 Yongmei Shen1,2†
Yongmei Shen1,2† Lei Zhang1,2†
Lei Zhang1,2† Yaqi Li3†Liying Yao1,2
Yaqi Li3†Liying Yao1,2 Jiasong Cao1,2
Jiasong Cao1,2 Qimei Lin1,2Maolin Nie3Hefei Wang3
Qimei Lin1,2Maolin Nie3Hefei Wang3 Rongxin Wei3
Rongxin Wei3 Ying Chang1,2,3,4*
Ying Chang1,2,3,4*- 1Tianjin Key Laboratory of Human Development and Reproductive Regulation, Tianjin Central Hospital of Gynecology Obstetrics, Tianjin, China
- 2Tianjin Institute of Obstetrics and Gynecology, Tianjin Central Hospital of Gynecology Obstetrics, Tianjin, China
- 3Obstetrics and Gynecology, Tianjin Medical University, Tianjin, China
- 4Medical School, Tianjin University, Tianjin, China
Renpenning syndrome is a rare X-linked genetic disorder caused by variants in the PQBP1 gene, but the information about its prenatal presentation is very limited. A 35-year-old woman experienced two male pregnancies with thickened nuchal translucency (NT) (5.5 mm and 5 mm). She went to our prenatal diagnosis center for the current natural conception during the second pregnancy. Trio-whole exome sequencing (TrioWES) of chorionic villus biopsy revealed a 666-bp genetic deletion (chrX:48755195-49760422) in the fetus, inherited from the mother, which included TIMM17B and PQBP1. The couple opted for termination of pregnancy. During the third pregnancy, systematic fetal screening was performed in early pregnancy. An ultrasound examination at 12+1 weeks revealed a thickened NT (6.5 mm), nasal bones abnormalities and a cleft palate. Ultrasound examination at 16 weeks showed ventricular septal defect (VSD), and mild enlargement of the lateral ventricles in the fetus. Chorionic villus biopsy samples were tested for Multiplex Ligation-dependent Probe Amplification (MLPA), showing a 666-bp genetic deletion, inherited from the mother. The couple opted for termination of pregnancy, and the male fetus had a sunken nose and cup-shaped ears leading to a diagnosis of Renpenning syndrome. In conclusion, this emphasized the importance of early systematic pregnancy screening. Increased NT in the first trimester, especially when present in conjunction with ultrasound structural abnormalities such as nasal bone abnormalities, VSD, and mild bilateral ventriculomegaly, emphasized the importance of genetic testing, including chromosome testing, genomic testing, and Whole-exome sequencing.
Introduction
Renpenning Syndrome is a rare X-linked genetic disorder caused by variants in the PQBP1 gene. Symptoms of this condition include intellectual disability (HP:0001249), microcephaly (HP:0000252), short stature (HP:0004322), small testes (HP:0008734), and facial dysmorphism (HP:0001999). Studies using different models have shown that PQBP1 plays a crucial role in neural development and function (Cheng et al., 2023). These patients showed moderate intellectual disability, poor autonomy, communication and social interaction disorders, learning difficulties, and obvious autistic behaviors (Redin et al., 2014). An increasing number of articles have documented Renpenning syndrome. For example, a recent Korean case study identified a novel mutation in the PQBP1(NM_001032381.2) gene, leading to the diagnosis of Renpenning syndrome. Similarly, a case of Renpenning syndrome has been reported in China, which was associated with the PQBP1(NM_001032381.2):c.28C>G (p.Arg10Gly) (Jeong et al., 2018; Germanaud et al., 2011; Pan et al., 2025). However, the lack of prenatal information poses a major challenge for the identification of Renpenning Syndrome.
Currently, international reports are available on Renpenning syndrome in adults with different physical features, making it easy to distinguish and diagnose. However, there is no information is available, and intelligence cannot be determined before delivery. Expanded noninvasive prenatal testing (NIPT-Plus) showed high sensitivity in detecting chromosomal aneuploidy and copy number variations (CNVs) exceeding 5M fragments. Nevertheless, its ability is limited to predefined CNV detection and is prone to false negatives and positives (Chen et al., 2025). NIPT for monogenic diseases is limited to specific genes, and WES is not a routine prenatal test without specific indications. Therefore, only relying on conventional chromosome screening may not be able to detect and diagnose genetic diseases such as Renpenning syndrome in time. The use of ultrasound imaging is essential for identifying fetal structural abnormalities during pregnancy. However, there is no international report on the prenatal ultrasound manifestations of Renpenning syndrome.
The first reported case of prenatal diagnosis of Renpenning Syndrome emphasized the importance of systematic screening during pregnancy.
Case report
A 35-year-old female, healthy and without a family history of genetic diseases, experienced two abnormal pregnancies. In the first pregnancy, the nuchal translucency (NT) measured 5.5 mm. The decision to terminate the pregnancy before 28 weeks was based on parental choice without chromosome screening. The fetus was male. The patient visited our prenatal diagnosis center for the second pregnancy. Systematic fetal screening was performed in the first trimester of pregnancy. TORCH (Toxoplasma/Other Agents/Rubella Virus/Cytomegalovirus/Herpes Simplex Virus) screening was negative. At 12 weeks, the patient received NIPT, which showed a low-risk outcome. Ultrasound examination showed an NT of 5 mm (HP:0010880) and nasal bones abnormalities (HP:0010937). At 13 weeks, chorionic villus sampling was performed, followed by trio-whole exome sequencing (Trio-WES) using samples of the parents and chorionic villus for family verification. WES was carried out using the Agilent’s SureSelect Human All Exome V6 Capture Kit to prepare libraries, followed by high-throughput sequencing based on the Illumina NovaSeq 6000 platform to obtain high-quality exome data. GRCh37/hg19 was used as the reference genome. CNV was analyzed using the GermlineCNVCaller module in the Genome Analysis Toolkit (GATK). This tool efficiently detected CNV events in WES data applying the sequencing depth information and the output of DetermineGermlineContigPloidy. Trio-WES analysis revealed a 666-bp gene deletion (chrX:48755195-49760422) in the fetus, including TIMM17B(NM_001167947.2) and PQBP1(NM_001032381.2), inherited from the mother, and no related gene variants or copy number variations were detected in the father. The parents chose to terminate the pregnancy and the fetus was male. According to the 2019 ClimGen/ACMG CNV interpretation guidelines, the pathogenicity classification was considered to be likely pathogenic.
During the patient’s third pregnancy, NIPT was performed at 11 weeks, showing a low-risk outcome. Subsequently, an ultrasound scan at 12 weeks showed increased NT (6.5 mm) (HP:0010880), nasal bone abnormalities (HP:0010937) and cleft palate (HP:0000175) at 12+1 weeks (Figures 1A–C). According to medical recommendations, chorionic villus sampling was performed, and MLPA analysis confirmed a 666 BP gene deletion (chrX:48755195-49760422) in the fetus inherited from the mother. An ultrasound scan at 16 weeks revealed the presence of ventricular septal defect (VSD, HP:0001629), and subsequently mild enlargement of bilateral lateral ventricles was observed at 17 weeks (Figures 1D,E). By applying PVS1 and PM2 criteria, prenatal ultrasound could help detect potentially pathogenic PQBP1 deletions associated with Renpenning syndrome. The pathogenicity level was classified as likely pathogenic according to the 2019 ClimGen/ACMG CNV interpretation guidelines. At 17 weeks of termination of pregnancy, the fetus showed obvious facial features, such as a depressed nose and cup-shaped ears (Figures 1F–H), indicating that the pathogenic PQBP1 deletion met the criteria PVS1, PM2, and PP4. Based on the results of the ultrasound scan, facial features and genetic testing, the pathogenicity classification was consistent with the 2019 ClimGen/ACMG CNV interpretation guidelines and is pathogenic. Despite the recommendation, the couple refused further pathological autopsy. The pedigree of the patient was shown in Figure 1I, and the schedule of pregnancy information was detailed in Table 1.
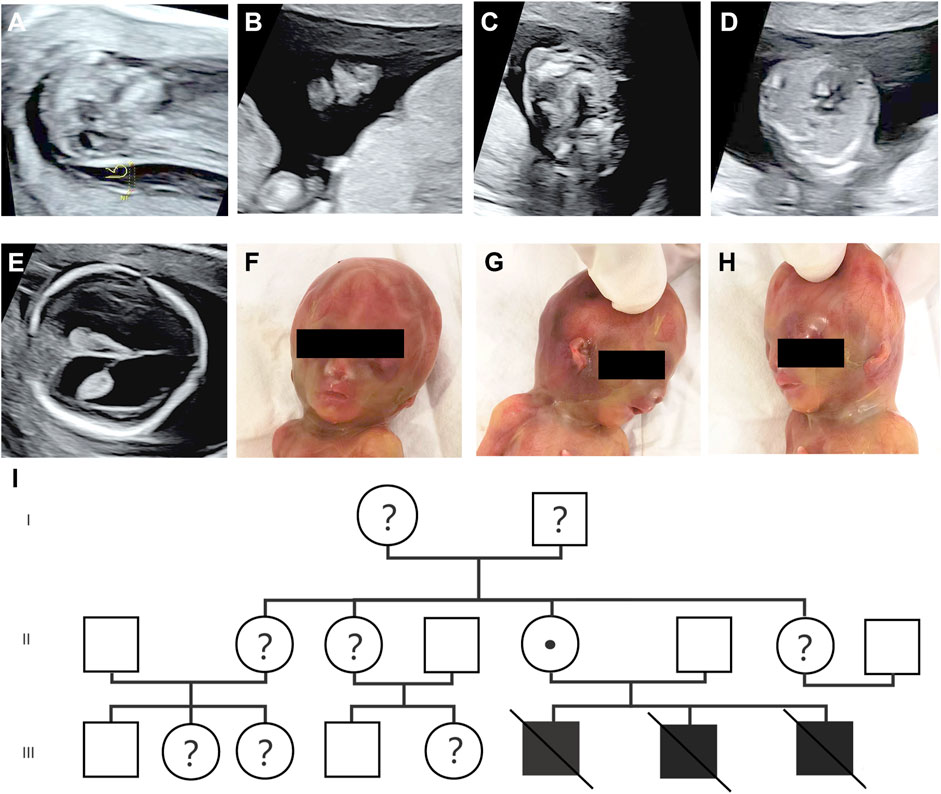
Figure 1. Prenatal ultrasound scan and fetal phenotype after delivery. (A–C) Prenatal ultrasound at 12+1 weeks of gestation showed thickened NT (6.5 mm) (A), nasal bone abnormalities (B), and cleft palate (C); (D,E) Prenatal ultrasound at 16+1 weeks of gestation showing VSD (D), and mildly enlarged bilateral lateral ventricles (E); (F–H) the image of fetus after delivery with a sunken nose and cup-shaped ears; (I) the lineage.
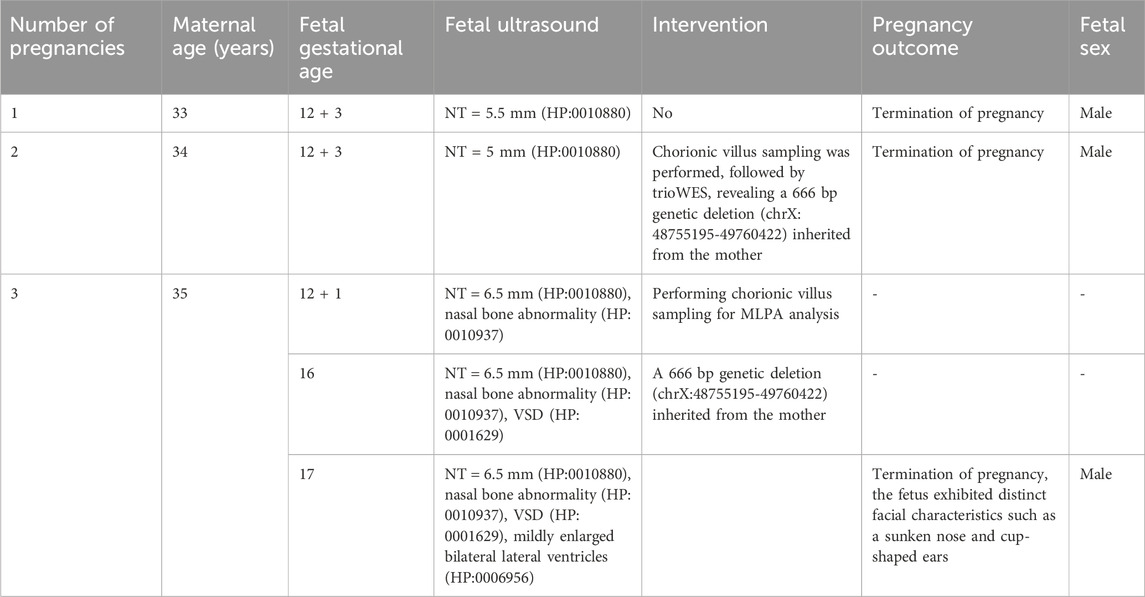
Table 1. The timeline of incorporating information on pregnancy.
The diagnostic challenges in this case included patient compliance, timely prenatal diagnosis, and the high level of expertise required by prenatal diagnostic specialists, such as timely recognition of abnormal ultrasound findings and selection of appropriate chromosome screening methods. After thorough explanation, it was found that the pregnant woman had a 666 BP gene deletion (chrx:48755195-49760422). Then, the probability of the next male fetus being affected was 50%, and the probability of the next female fetus being normal was 50%. As a result, Preimplantation Genetic Testing for Monogenic disorders (PGT-M) was considered. It was recommended to carry out systematic screening in the early stage of the next pregnancy and chromosome screening of chorionic villi at about 12 weeks.
Discussion and conclusion
At present, international reports mainly focus on Renpenning syndrome in adults, which is characterized by distinct physical features that facilitate differentiation and diagnosis. Most variants in PQBP1 associated with Renpenning syndrome are frameshift variants in the PRD (POU-specific domain) and CTD (C-terminal domain) regions, leading to premature termination (Kalscheuer et al., 2003; Stevenson et al., 2005). Some known PQBP1 variants, such as c.727 C>T (p.Arg243Trp) and c.459_462del (p.Arg153SerfsTer41), involve heterozygous variants, deletion frameshift nonsense variants, point missense variants, and duplication variants within the PQBP1 gene (Pan et al., 2025; Lopez-Martín et al., 2022; Fan et al., 2025). The pathogenesis of Renpenning syndrome involves the regulation of various cellular processes, such as neural progenitor cell proliferation, neural projection, synaptic growth, neuronal survival, and cognitive function, through mRNA transcription and splicing-dependent or -independent mechanisms (Cheng et al., 2023). Moreover, the literature suggests that it is associated with a unique DNA methylation profile (Haghshenas et al., 2023). Renpenning syndrome has been observed only in males, and female carriers are usually asymptomatic. Nevertheless, one study showed that a significant proportion of female carriers in Renpenning syndrome families exhibited skewed X-chromosome inactivation, which complicated the presentation of the syndrome in carriers (Cho et al., 2020).
TIMM17B is a gene located on the X chromosome that encodes a translocase present on the inner mitochondrial membrane in humans. It is the only gene in Xp11.23 that shows structural conservation and functional homology on autosomes. This gene encodes a multipass transmembrane protein, which is an important component of the mitochondrial translocase TIM23 complex and is responsible for promoting the transport of proteins containing transport peptides across the inner mitochondrial membrane (Bauer et al., 1999). High expression of TIMM17B has been identified as a potential diagnostic and prognostic marker for breast cancer (Mingting et al., 2023). Furthermore, a recent study comprehensively analyzed the genetics of ChrX involvement in autoimmune liver disease and identified a novel genome-wide significant locus associated with primary biliary cholangitis, especially rs7059064. This locus contains seven genes: TIMM17B(NM_001167947.2), PQBP1(NM_001032381.2), PIM2(NM_006875.4), SLC35A2(NM_006875.4), OTUD5(NM_006875.4), KCND1(NM_006875.4), and GRIPAP1(NM_006875.4), as well as a super-enhancer (GH0XJ048933 in OTUD5) that regulates all of these genes (Asselta et al., 2021). Of note, a 666bp deletion (chrX:48755195-49760422) involving TIMM17B(NM_001167947.2) and PQBP1(NM_001032381.2) was found in this case, which raised doubts about its relevance to primary biliary cholangitis, especially that no biliary abnormalities were detected during ultrasound examination. Although disorders associated with TIMM17B-related genes have rarely been reported, the observed fetal phenotype does not definitively exclude a potential influence of TIMM17B deficiency, independent of the syndrome caused by PQBP1 variants.
In this case, a pregnant woman presented with three pregnancies, showing thickened nuchal translucency and nasal bone abnormalities. In addition, prenatal ultrasound revealed VSD (HP:0001629), suggesting cardiac impact. The integration of ultrasound findings, facial features, and genetic testing led to the diagnosis of Renpenning syndrome in the fetus. This case highlighted the importance of early systematic fetal screening. However, there are some limitations to consider. Renpenning syndrome caused by PQBP1 variants exhibits considerable individual variation. In this case, the fetus had a 666-bp deletion, resulting in a complete loss of PQBP1. It is unclear whether other PQBP1 variants, such as deletion frameshift nonsense variants or point missense variants, could manifest prenatal ultrasound abnormalities. Moreover, the effect of TIMM17B on fetal ultrasound findings lacks validation of relevant in vitro experiments. Finally, diseases such as Down syndrome may show prenatal ultrasound features similar to Renpenning syndrome. Although the risk of NIPT in this case was low, there were false negatives and false positives in NIPT. In addition to Trio-WES, karyotype analysis was required for differential diagnosis during chromosome screening.
Timely detection and diagnosis of chromosomal diseases during pregnancy is a major challenge. Systematic early screening of fetal development plays a crucial role in identifying genetic conditions that may affect fetal growth. It is recommended to closely monitor fetal ultrasound in the case of multiple adverse pregnancies or obvious genetic susceptibility. If abnormalities are observed, such as increased NT or abnormal ultrasound examinations, prenatal diagnosis should be considered. This may involve chromosomal testing, genomic testing, and WES through procedures such as chorionic villus sampling or amniocentesis to prevent missed diagnosis. Early genetic diagnosis could be carried out to appropriate clinical management and counseling, facilitating informed decision-making and timely clinical intervention.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of Tianjin Central Hospital of Obstetrics and Gynecology. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing. LZ: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Supervision, Writing – original draft, Writing – review and editing. YL: Data curation, Investigation, Software, Writing – original draft. LY: Conceptualization, Formal Analysis, Investigation, Methodology, Software, Writing – original draft. JC: Conceptualization, Data curation, Formal Analysis, Visualization, Writing – review and editing. QL: Formal Analysis, Investigation, Methodology, Visualization, Writing – review and editing. MN: Methodology, Software, Validation, Writing – original draft. HW: Methodology, Project administration, Visualization, Writing – original draft. RW: Methodology, Validation, Visualization, Writing – original draft. YC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This report was supported by Natural Science Foundation of Tianjin (22JCYBJC01110), Tianjin Health Research Project (TJWJ2024RC017), the 2024 Medical Service and Guarantee Capacity Improvement Project for Improving the Diagnosis and Treatment Capacity of Critically Ill Pregnant Women and Newborns.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1575378/full#supplementary-material
References
Asselta, R., Paraboschi, E. M., Gerussi, A., Cordell, H. J., Mells, G. F., Sandford, R. N., et al. (2021). X chromosome contribution to the genetic architecture of primary biliary cholangitis. Gastroenterology 160 (7), 2483–2495.e26. doi:10.1053/j.gastro.2021.02.061
Bauer, M. F., Gempel, K., Reichert, A. S., Rappold, G. A., Lichtner, P., Gerbitz, K. D., et al. (1999). Genetic and structural characterization of the human mitochondrial inner membrane translocase. J. Mol. Biol. 289 (1), 69–82. doi:10.1006/jmbi.1999.2751
Chen, M., Chen, P., Yu, S., Ai, L., Yu, X., Wang, R., et al. (2025). Retrospective study on NIPT or NIPT plus combined with ultrasound in screening fetal chromosomal abnormalities. Sci. Rep. 15 (1), 12859. doi:10.1038/s41598-025-97230-w
Cheng, S., Liu, X., Yuan, L., Wang, N., Zhang, Z. C., and Han, J. (2023). The role of PQBP1 in neural development and function. Biochem. Soc. Trans. 51 (1), 363–372. doi:10.1042/BST20220920
Cho, R. Y., Peñaherrera, M. S., Du Souich, C., Huang, L., Mwenifumbo, J., Nelson, T. N., et al. (2020). Renpenning syndrome in a female. Am. J. Med. Genet. A 182 (3), 498–503. doi:10.1002/ajmg.a.61451
Fan, Y., Zhao, J., Chen, Q., Huang, X., Li, F., and Qiao, J. (2025). Clinical analysis of a child with heterotopic ventricular gray matter renpenning syndrome caused by PQBP1 gene mutation and a literature review. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 42 (3), 314–321. doi:10.3760/cma.j.cn511374-20240923-00502
Germanaud, D., Rossi, M., Bussy, G., Gérard, D., Hertz-Pannier, L., Blanchet, P., et al. (2011). The renpenning syndrome spectrum: new clinical insights supported by 13 new PQBP1-mutated males. Clin. Genet. 79 (3), 225–235. doi:10.1111/j.1399-0004.2010.01551.x
Haghshenas, S., Foroutan, A., Bhai, P., Levy, M. A., Relator, R., Kerkhof, J., et al. (2023). Identification of a DNA methylation signature for renpenning syndrome (RENS1), a spliceopathy. Eur. J. Hum. Genet. 31 (8), 879–886. doi:10.1038/s41431-023-01313-z
Jeong, H. I., Yang, A., Kim, J., Jang, J. H., Cho, S. Y., and Jin, D. K. (2018). First Korean case of renpenning syndrome with novel mutation in PQBP1 diagnosed by targeted exome sequencing, and literature review. Ann. Clin. Lab. Sci. 48 (4), 522–527.
Kalscheuer, V. M., Freude, K., Musante, L., Jensen, L. R., Yntema, H. G., Gécz, J., et al. (2003). Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 35 (4), 313–315. doi:10.1038/ng1264
Lopez-Martín, S., Albert, J., Peña Vila-Belda, M. D. M., Liu, X., Zhang, Z.-C., Han, J., et al. (2022). A mild clinical and neuropsychological phenotype of renpenning syndrome: a new case report with a maternally inherited PQBP1 missense mutation. Appl. Neuropsychol. Child. 11 (4), 921–927. doi:10.1080/21622965.2021.1970551
Mingting, D., Yun, R., Jiwen, Z., Tingting, Z., Yanhong, W., and Hongyan, J. (2023). High expression of TIMM17B is a potential diagnostic and prognostic marker of breast cancer. Cell Mol. Biol. (Noisy-le-grand) 69 (3), 169–176. doi:10.14715/cmb/2023.69.3.25
Pan, J., Chia, H., Kusnadi, J., Li, Z., and Yu, L. (2025). Renpenning syndrome related to a missense variant in polyglutamine-binding protein 1 (PQBP1): two pediatric cases from a Chinese family and literature review. Appl. Neuropsychol. Child., 1–9. doi:10.1080/21622965.2025.2457990
Redin, C., Gérard, B., Lauer, J., Herenger, Y., Muller, J., Quartier, A., et al. (2014). Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 51 (11), 724–736. doi:10.1136/jmedgenet-2014-102554
Keywords: PQBP1, Renpenning syndrome, nuchal translucency, ultrasound, prenatal diagnosis
Citation: Shen Y, Zhang L, Li Y, Yao L, Cao J, Lin Q, Nie M, Wang H, Wei R and Chang Y (2025) Ultrasound combined with molecular genetics to diagnose hereditary Renpenning syndrome in early pregnancy: a case report. Front. Genet. 16:1575378. doi: 10.3389/fgene.2025.1575378
Received: 14 February 2025; Accepted: 10 July 2025;
Published: 21 July 2025.
Edited by:
Milind Ratnaparkhe, ICAR Indian Institute of Soybean Research, IndiaReviewed by:
Wanju Wi, Changhua Christian Hospital, TaiwanNathalie Vanden Eynde, Vrije Universiteit Brussel (VUB), Belgium
Copyright © 2025 Shen, Zhang, Li, Yao, Cao, Lin, Nie, Wang, Wei and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Chang, Y2hhbmd5aW5nNDQ3MEBzaW5hLmNvbQ==
†These authors have contributed equally to this work