi, He, Xia, Duan, Ye, Yi, Lan, Kong and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingyu Kong, NjA0MzM1MTEwQHFxLmNvbQ==; Zhanqi Hu, aHV6aGFucWkxOTgzQGFsaXl1bi5jb20=
†These authors have contributed equally to this work