 Qian Sun
Qian Sun Yaqiong Ren
Yaqiong Ren Yue Cao2
Yue Cao2 Xiaodong Wang
Xiaodong Wang Hongying Wang
Hongying Wang- 1Department of Orthopedics, Children’s Hospital of Soochow University, Suzhou, China
- 2Laboratory of Pediatric Research, Children’s Hospital of Wujiang District, Suzhou, China
- 3Department of Clinical Laboratory, Children’s Hospital of Soochow University, Suzhou, China
Background: Nail-patella syndrome (NPS) is an autosomal dominant disorder caused by the variants of the LMX1B gene, affecting several systems, including musculoskeletal, renal, and ocular systems. Despite the well-established genetic basis, the complicated relationship between genotype and phenotype still remains unclear. This study aimed to identify the genetic cause of NPS in a Chinese family and elucidate its potential contribution to the disease’s phenotypic spectrum.
Methods: Clinical data and peripheral blood samples were collected from the affected family. Whole-exome sequencing (WES) was conducted to identify potential pathogenic variants, followed by Sanger sequencing to validate the candidate variant. Bioinformatic tools were employed to predict the 3D structure alterations and pathogenicity of the variant. Wild-type and mutant LMX1B overexpression plasmids were constructed to investigate the functional consequences of the variant. Western blotting and immunofluorescence were conducted to measure the expression and localization of the protein.
Results: The proband presented with clinical manifestations, including nail malformation, patella dysplasia, restricted elbow movement, and pes planus. Both his mother and sister exhibited symptoms related to the skeletal system. WES identified a novel c.812G>C (p.R271T) variant in the affected family members. Bioinformatic analyses revealed structural modification in the protein and predicted functional impairment. Western blotting showed no significant difference in the expression level between wild-type and mutant protein. However, immunofluorescence demonstrated distinct changes in the subcellular localization of c.812G>C mutant.
Conclusion: NPS is a rare multisystem disorder with variable clinical presentations. In this family, the skeletal system was mainly involved with variations among different members. Our study identified a novel c.812G > c variant in the LMX1B gene, changing the nuclear localization of the protein.
Introduction
Nail-patella syndrome (NPS; OMIM#161200), also known as hereditary osteo-onycho-dysplasia, is a rare autosomal dominant disease characterized by nail hypoplasia, hypoplastic patella, radial head dislocation, and iliac horns, with a reported incidence of 1 in 50,000 (Sweeney, 2003). Regional epidemiological studies have reported a wide variation in prevalence (22 per million in England and 4.5 per million in the United States) (Guidera et al., 1991). The disease predominantly manifests as skeletal abnormalities, ocular complications (particularly glaucoma), and renal dysfunction. However, the involvement of the auditory, neurological, dental, and gastrointestinal systems has also been documented (Bongers E. M. et al., 2005). Despite extensive research, a clear association between genotype and phenotype among patients with NPS remains elusive. Significant phenotypic heterogeneity has been observed at the individual, intrafamilial, and interfamilial levels (Lin et al., 2008).
NPS is caused by heterozygous loss-of-function mutations in the LMX1B gene (OMIM#602575), which encodes the LIM-homeodomain transcription factor that plays a critical role in embryonic development. In patients with NPS, LMX1B mutations disrupt the normal development of the dorsal-ventral axis in limbs, leading to defects in dorsal structures, such as nails and patella, and causing musculoskeletal symptoms. The variants also affect the expression of collagen IV chains and podocyte differentiation, leading to glomerular basement membrane abnormalities and renal diseases (Chen et al., 1998). These variants lead to altered transcriptional activity and affect the DNA-binding ability of the LMX1B protein, finally resulting in multisystem symptoms in patients with NPS (Dreyer et al., 1998; Dreyer, 2000).
In this study, we reported a Chinese family whose 3 members had NPS, including the proband, his younger sister, and his mother. We also identified a novel c.812G>C missense variant in the LMX1B gene, which has never been reported in the literature. We expand the genotypic and phenotypic spectrum of NPS and provide a better understanding of genotype-phenotype correlations in this disease.
Materials and methods
Subjects and clinical assessment
This two-generation family from eastern China comprised three affected individuals (Figures 3A, I–2, II-2, and II-2). A thorough assessment of their natural history and physical condition was conducted on all family members. The affected members underwent additional radiological and laboratory examinations. X-ray was conducted to assess their skeletal deformities, while renal function was measured through urinalysis and hematologic tests. Additionally, intraocular pressure measurement using the non-contact tonometer method was conducted. Peripheral blood samples of all members of the family were obtained for further analysis. This study was reviewed and approved by the Institutional Review Board of the Children’s Hospital of Soochow University (approval number: 2024CS056). Written informed consent was obtained from all participants or their legal guardians, authorizing both the standardized collection of clinical records and peripheral blood samples. The subsequent use of genetic testing and scientific dissemination was conducted in compliance with the Declaration of Helsinki.
Whole-exome sequencing
Whole-exome sequencing (WES) was conducted using the following protocol. Firstly, DNA was extracted from peripheral blood using a Qiagen DNA Blood Midi/Mini kit (Qiagen GmbH, Hilden) following the manufacturer’s protocol. Then, DNA was interrupted to approximately 200bp and then was end-repaired. Subsequently, the DNA fragments were ligated, and fragments with nearly 320bp were collected. After PCR amplification, DNA fragments were hybridized and captured using Berry’s NanoWES Human Exome V1.0 (Berry Genomics) following the manufacturer’s protocol. Next, the libraries were measured using qPCR, and size distribution was determined using an Agilent Bioanalyzer 2,100 (Agilent Technologies). Finally, the Novaseq6000 platform (Illumina), with 150 bp pair-end sequencing mode, was employed for sequencing the captured exome library. The sequencing reads were aligned to the human reference genome (hg38/GRCh38) utilizing Burrows–Wheeler Aligner tool. PCR duplicates were deleted using Picard v1.57 (http://picard.sourceforge.net/). Verita Trekker® Variants Detection System by Berry Genomics and the third-party software GATK (https://software.broadinstitute.org/gatk/) were employed for variant calling. Variant annotation and interpretation were conducted using ANNOVAR (Wang et al., 2010) and the Variants Annotation Interpretation System. Human population databases, such as 1,000 Genome Project (http://browser.1000genomes.org), were employed to exclude high-frequency variants.
Variants with minor allele frequency (MAF) ≥1% in public databases or variants existing in in-house controls were excluded. Only variants in exonic regions or with potential splicing effects were retained. In silico tools, including REVEL, SIFT, Polyphen2, Variant Taster, CADD, and PROVEAN, were employed to predict the pathogenicity of the remaining variants. Disease and phenotype databases, such as OMIM (http://www.omim.org), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), and HGMD (http://www.hgmd.org), were utilized for further assessment. In trio analysis, the variants were filtered to retain those that fit the expected inheritance pattern for the disease. The candidate variants were then classified following the ACMG guidelines (Richards et al., 2015) to determine their pathogenicity. Thereafter, further bioinformatics analysis and Sanger sequencing were employed.
Bioinformatics analysis
The three-dimensional structure of the wild-type LMX1B protein was obtained from the AlphaFold Protein Structure Database to analyze the potential functional effect of the identified genetic variants. The three-dimensional conformation of both wild-type and mutant LMX1B proteins and their target DNA sequence FLAT-E (5′-TAATTA-3′) (German et al., 1992) were modeled using AlphaFold 3. Next, molecular docking simulations were conducted using PyDockDNA (Rodríguez-Lumbreras et al., 2022). The predicted structural models and protein-DNA docking complexes were visualized and analyzed using PyMOL (Version 2.5; Schrödinger, LLC). NCBI HomoloGene was employed to analyze the conservation of the variant amino acid residue among different species.
Sanger sequencing
Sanger sequencing was conducted to validate the c.812 G>C variant identified by WES. Primers were designed using Primer5 using the following sequences: forward: 5′- ACCCACCATCTCCCCGTT -3′ and reverse: 5′- CGCCAGCTTCTTCATCTGC -3′. Phanta® Max Super-Fidelity DNA Polymerase (Nanjing Novogene Bioinformatics Technology Co., Ltd.) was employed for PCR amplification. Following purification, the PCR product was sequenced using an AB1 3500DX instrument (Applied Biosystems, United States).
Plasmid construction
The full-length wild-type human LMX1B cDNA (NM_001174147.2) was cloned into the C-terminal region of pcDNA3.1-3×Flag (EGFP + PURO) expression vector to generate a recombinant plasmid for overexpression studies. Using a site-directed mutagenesis kit (TIANGEN, KM101, and Beyotime, D0206S), mutagenesis was conducted to introduce the specific point variants c.737G>A and c.812G>C into the LMX1B-coding sequence. The following mutagenic primers were utilized: c.737G>A-forward: 5′- CTCGTCGAAGCCTTGCCAAAAGGTCCGAGA-3′, c.737G>A-reverse: 5′-TGGCAAGGCTTCGACGAGACCTCGAAGGAGG-3'; c.812G>C-forward: 5′-CCAGGTCTGGTTTCAGAACCAAACAGCAAAGATGAAGAAGC-3′, c.812G>C- reverse: 5′-GCTTCTTCATCTTTGCTGTTTGGTTCTGAAACCAGACCTGG-3'.
Cell culture and transfection
HEK293T and HeLa cell lines were cultured in Dulbecco’s Modified Eagle Medium (Gibco, C11995500BT) containing 10% fetal bovine serum (Vivacell, C04001), 1% penicillin-streptomycin (Gibco, 15140122), and 1% L-glutamine (Solarbio, G0200). Cells were seeded into culture plates 1 day before transfection. After reaching 60%–70% cell confluence, transfection was conducted using Lipofectamine 3,000 reagent (Invitrogen, L3000015) following the manufacturer’s instructions. Briefly, DNA-lipid complexes were prepared by mixing the appropriate amount of plasmid DNA with Lipofectamine 3,000 reagent in a serum-free medium and incubating the mixture at room temperature for 10–15 min. The transfection complexes were gently added to the cells, followed by incubation under standard culture conditions (37°C, 5% CO2). The cells were harvested for protein extraction or immunofluorescence staining 48 h after transfection.
Western blotting
Total proteins were extracted from HEK293T and HeLa cells using RIPA lysis buffer (Beyotime, P0013B) containing protease inhibitor cocktail (apexbio, K1007) and phosphatase inhibitor cocktail (apexbio, L1015). The concentration of proteins was measured using the BCA protein assay kit (Servicebio, G2026) following the manufacturer’s instructions. Equal amounts of protein (10 μg) were separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, IPFL00010). The membranes were blocked with 5% non-fat milk in TBST (TBS containing 0.1% Tween-20) at room temperature for 1 h. Subsequently, the membranes were incubated with primary antibodies against Flag (ABclonal, AE092; 1:10,000) and GAPDH (Cell Signaling Technology, 2118S; 1:6,000) at 4°C overnight. After three washes with TBST, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (ZSGB-BIO, ZB-2301; 1:5,000) at room temperature for 1 h. Protein bands were visualized using enhanced chemiluminescence (ECL) reagent (Servicebio, G2074) and detected using an automated chemiluminescence imaging system (Tannon, 4,160).
Immunofluorescence
The cells were washed three times with phosphate-buffered saline (PBS) at room temperature. Subsequently, the cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) at room temperature for 15 min, followed by three rounds of 5-minute washes with PBS. The cells were blocked with the blocking buffer containing 5% normal goat serum (Jackson ImmunoResearch, 005–000–121) and 0.3% Triton X-100 (biosharp, BS084) in PBS at room temperature for 1 h to minimize non-specific binding. After blocking, the cells were incubated with primary anti-Flag antibody (ABclonal, AE092; 1:200) at 4°C overnight. The next day, the cells were washed three times with PBST (PBS containing 0.02% Tween-20) and three times with PBS, with each wash lasting 5 min. Then, the cells were dark incubated with CoraLite488-conjugated goat anti-rabbit secondary antibody (Proteintech, SA00013-2) at room temperature for 1 h. Nuclear staining was conducted using DAPI (Servicebio, G1012) for 10 min. Finally, the slides were mounted using an anti-fade mounting medium (Yeasen, 36307ES08). Under consistent imaging parameters, all images were captured using a confocal microscope (ZEISS, LSM 800).
Results
Clinical assessment revealed that the three affected members in this family mainly exhibited musculoskeletal malformations
The proband was an eleven-year-old boy with bilateral elbow dysplasia, nail deformities, and a right flat foot. He suffered from bilateral flexion contractures of the elbows, with complete loss of pronation and supination. The range of motion of both elbows was severely limited, with only approximately 40° of mobility. Antecubital pterygia was also observed (Figures 1A, B). Nail bed shortening and longitudinal ridging were observed on the hands, particularly on the second to fourth fingers (Figure 1C). The lunula of all nails was absent. The first toenails on both feet were hypoplastic, while the morphology of the remaining toenails was relatively normal (Figure 1D). Pes planus of the right foot was also observed (Figures 1E, F). Radiographic images demonstrated dislocation and hypoplasia of the radial heads (Figure 2A). Additionally, bilateral patellar hypoplasia and dislocation were observed; however, the proband was asymptomatic (Figure 2B). Consistent with pes planus, radiographic examination revealed eversion of the first metatarsal bone in the right foot (Figures 2C, D). Notably, iliac horns, a characteristic feature of NPS, were evident on radiographic examination (Figure 2E).
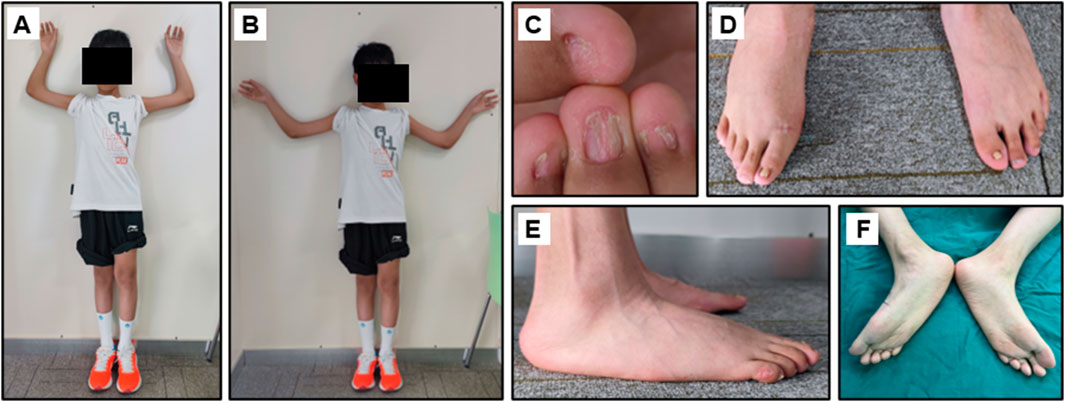
Figure 1. Clinical manifestation of the proband. (A, B) Limited range of motion of the elbows and the hypoplastic patella. (C) Dysplastic fingernails. (D–F) Hypoplastic thumbnails of the feet and the Pes planus of the feet.
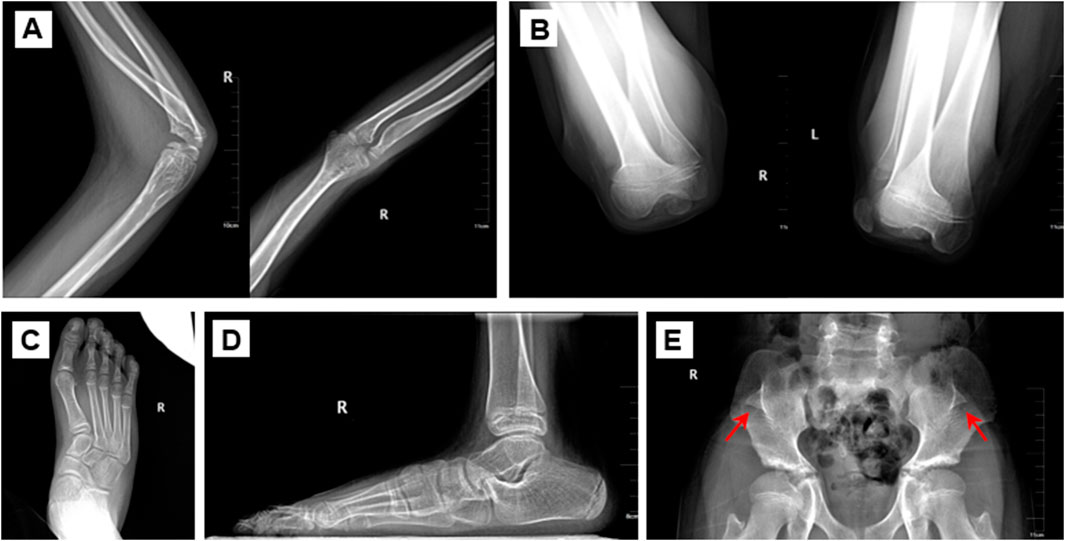
Figure 2. Radiological assessment of the proband. (A) Dislocated radial head (B) Bilateral patellar dysplasia and displacement. (C, D) Pes planus of the feet. (E) Iliac horns. The arrow refers to the iliac horns.
The proband’s mother, a 38-year-old woman presented with milder manifestations. Physical examination exhibited mildly restricted flexion, extension, pronation, and supination of both elbows (Supplementary Figure S1A). Additionally, hypoplasia of the nails was limited to bilateral thumbnails. Notably, there was a loss of creases in the skin overlying the dorsal surface of the distal interphalangeal joints (Supplementary Figure S1B). The proband’s 4-month-old sister was also affected, exhibiting flexion contractures of the bilateral elbows and nail hypoplasia. Her nails displayed typical triangular lunulae and longitudinal ridging (Supplementary Figure S1C). Physical examination did not reveal pes planus or patellar dysplasia in this infant. Subluxation of the right radial head and dislocation of the left radial head were evident on radiographic imaging (Supplementary Figure S2A). Bilateral hypoplastic patella was also identified on imaging, without patellar dislocations (Supplementary Figure S2B). In radiological assessment, iliac horns were visible only on the right side. Furthermore, bilateral acetabular dysplasia was detected, with more extensive involvement on the left side (Supplementary Figure S2C).
Renal dysfunction was not detected in any of the affected family members. Similarly, glaucoma and ocular hypertension were not detected (Supplementary Table S1). The clinical manifestations of the patients are summarized in Table 1.
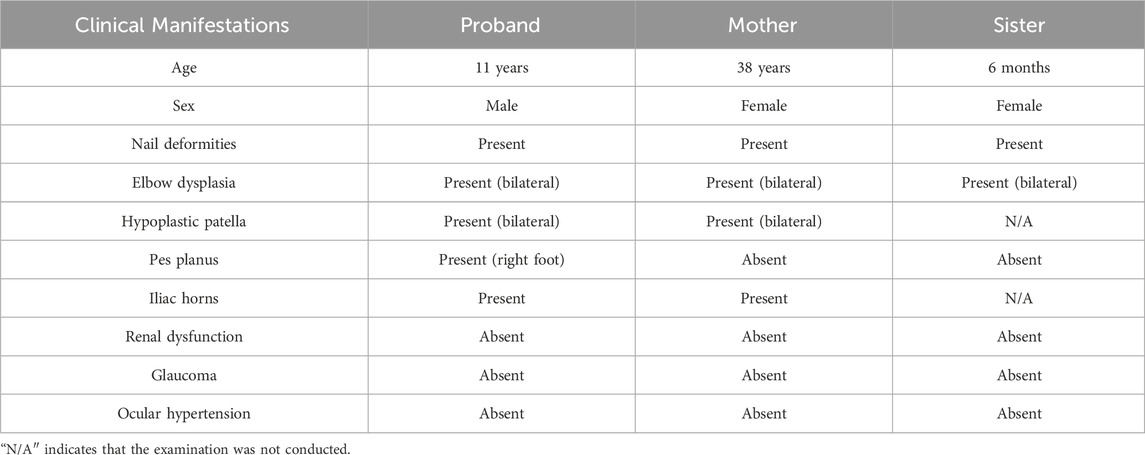
Table 1. Clinical manifestations of the affected members.
Genetic analysis identified a novel c.812 G>C missense variant in the LMX1B gene
The two-generation pedigree of the family with NPS comprised three affected individuals, with an autosomal recessive inheritance pattern (Figure 3A). WES revealed a heterozygous c.812G>C (p.R271T) variant in exon 5 of the LMX1B gene (NM_001174147.2), segregated in all three affected family members. The variant was absent in the proband’s unaffected father, which was confirmed by Sanger sequencing (Figure 3B). This variant was absent in major population databases (Exome Aggregation Consortium and 1000 Genomes Project) and the HGMD database. Computational pathogenicity analysis using REVEL and other five prediction algorithms unanimously classified the c.812G>C variant as a deleterious variant (Table 2). Additionally, this variant was classified as “Likely Pathogenic” based on the 2015 ACMG/AMP guideline (Richards et al., 2015). The c.812G>C variant localized to the homeodomain (HD) of the LMX1B gene (Figure 3C), a conserved DNA-binding domain involved in transcriptional regulation. Conservation analysis across 10 representative vertebrate species revealed complete preservation of the arginine residue at position 271 (Figure 3D).
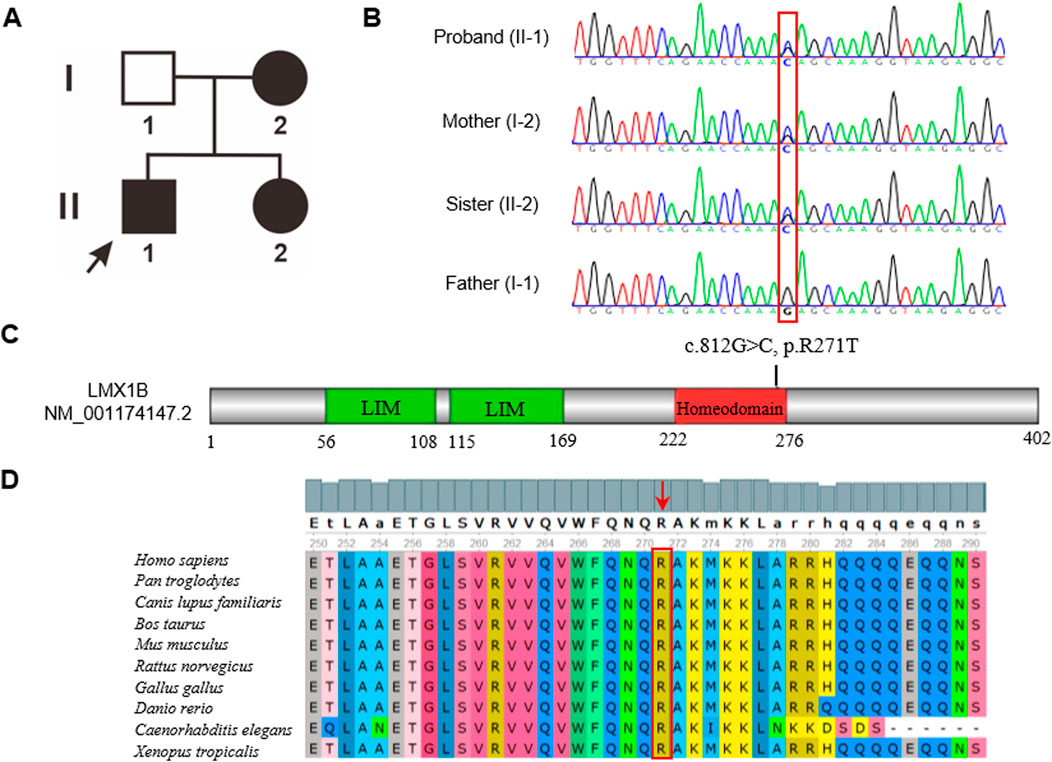
Figure 3. (A) The pedigree of this family. Patients are indicated by solid black. The black arrow shows the proband. (B) Sanger sequence analysis of the family. Variant c.812G>C (p.R271T) was found in the affected members. While the proband’s father had the wild-type variant. (C) Schematic representation of LMX1B. The variant in our study was located in the homeodomain. (D) The amino acid in the 271 position was highly conserved among different species.

Table 2. Bioinformatics predictions of pathogenicity for variant p.R271T. All analyses indicated that the variant is pathogenic.
Structural modeling identified hydrogen-bond network rearrangement and DNA-binding interface destabilization in the LMX1B c.812G>C variant. Structural modeling employing AlphaFold (AF-O60663-F1-v4) indicated critical hydrogen-bonding interactions involved in protein stability. In wild-type LMX1B, Arg271 formed three essential bonds: a 2.9Å bond with Glu242 and two 3.0Å bonds with Asp267 and Thr274 (Figures 4A, B). The c.812G>C variant disrupted this network, eliminating the Glu242 and Thr274 interactions while forming aberrant 3.01Å and 3.0Å bonds respectively between the mutant threonine and Leu267 and Val275 (Figure 4C), resulting in helical structure changes in the homeodomain.
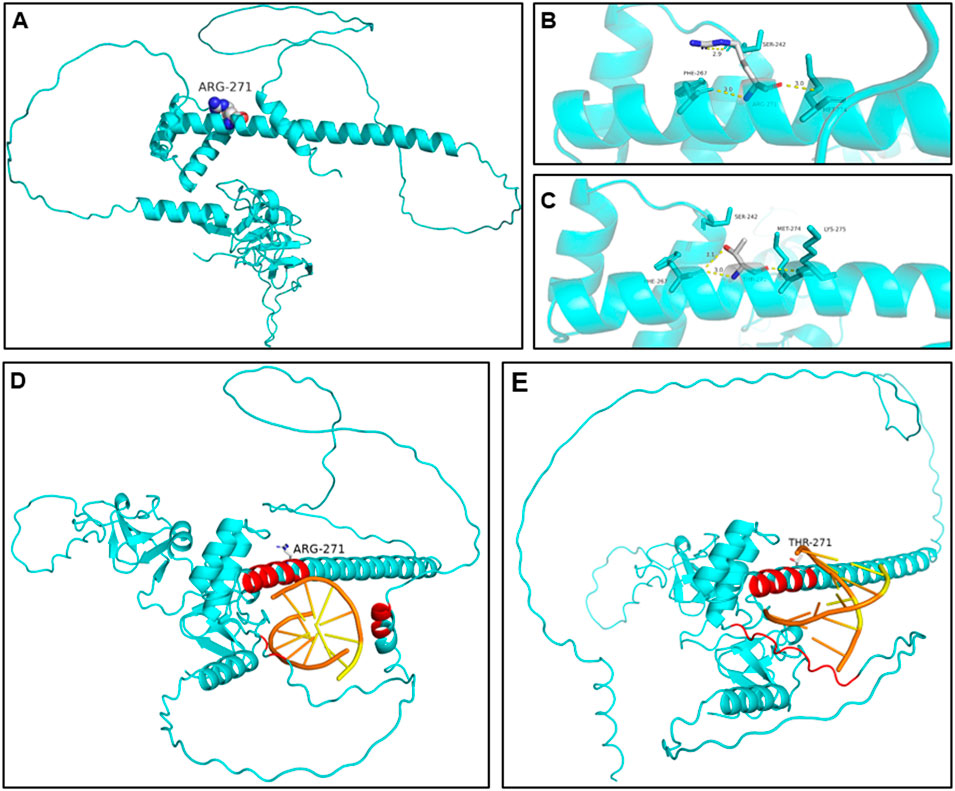
Figure 4. The predicted structural alterations of the mutated protein. (A, B) 3D structure of the wild-type LMX1B protein. (C) 3D structure of the variant type protein. Hydrogen bonds between positions 271 and 242 and 274 were disrupted. New hydrogen bonds were formed between positions 271 and 267 and 275. The rearrangement of the hydrogen disturbed the structure of the homeodomain, impairing its ability to bind to DNA. (D) Molecular docking simulation of wild-type LMX1B-DNA. Effective interaction pairs involve amino acid residues 261–269 and 271–273. (E) Molecular docking simulation of variant LMX1B-DNA. Effective interaction pairs involve amino acid residues 261–266, 268–269, and 272. Effective interaction pairs are labeled as red for amino acid residues and as orange for DNA nucleotides.
We utilized PyDockDNA to simulate the protein-DNA docking interactions. Figure 4D and E depict the results of the docking simulation. The structure in the protein-DNA binding region with an atom distance less than 7Å was recognized as effective interacting pairs (Baek et al., 2024), labeled as red on the protein and orange on the DNA. For wild-type LMX1B, the effective interface region consisted of amino acid residues 261–269 and 271–273. The variant protein exhibited a distinct interaction pattern, involving residues 261–266, 268–269, and 272 at the binding interface. The direction and binding position of the DNA site was altered in the variant. Although the overall PyDockDNA binding scores were not significantly different between the wild-type and mutant proteins (−97.692 vs. −100.056, Table 3), the variant LMX1B exhibited enhanced van der Waals forces (−21.801 for wild-type and 14.236 for the mutant variant), suggesting impaired binding functionality.

Table 3. PyDockDNA simulation of the wild-type and variant LMX1B protein-DNA docking.
In vitro assays revealed altered subcellular localization caused by the LMX1B c.812G>C variant
Western blotting and immunofluorescence were conducted to measure the expression and localization of wild-type and mutant LMX1B protein and investigate the pathogenicity of the c.812G>C variant. The c.737G>A variant, a known pathogenic variant (Andeen et al., 2017), was employed as a positive control. Western blotting revealed no significant difference in the expression of mutant proteins compared to the wild-type protein (Figures 5A, B). Compared to the wild-type protein and c.737G>A mutant protein, which showed the nuclear localization of LMX1B, immunofluorescence staining revealed a cell type-specific mislocalization of the c.812G>C mutant. This variant induced aberrant cytoplasmic distribution of LMX1B in HeLa cells, but in HEK293T cells, it induced pathological nuclear aggregation, suggesting the potential pathogenic effects of this variant by disrupting normal protein localization (Figure 5C).
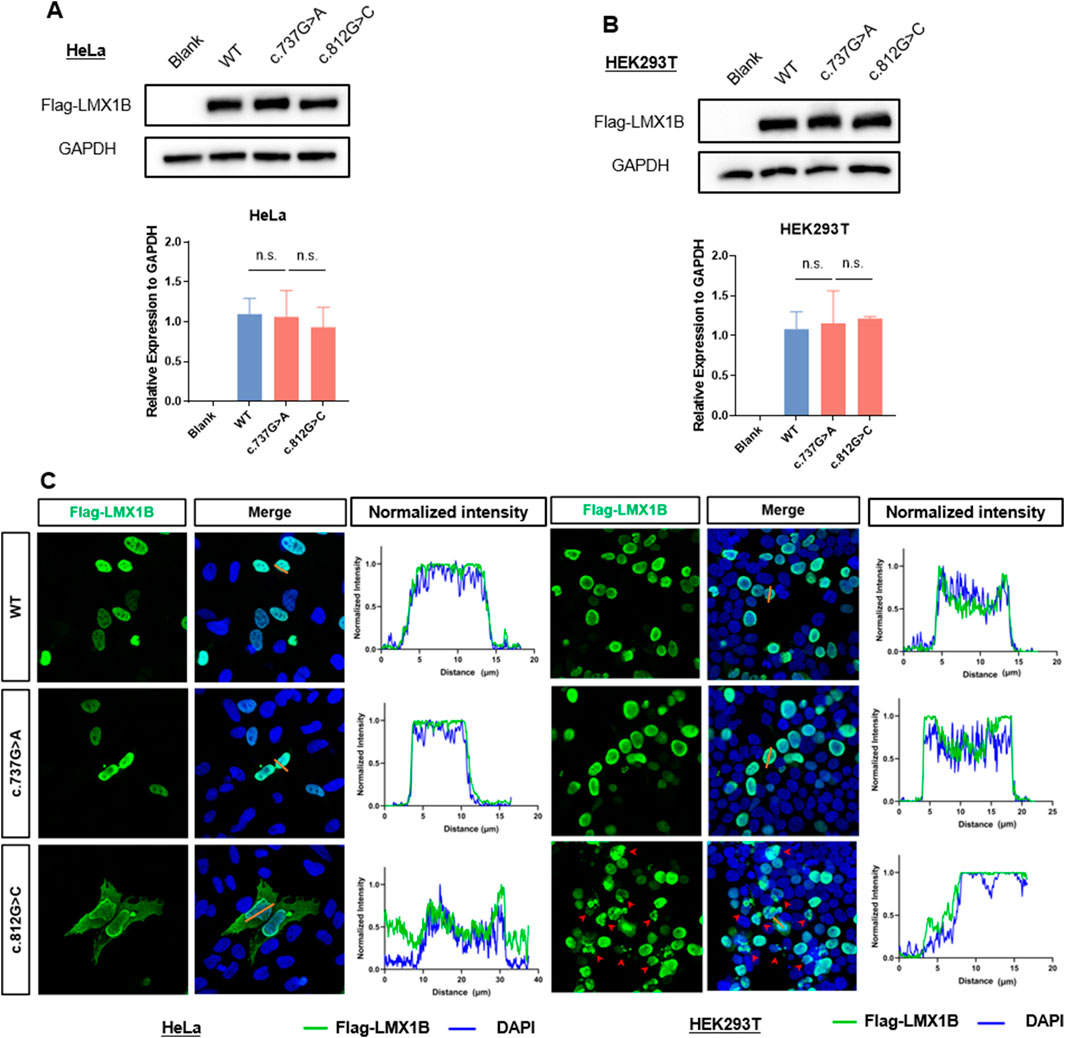
Figure 5. In vitro experiment analysis the expression level and subcelluar distribution of variation. (A) The protein expression level of LMX1B in HeLa cells overexpressing wild-type LMX1B, c.737G>A variant, and c.812G>C variant. (B) The protein expression level of LMX1B in HEK293T cells overexpressing wild-type LMX1B, c.737G>A variant, and c.812G>C variant. (C) Immunofluorescence staining showed subcellular localization of LMX1B variants in HeLa and HEK293T cells. In HeLa cells, the wild-type and c.737G>A mutant proteins were predominantly localized in the nucleus, whereas the c.812G>C mutant exhibited aberrant cytoplasmic distribution. In HEK293T cells, the wild-type and c.737G>A mutant proteins also showed nuclear localization, but the c.812G>C mutant induced pathological nuclear aggregation (indicated by red arrows), suggesting a cell type-specific mislocalization pattern. The normalized fluorescence intensities were quantified on the right side of the figure. The corresponding normalized fluorescence intensities of the straight orange lines in are displayed on the right side of merge image.
Discussion
NPS is a complex hereditary disorder affecting multiple systems. It is primarily characterized by nail and patellar abnormalities and renal dysfunction (Harita et al., 2016). Additionally, the ocular (Pallozzi Lavorante et al., 2022), nervous, dental, and digestive systems can simultaneously exist (Ghoumid et al., 2015; Witzgall, 2017). In this study, the patients carried the LMX1B c.812G>C variant and mainly suffered from musculoskeletal symptoms, including elbow malformation, nail deformities, and patella dysplasia.
The LMX1B protein contains two zinc-binding LIM domains at the NH2-terminus and a DNA-binding homeodomain. The LIM domains are responsible for protein-protein interactions (Kadrmas and Beckerle, 2004). The homeodomain is a highly conserved domain constituting a sequence-specific DNA-binding motif involved in the modulation of gene transcription. More than 200 pathogenic variants of LMX1B have been recognized so far (Castilla-Ibeas et al., 2024).
Variants located in the LIM domain are primarily associated with abnormal protein-protein interactions, whereas variants in the homeodomain predominantly impair protein-DNA binding. Some studies have suggested that homeodomain variants may be associated with renal dysfunction (Bongers E. M. et al., 2005). Some variants located in the non-coding region of the LMX1B gene only lead to musculoskeletal symptoms (Haro et al., 2021). Apart from the above-mentioned examples, no clear association has been identified between mutation sites and patient phenotypes.
German et al. identified FLAT-E and FLAT-F as the binding sites of the homeodomain (German et al., 1992). Sandra et al. (Dreyer et al., 2000) also confirmed that LMX1B protein regulates transcription by binding to these sequences, and loss-of-function variants in LMX1B can lead to the development of NPS. In our study, AlphaFold simulations revealed structural alterations in the helical conformation and hydrogen bonding patterns within the homeodomain. Molecular docking simulations visualized the protein-DNA effective interacting pair modification, indicating pathogenic changes in protein structure. The similarity in binding scores between wild-type and variant proteins can be attributed to two key factors. First, molecular simulations predominantly analyze structural and spatial interactions but do not assess biological function. Second, parameter constraints in these models may not reproduce physiologically relevant binding dynamics. However, based on other functional predictions and the higher van der Waals forces, we believe that the predictive model still supports the pathogenicity of the mutation.
Due to its well-established pathogenicity, proximity to the mutation site (both were located within the homeodomain), and shared pathogenic mechanisms (impaired DNA-binding activity), the c.737G>A variant was selected as a positive control in our in vitro experiments. This alignment ensures comparability in assessing the functional effects of homeodomain disruptions.
The cell type-dependent mislocalization patterns of the LMX1B c.812G>C variant suggests a previously unrecognized layer of complexity in nuclear protein homeostasis. While HeLa cells exhibited nucleocytoplasmic redistribution, the aberrant nuclear aggregation in HEK293T cells suggests distinct and cell-specific pathogenic mechanisms. This dichotomy may be due to the differential expression of nuclear transport chaperones between epithelial-derived HeLa and embryonic kidney-derived 293T cells. As a transcription factor, LMX1B typically shuttles between the cytoplasm and nucleus in physiological conditions. The wild-type protein binds to DNA to regulate gene transcription (Zhou and Qin, 2012). However, the variant protein displayed decreased DNA-binding capacity, resulting in impaired subcellular distribution and functional activity. This observation aligns with the findings of Wang et al. (2014), who reported a family with NPS carrying the LMX1B-R198X variant. Similarly, in that family, complete deletion of the DNA-binding domain led to abnormal cytoplasmic-nuclear protein localization. In their study, immunofluorescence analysis confirmed this mislocalization. Furthermore, studies employing electrophoretic mobility shift assay and luciferase reporter assays have demonstrated that pathogenic variants in the homeodomain can impair the DNA-binding capacity and transcriptional activity of LMX1B, confirming functional disruptions at the molecular level (Sato et al., 2005). These findings collectively validate the bioinformatic predictions of pathogenicity for this variant.
NPS is an autosomal dominant disorder primarily caused by haploinsufficiency. Affected individuals are all heterozygous, and no homozygous variants have been reported in humans so far, suggesting that complete loss of LMX1B may be embryonically lethal. In contrast, homozygous Lmx1b knockout mice (Lmx1b−/−) exhibited severe phenotypes, while heterozygous mice (Lmx1b+/−) did not show overt manifestations of NPS (Chen et al., 1998). Notably, Lmx1b+/− mice demonstrated decreased compensatory kidney growth after unilateral nephrectomy compared to wild-type mice (Lmx1b+/+) (Endele et al., 2007). These findings underscore the critical role of LMX1B dosage in normal renal development and suggest that NPS phenotypes originate from insufficient functional LMX1B protein. However, despite all NPS patients being heterozygous and the observed uncertainty in genotype-phenotype correlations, the association between the protein levels of LMX1B and specific clinical manifestations remains unclear.
A distinct genotype-phenotype correlation could not be established in patients with NPS. Even significant phenotypic heterogeneity is observed within the same family. The role of LMX1B in limb development is highly complex, involves several downstream genes, and is regulated by various transcription factors. Notably, approximately 10% of patients with NPS do not exhibit variants in the LMX1B exons (Bongers E. M. H. F. et al., 2005). Therefore, establishing a clear genotype-phenotype relationship remains challenging. However, our patients primarily presented with skeletal manifestations and normal kidney function. This suggests that in our patients, LMX1B dysfunction in regulating limb development was the predominant cause of the observed clinical features.
LMX1B is essential for dorsal-ventral patterning during limb development (Vogel et al., 1995), which possibly leads to musculoskeletal symptoms. LMX1B is responsible for establishing dorsal identity in the limb mesoderm, where its expression is induced by the dorsal ectodermal signal Wnt7a (Riddle et al., 1995) The expression pattern of LMX1B in undifferentiated dorsal mesoderm remains uniform across the tissue and is typically considered an all-or-none phenomenon. This expression pattern implies that LMX1B is activated through a threshold mechanism, where a critical level of dorsal ectodermal signaling triggers its induction. Loss of Lmx1b in mice can lead to defective patterning of dorsal autopod (hand/foot) and zeugopod (forearm/leg). Autopods show symmetrical ventral characteristics, including dorsal-type footpads, absent dorsal hair follicles and nails, and matching ventral-type muscles, tendons, and ligaments. Variants in LMX1B impair dorsalization during limb development, leading to the hypoplasia of dorsal structures, such as nails and patella. Our patients all exhibited elbow joint malformation. The proband also had antecubital pterygium, which refers to skin webbing across the anterior elbow. Given the restricted joint mobility, surgical interventions, such as soft tissue release and radial head excision, are planned to address functional limitations. Radiographic imaging was not conducted due to the young age of the proband’s sister. Given the infant’s developmental immaturity, longitudinal monitoring is essential to observe potential manifestations that may emerge with age. We also identified hip dysplasia in the proband’s mother, a relatively rare finding among patients with NPS (Jacofsky et al., 2003). The mechanism by which LMX1B affects hip joint development remains unclear, but it is well-known that LMX1B plays a crucial role in skeletal development. Although direct evidence is lacking, hip dysplasia seems to be a phenotype of NPS rather than being two separate conditions manifesting coincidentally.
Identification of potential genotype-phenotype correlations represents a pivotal research direction in NPS. Further studies are needed to illuminate the upstream regulators and downstream effectors affecting the function of LMX1B. For instance, Haro et al. identified two LMX1B-associated regulatory modules, namely, LARM1 and LARM2, which bind to LMX1B to potentiate its expression during limb dorsalization in discrete spatial patterns. Ablation of these elements exclusively disrupted dorsalization and did not affect other LMX1B-mediated processes (Lemley, 2009; Haro et al., 2021). Other studies have delineated that E47 upregulates the transcriptional activity of LMX1B (Dreyer, 2000). LDB1 downregulates the activity of LMX1B through E47. As a transcription factor involved in different physiological pathways, substantial studies are needed to unravel the complex roles of LMX1B and the pathogenesis of NPS.
Conclusion
In this article, we presented a family with NPS. Through genetic testing, we identified a novel missense variant of the LMX1B gene, c.812G>C (p.R271T), located on the exon 5. Further bioinformatic analysis and in vitro experiments unveiled the structural modification and abnormal distribution of the mutated protein and its pathogenic effects.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://db.cngb.org/search/project/CNP0005417/, CNP0005417.
Ethics statement
The studies involving humans were approved by Institutional Review Board of Children’s Hospital of Soochow University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
QS: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review and editing. YR: Investigation, Methodology, Writing – review and editing. YC: Data curation, Writing – review and editing. WZ: Data curation, Writing – review and editing. GS: Data curation, Funding acquisition, Writing – review and editing. XW: Funding acquisition, Supervision, Writing – review and editing. HW: Conceptualization, Funding acquisition, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Project of Jiangsu Province Key Research and Development Plan (Social Development) (BE2022732), the Maternal and Child Healthcare Program of Jiangsu Province (F202119), and Suzhou City Youth Science and Technology Project for the Promotion of Health and Education (KJXW2023083).
Acknowledgments
We thank the patient and his family for their participation and cooperation. And we would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1574076/full#supplementary-material
Abbreviations
NPS, Nail-patella syndrome; LMX1B, LIM homeobox transcription factor 1 beta; HD, Homeodomain; WES, Whole-exome sequencing; HGMD, Human gene mutation database; PCR, Polymerase chain reaction.
References
Andeen, N., Schleit, J., Blosser, C., Dorschner, M., Hisama, F., and Smith, K. D. (2017). LMX1B-Associated nephropathy with type III collagen deposition in the glomerular and tubular basement membranes. Am. J. Kidney Dis. 72 (2), 296–301. doi:10.1053/j.ajkd.2017.09.023
Baek, M., McHugh, R., Anishchenko, I., Jiang, H., Baker, D., and DiMaio, F. (2024). Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA. Nat. Methods 21 (1), 117–121. doi:10.1038/s41592-023-02086-5
Bongers, E. M., Huysmans, F. T., Levtchenko, E., de Rooy, J. W., Blickman, J. G., Admiraal, R. J., et al. (2005a). Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur. J. Hum. Genet. 13 (8), 935–946. doi:10.1038/sj.ejhg.5201446
Bongers, E. M. H. F., Huysmans, F. T., Levtchenko, E., de Rooy, J. W., Blickman, J. G., Admiraal, R. J. C., et al. (2005b). Genotype–phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur. J. Hum. Genet. 13 (8), 935–946. doi:10.1038/sj.ejhg.5201446
Castilla-Ibeas, A., Zdral, S., Oberg, K. C., and Ros, M. A. (2024). The limb dorsoventral axis: Lmx1b's role in development, pathology, evolution, and regeneration. Dev. Dyn. 253 (9), 798–814. doi:10.1002/dvdy.695
Chen, H., Lun, Y., Ovchinnikov, D., Kokubo, H., Oberg, K. C., Pepicelli, C. V., et al. (1998). Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat. Genet. 19 (1), 51–55. doi:10.1038/ng0598-51
Dreyer, S. D., Morello, R., German, M., Zabel, B., Winterpacht, A., Lunstrum, G. P., et al. (2000). LMX1B transactivation and expression in nail-patella syndrome. Hum. Mol. Genet. 9 (7), 1067–1074. doi:10.1093/hmg/9.7.1067
Dreyer, S. D., Morello, R., German, M. S., Zabel, B., Winterpacht, A., Lunstrum, G. P., et al. (2000). LMX1B transactivation and expression in nail-patella syndrome. Hum. Mol. Genet. 9 (7), 1067–1074. doi:10.1093/hmg/9.7.1067
Dreyer, S. D., Zhou, G., Baldini, A., Winterpacht, A., Zabel, B., Cole, W., et al. (1998). Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat. Genet. 19 (1), 47–50. doi:10.1038/ng0598-47
Endele, S., Klein, S., Richter, S., Molter, T., Amann, K., Klanke, B., et al. (2007). Renal phenotype in heterozygous Lmx1b knockout mice (Lmx1b+/-) after unilateral nephrectomy. Transgenic Res. 16 (6), 723–729. doi:10.1007/s11248-007-9118-7
German, M. S., Wang, J., Chadwick, R. B., and Rutter, W. J. (1992). Synergistic activation of the insulin gene by a LIM-homeo domain protein and a basic helix-loop-helix protein: building a functional insulin minienhancer complex. Genes. and Dev. 6 (11), 2165–2176. doi:10.1101/gad.6.11.2165
Ghoumid, J., Petit, F., Holder-Espinasse, M., Jourdain, A.-S., Guerra, J., Dieux-Coeslier, A., et al. (2015). Nail–Patella Syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity. Eur. J. Hum. Genet. 24 (1), 44–50. doi:10.1038/ejhg.2015.77
Guidera, K. J., Satterwhite, Y., Ogden, J. A., Pugh, L., and Ganey, T. (1991). Nail patella syndrome: a review of 44 orthopaedic patients. J. Pediatr. Orthop. 11 (6), 737–742. doi:10.1097/01241398-199111000-00007
Harita, Y., Kitanaka, S., Isojima, T., Ashida, A., and Hattori, M. (2016). Spectrum of LMX1B mutations: from nail–patella syndrome to isolated nephropathy. Pediatr. Nephrol. 32 (10), 1845–1850. doi:10.1007/s00467-016-3462-x
Haro, E., Petit, F., Pira, C. U., Spady, C. D., Lucas-Toca, S., Yorozuya, L. I., et al. (2021). Identification of limb-specific Lmx1b auto-regulatory modules with Nail-patella syndrome pathogenicity. Nat. Commun. 12 (1), 5533. doi:10.1038/s41467-021-25844-5
Jacofsky, D. J., Stans, A. A., and Lindor, N. M. (2003). Bilateral hip dislocation and pubic diastasis in familial nail-patella syndrome. Orthopedics 26 (3), 329–330. doi:10.3928/0147-7447-20030301-16
Kadrmas, J. L., and Beckerle, M. C. (2004). The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell. Biol. 5 (11), 920–931. doi:10.1038/nrm1499
Lemley, K. V. (2009). Kidney disease in nail-patella syndrome. Pediatr. Nephrol. 24 (12), 2345–2354. doi:10.1007/s00467-008-0836-8
Lin, Y., Zhao, J., Chen, S., Zeng, X., Du, Q., Yang, Y., et al. (2008). A novel mutation in LMX1B gene causes nail-patella syndrome in a large Chinese family. Bone 43 (3), 591–595. doi:10.1016/j.bone.2008.04.025
Pallozzi Lavorante, N., Iester, M., Bonzano, C., Bagnis, A., Traverso, C. E., and Cutolo, C. A. (2022). Nail-patella syndrome and glaucoma: a case report and review of the literature. Case Rep. Ophthalmol. 13 (3), 984–990. doi:10.1159/000527234
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riddle, R. D., Ensini, M., Nelson, C., Tsuchida, T., Jessell, T. M., and Tabin, C. (1995). Induction of the LIM homeobox gene Lmx1 by WNT7a establishes dorsoventral pattern in the vertebrate limb. Cell. 83 (4), 631–640. doi:10.1016/0092-8674(95)90103-5
Rodríguez-Lumbreras, L. A., Jiménez-García, B., Giménez-Santamarina, S., and Fernández-Recio, J. (2022). pyDockDNA: a new web server for energy-based protein-DNA docking and scoring. Front. Mol. Biosci. 9, 988996. doi:10.3389/fmolb.2022.988996
Sato, U., Kitanaka, S., Sekine, T., Takahashi, S., Ashida, A., and Igarashi, T. (2005). Functional characterization of LMX1B mutations associated with nail-patella syndrome. Pediatr. Res. 57 (6), 783–788. doi:10.1203/01.Pdr.0000157674.63621.2c
Sweeney, E., Fryer, A., Mountford, R., Green, A., and McIntosh, I. (2003). Nail patella syndrome: a review of the phenotype aided by developmental biology. J. Med. Genet. 40 (3), 153–162. doi:10.1136/jmg.40.3.153
Vogel, A., Rodriguez, C., Warnken, W., and Izpisúa Belmonte, J. C. (1995). Dorsal cell fate specified by chick Lmx1 during vertebrate limb development. Nature 378 (6558), 716–720. doi:10.1038/378716a0
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, W.-M., Chen, J.-F., Liu, S.-T., Hsu, Y.-J., Liu, Y.-C., and Huang, S.-M. (2014). Biochemical properties of the recurrent LMX1b truncated mutant carried in a Taiwanese family with nail-patella syndrome. Br. J. Dermatology 171 (2), 356–362. doi:10.1111/bjd.13051
Witzgall, R. (2017). Nail-patella syndrome. Pflügers Archiv - Eur. J. Physiology 469 (7-8), 927–936. doi:10.1007/s00424-017-2013-z
Keywords: nail-patella syndrome, LMX1B, whole-exome sequencing, gene variant, musculoskeletal deformity
Citation: Sun Q, Ren Y, Cao Y, Zheng W, Su G, Wang X and Wang H (2025) Identification of a novel missense variant in the LMX1B gene associated with nail-patella syndrome in a Chinese family. Front. Genet. 16:1574076. doi: 10.3389/fgene.2025.1574076
Received: 17 February 2025; Accepted: 02 April 2025;
Published: 12 May 2025.
Edited by:
Yang Guohua, Wuhan University, ChinaReviewed by:
Niu Li, International Peace Maternity and Child Health Hospital, ChinaCong Li, Southern Medical University, China
Copyright © 2025 Sun, Ren, Cao, Zheng, Su, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongying Wang, d2h5OTIzODExQHNpbmEuY29t; Xiaodong Wang, b3J0aG93eGRAMTYzLmNvbQ==
†These authors have contributed equally to this work