 Tanguy Demaret1*†
Tanguy Demaret1*† Damien Feret1Barbara Lambert1†Delphine Pranger2Jean-Louis Dargent3†Maria Dolores Martin Martinez3Antonio Renda4
Damien Feret1Barbara Lambert1†Delphine Pranger2Jean-Louis Dargent3†Maria Dolores Martin Martinez3Antonio Renda4 Isabelle Maystadt1,5†
Isabelle Maystadt1,5†- 1Centre de Génétique Humaine, Institut de Pathologie et Génétique, Gosselies, Belgium
- 2Service d’Hématologie, Grand Hôpital de Charleroi (GHdC), Charleroi, Belgium
- 3Centre d’Anatomopathologie, Institut de Pathologie et Génétique, Gosselies, Belgium
- 4Service d’Urologie, Grand Hôpital de Charleroi (GHdC), Charleroi, Belgium
- 5Département de Médecine, Unité de Recherche en Physiologie Moléculaire (URPhyM), Université de Namur (UNamur), Namur, Belgium
CEBPA-associated familial acute myeloid leukemia (AML) is an autosomal dominant leukemia predisposition syndrome associated with germline variants in the CEBPA gene. Werner syndrome (WS) is an autosomal recessive progeroid syndrome causing premature aging and malignancies (e.g., AML). We report a 41-year-old man referred for medical genetic evaluation because of 3 synchronous tumors (colon, kidney, and thyroid) and premature aging. He underwent hematopoietic stem cell transplantation (HSCT) at 12 years of age because of AML diagnosed 3 years earlier. His sister (donor for the HSCT) and his brother later developed AML, as did two of her sister’s three children. The patient met the clinical criteria for a “probable” WS, but duo-based (urine and blood DNA) whole exome sequencing did not confirm this diagnosis. A heterozygous c.350del p.(Gly117Alafs*43) pathogenic variant in the CEBPA gene was found in the proband’s urine and blood DNA, and in his affected relatives. We postulate that AML management led to adverse effects in the proband, mimicking a WS phenotype (phenocopy). To our knowledge, this is the first report of a leukemia predisposition syndrome mimicking a progeroid syndrome. The diagnosis allowed for personalized medicine (i.e., lifelong regular complete blood count check-up) in the proband and his affected relatives.
1 Introduction
CEBPA-associated familial acute myeloid leukemia (AML) is an autosomal dominant predisposition to AML confered by germline (likely) pathogenic variant affecting the N-terminal extremity of the protein (upstream of the codon 120) (Smith et al., 2004; Tawana and Fitzgibbon, 1993). The CEBPA gene encodes the CCAAT/enhancer-binding protein alpha, a transcription factor crucial for myeloid differentiation (Avellino and Delwel, 2017). CEBPA mRNA has two translation initiation sites at codons 1 and 120, resulting in the synthesis of a full-length (42 kDa, p42) and a shorter (30 kDa, p30) isoform of C/EBPα, respectively. p42 and p30 homo- and hetero-dimerize, and their ratio influences cell fate: p42 inhibits cell proliferation and promotes differentiation, whereas p30 preponderance is associated with an immature cell state and inhibition of terminal differentiation (Schmidt et al., 2020). The proposed leukemogenesis mechanism is based on a “second hit” model (Knudson, 1971): the germline variant frequently affects the p42 isoform (the “cell differentiation” isoform, upstream of codon 120) while the somatic variant (downstream of codon 120) affects both isoforms, leaving intact only one copy of p30 (the “anti-cell differentiation” isoform) on the allele affected by the germline variant. The final consequence is thought to be a gain-of-function mechanism induced by a dramatically decreased p42/p30 ratio (Schmidt et al., 2020). CEBPA-associated familial AML is a highly penetrant predisposition with favorable long-term outcomes (Pan et al., 2024; Tawana et al., 2015). Carrier surveillance is based on complete blood count (CBC) every 6–12 months, followed by bone marrow examination in case of CBC anomalies.
Werner syndrome (WS) is an autosomal recessive progeroid syndrome caused by germline bi-allelic (likely) pathogenic variants in RECQL2, encoding the WRN protein, which is important for DNA repair and telomere maintenance (Rossi et al., 2010). Patients develop normally (until adolescence) before presenting features of accelerated aging: skin atrophy, wrinkles, bilateral cataracts, hypogonadism, early loss of fertility, short stature, high-pitched hoarse voice, osteoporosis, type 2 diabetes, and indolent ulceration around the Achilles tendon (Oshima et al., 2017). Early death (around 60 years) mostly results from myocardial infarction (secondary to premature atherosclerosis) or malignancies (leukemia, sarcoma, thyroid follicular carcinomas, - among others).
For the first time, we report a patient presenting with a possible clinical diagnosis of WS (bilateral cataracts, premature aging, short stature, hypogonadism, and multiple malignancies) who was finally diagnosed with CEBPA-associated familial AML through whole exome sequencing. This diagnosis allowed for the implementation of personalized medicine strategies.
2 Case description
Aged 41, the proband presented with 3 synchronous tumors, detected by a full-body positron emission tomography and computed tomography (PET-CT) performed during the work-up of anemia revealed by pallor and fatigue. A tubulovillous adenoma (high-grade dysplasia) of the right colic flexure, associated with multiple colon polyps (low-grade dysplasia), and an unclassified renal cell carcinoma were resected by right colectomy and partial right nephrectomy, respectively. Additionally, the patient declined the resection of a hypermetabolic nodule located inside a nodular goiter, also detected during PET-CT.
His medical history was marked by an AML, at the age of 9, managed with chemotherapy (Figure 1A). Three years later, at 12 years, he received hematopoietic stem cell transplantation (HSCT, from his older sister) to manage disease recurrence. The HSCT was pre-conditioned by total body irradiation and was complicated by a graft-versus-host-disease (GVHD), which subsequently resolved. Complete donor chimerism was confirmed on a bone marrow sample at 28 years. He further developed bilateral cataracts (surgically corrected at 26 years), hypergonadotropic hypogonadism (intermittently treated with intramuscular injections of androgens), pulmonary emphysema (in the context of tobacco product exposure), osteoporosis, hepatic steatosis, hypercholesterolemia, hypertriglyceridemia, and depression.
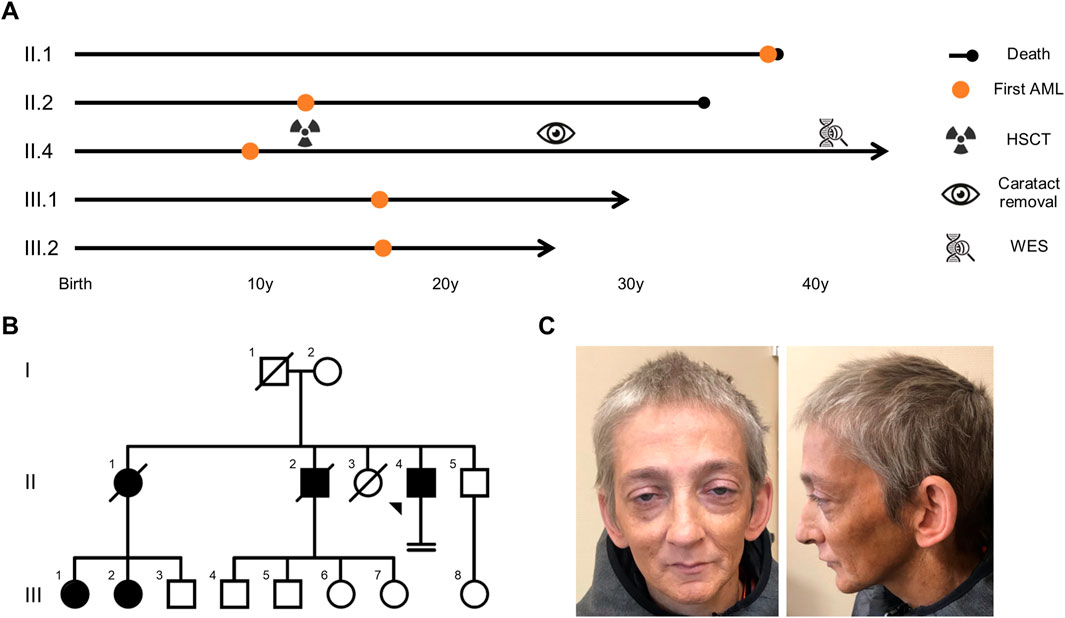
Figure 1. AML segregated over two generations in the proband’s pedigree, with an age of onset comprised between 9 and 37 years of age. (A) Overview of the proband’s and his relatives’ medical journey. (B) Family tree presenting females (circles) and males (squares) affected (filled symbols) by AML. Arrow: proband, crossed symbol: deceased, horizontal double lines: infertility. (C) Proband’s face and profile photographs (taken at 41 years) highlighting premature aging and bilateral ptosis. AML: acute myeloid leukemia, HSCT: hematopoietic stem cell transplantation, WES: whole exome sequencing.
He is the fourth child of unrelated parents originating from Belgium with no known hematological malignancies in their ancestors. Their two eldest children died of AML at the ages of 38 years (AML diagnosed at 37) and 34 years (AML diagnosed at 12), respectively (Figure 1B). The oldest of the two was the donor for the proband’s HSCT. She had 3 children, two of whom developed AML before the age of 18.
Clinical examination showed a progeroid appearance (Figure 1C) characterized by premature graying of hair, bilateral ptosis, a beaked nose, wrinkles, a prematurely aged face, and short stature (156.5 cm and 35 kg, corresponding to a body-mass index of 14.3 kg/m2). The patient also presented with a hoarse high-pitched voice.
3 Diagnostic assessment
Based on the multiple tumors, the proband’s urologist requested the sequencing of a gene panel including 38 genes (Supplementary Table S1) involved in hereditary cancer syndromes (Roche KapaHypercapV3 capture followed by Illumina sequencing, mean coverage 3582x). Testing was performed on DNA extracted from blood (patient’s sister DNA), urine, and buccal swab (patient’s own DNA). The analysis revealed no (likely) pathogenic variant explaining the proband’s phenotype (WRN and CEBPA genes were not included, see below).
The patient was referred to our center for further medical genetic evaluation. Based on the patient’s phenotype, the hypothesis of WS was considered. The patient met the clinical criteria for a “probable” WS (Table 1 (Oshima et al., 2017)). A duo-based (DNA isolated from urine and blood, the latter being the patient’s sister DNA) whole exome sequencing (Roche KAPA HyperExome capture followed by Illumina sequencing, mean coverage 300x) revealed a heterozygous c.350del p.(Gly117Alafs*43) variant in the CEBPA gene (NM_004364.5) (Figure 2) which was confirmed by Sanger sequencing (ClinVar SCV005387846.1). The variant was not found in large databases (gnomAD and ClinVar) or in our in-house exome database. It affects the N-terminal end of the protein and segregates with the disease (see below). According to the American College of Medical Genetics criteria, this variant was classified as pathogenic (class 5: PVS1, PS3, PM1, PM2, and PP1) (Richards et al., 2015). No class 3 variant (or higher) was detected in the WRN gene, thus providing no molecular evidence for WS to explain the patient’s phenotype (phenocopy).

Table 1. Werner syndrome diagnostic criteria (Oshima et al., 2017).
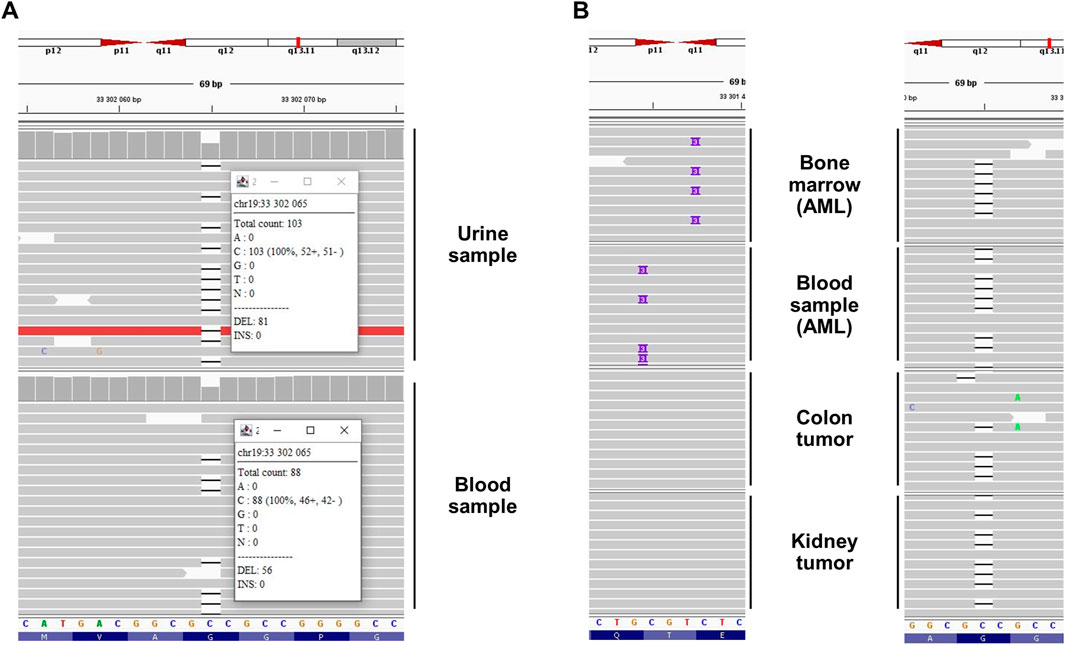
Figure 2. Next-generation sequencing results highlighted a germline familial c.350del CEBPA variant related to CEBPA-associated familial AML in (A) the proband’s urine (own DNA) and blood (sister’s DNA, due to HSCT) and (B) the proband’s (colon and kidney tumors) and relatives’ (bone marrow and blood) samples. In the bone marrow and blood samples, somatic variants (c.925_927dup and c.930_931insAAG, respectively) affecting the C-terminal extremity of the protein were detected (“second hit”) in addition to the germline variant. Integrative Genomics Viewer screenshots with black dash and purple rectangle representing deletion of one nucleotide and insertion of three nucleotides, respectively. Note that CEBPA is located on the minus strand of the chromosome. Thus the 5′ extremity is presented on the right side of the figure. AML: acute myeloid leukemia, HSCT: hematopoietic stem cell transplantation.
The molecular mechanism leading to AML associated with a germline CEBPA variant is based on a somatic second hit at the C-terminal extremity of the protein (after codon 120) (Pabst et al., 2008; Pabst and Mueller, 2009). This mechanism was confirmed in blood and bone marrow samples from two relatives (Individuals II.2 and III.1) during AML (Table 2, Figure 2). We hypothesized that this mechanism could also underlie the tumorigenesis of the colon adenoma and the unclassified renal cell carcinoma. However, both tumor DNA sequencing analyses (Archer VARIANTPlex Core Myeloid amplification followed by Illumina sequencing, mean CEBPA coverage 2080x and 3560x) detected no second variant in the CEBPA gene (Table 2, Figure 2), and could not support our hypothesis for solid tumorigenesis in the proband. Tumor DNA methylation profiling array (Infinium MethylationEPIC BeadChip from Illumina) detected partial chromosomal losses and amplifications (Supplementary Figures S1, 2) but failed to provide information about tumor classification and/or tumorigenesis mechanisms. MGMT promoter was not found to be methylated (Bady et al., 2016).
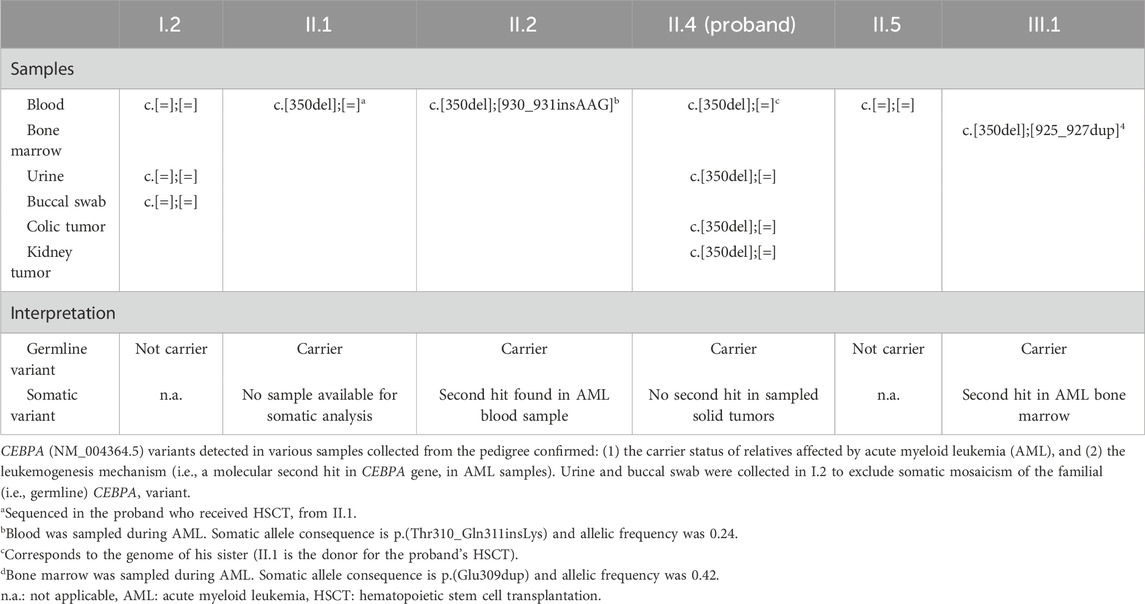
Table 2. CEBPA sequencing results.
Maternal follow-up testing in three different samples (blood, urine, and buccal swab) did not detect the familial CEBPA variant. The father could not be tested (deceased from a non-malignant cause at the age of 66) and there was no history of hematological malignancy on his side of the family. The recurrence of the pathogenic variant in 3 out of 5 siblings suggests the hypothesis of parental germline mosaicism as a potential explanation, especially considering that maternal screening was negative and both the father’s personal medical history and family history were unremarkable for hematological malignancies.
4 Discussion
We report an adult patient presenting with three synchronous tumors (colon, kidney, and thyroid) following childhood (ages 9–12) AML management with chemotherapy, total body irradiation, and HSCT received from his deceased sister (who died of AML at 38 years). WS was clinically suspected but not confirmed by WES which revealed a pathogenic (class 5) truncating variant in the CEBPA gene, associated with familial AML, in the patient’s urine (own DNA) and blood (sister’s DNA). Blood and bone marrow sequencing confirmed the leukemogenesis mechanism by the presence of a second hit in the CEBPA gene in two relatives affected by AML. No molecular second hit was found in the two solid tumors resected from the proband, thus making the tumorigenesis mechanism unlikely related to the germline CEBPA variant.
The germline origin of the c.350del p.(Gly117Alafs*43) CEBPA variant has been confirmed on three different samples collected from the proband (urine, kidney and colon) and on three relatives’ blood/bone marrow samples (i.e., familial inheritance of the variant). These results allow to firmly confirm the germline origin of the variant without requiring skin fibroblast DNA sequencing [i.e., the gold-standard method to confirm germline origin (Desai et al., 2017)].
The occurrence of a somatic variant in the CEBPA gene, known as the “second hit”, is the mechanism leading to AML in patients carrying a germline CEBPA variant, as confirmed in this pedigree. However, we found no evidence to support the same mechanism explaining the colon and kidney tumorigenesis in the patient. Interestingly, a 44-year-old patient carrying a germline CEBPA variant died of rectal cancer, without prior AML treatment (Pathak et al., 2016). This case suggests a potential predisposition to solid tumor associated with germinal CEBPA variants. Although this association has not been confirmed, it warrants further evaluation in future studies, given that CEBPA downregulation has been observed in solid tumors and is a potential therapeutic target (Chan et al., 2019; Koschmieder et al., 2008).
The proband presented with several signs and symptoms reported in WS (Table 1), yet no molecular etiology could be found to support this diagnosis. It is likely that all the harbored WS criteria are late effects of the leukemia management (alkylating agents and corticosteroid leading to cataracts, TBI causing dyslipidemia, gonadal dysfunction, and second cancers) (Chow et al., 2016; Inamoto and Lee, 2017) or their consequences (dermatological pathology can be secondary to GVHD, and hypogonadism can explain the short stature, premature hair graying and progeroid appearance) (Phelan et al., 2022).
Xiao and colleagues reported a 36-year-old patient who presented with donor cell leukemia (DCL) 13 months after having received HSCT (from his healthy sister) to manage AML (Xiao et al., 2011). CEBPA gene sequencing revealed the presence of a germline variant in the patient and the donor (his sister), meaning that both were predisposed to AML. In our case, the proband faces the same risk as the patient reported in this case report, because both have received HSCT from a donor carrying a germline CEBPA variant. Consequently, these findings suggest that CEBPA gene screening should be considered prior to any intrafamilial HSCT in the context of AML, to prevent the use of donors carrying germline CEBPA variants.
One of the limitations of this study is that we were not able to obtain precise information about the initial AML management in the proband (dose and duration of chemotherapy, dose of irradiation, etc.), which impeded drawing a precise correlation between the intensity of the management and the adverse effects presented by the patient. In addition, we had no access to the proband’s AML sample, which precluded establishing an unequivocal link between the AML of the proband and the CEBPA gene AML predisposition. However, this link has been confirmed in two relatives (individuals II.2 and III.1, Table 2). Finally, the fact that we had no thyroid tumor sample, and that WES was not performed on all samples (due to funding limitations), is restraining further the deciphering of tumorigenesis mechanisms.
Thanks to WES, we were able to identify the etiology of AML segregating in a pedigree, which allowed for personalized medicine. Further genetic testing (i.e., tumor CEBPA sequencing) provided no evidence supporting a link between the solid tumors presented by the proband and the AML predisposition. The diagnosis provided a recurrence risk of AML and a personalized surveillance plan for the proband and his relatives. It will also prevent using a CEBPA variant carrier as familial donor for HSCT in the future.
5 Patient perspective
The genetic diagnosis was able to exclude the severe diagnosis of WS in the proband. Yet, the AML treatment by HSCT received from his sister who later developed and died of AML, made him aware that he was at risk for DCL well before the genetic diagnosis was known. He told us that the genetic diagnosis (i.e., CEBPA-associated familial AML) was a formal confirmation of what he had already guessed some years ago.
The proband has been advised to do a check-up with CBC every 6 months.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The requirement of ethical approval was waived by Comité d'Ethique de l’Institut de Pathologie et Génétique for the studies involving humans because it is a retrospective study based on samples collected during clinical care. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
TD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Writing – original draft, Writing – review and editing. DF: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review and editing. BL: Investigation, Methodology, Writing – original draft, Writing – review and editing. DP: Data curation, Formal Analysis, Writing – original draft, Writing – review and editing. J-LD: Formal Analysis, Methodology, Writing – original draft, Writing – review and editing. MMM: Formal Analysis, Methodology, Writing – original draft, Writing – review and editing. AR: Formal Analysis, Investigation, Methodology, Writing – original draft, Writing – review and editing. IM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Funding covering publication fees has been granted by the Conseil Scientifique from the Institut de Pathologie et Génétique.
Acknowledgments
The authors thank the patient and his family for their collaboration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1575651/full#supplementary-material
References
Avellino, R., and Delwel, R. (2017). Expression and regulation of C/EBPα in normal myelopoiesis and in malignant transformation. Blood 129, 2083–2091. doi:10.1182/blood-2016-09-687822
Bady, P., Delorenzi, M., and Hegi, M. E. (2016). Sensitivity analysis of the MGMT-STP27 model and impact of genetic and epigenetic context to predict the MGMT methylation status in gliomas and other tumors. J. Mol. Diagnostics 18, 350–361. doi:10.1016/j.jmoldx.2015.11.009
Chan, E. M., Shibue, T., McFarland, J. M., Gaeta, B., Ghandi, M., Dumont, N., et al. (2019). WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556. doi:10.1038/s41586-019-1102-x
Chow, E. J., Anderson, L., Baker, K. S., Bhatia, S., Guilcher, G. M. T., Huang, J. T., et al. (2016). Late effects surveillance recommendations among survivors of childhood hematopoietic cell transplantation: a Children’s oncology group report. Biol. Blood Marrow Transpl. 22, 782–795. doi:10.1016/j.bbmt.2016.01.023
Desai, A. V., Perpich, M., and Godley, L. A. (2017). Clinical assessment and diagnosis of germline predisposition to hematopoietic malignancies: the university of Chicago experience. Front. Pediatr. 5, 252. doi:10.3389/fped.2017.00252
Inamoto, Y., and Lee, S. J. (2017). Late effects of blood and marrow transplantation. Haematologica 102, 614–625. doi:10.3324/haematol.2016.150250
Knudson, A. G. (1971). Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U. S. A. 68, 820–823. doi:10.1073/pnas.68.4.820
Koschmieder, S., Halmos, B., Levantini, E., and Tenen, D. G. (2008). Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J. Clin. Oncol. 27, 619–628. doi:10.1200/JCO.2008.17.9812
Oshima, J., Sidorova, J. M., and Monnat, R. J. (2017). Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 33, 105–114. doi:10.1016/j.arr.2016.03.002
Pabst, T., Eyholzer, M., Haefliger, S., Schardt, J., and Mueller, B. U. (2008). Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J. Clin. Oncol. 26, 5088–5093. doi:10.1200/jco.2008.16.5563
Pabst, T., and Mueller, B. U. (2009). Complexity of CEBPA dysregulation in human acute myeloid leukemia. Clin. Cancer Res. 15, 5303–5307. doi:10.1158/1078-0432.CCR-08-2941
Pan, L., Li, Y., Gao, H., Lai, X., Cai, Y., Chen, Z., et al. (2024). Clinical features and management of germline CEBPA-mutated carriers. Leuk. Res. 138, 107453. doi:10.1016/j.leukres.2024.107453
Pathak, A., Seipel, K., Pemov, A., Dewan, R., Brown, C., Ravichandran, S., et al. (2016). Whole exome sequencing reveals a C-terminal germline variant in CEBPA-associated acute myeloid leukemia: 45-year follow up of a large family. Haematologica 101, 846–852. doi:10.3324/haematol.2015.130799
Phelan, R., Im, A., Hunter, R. L., Inamoto, Y., Lupo-Stanghellini, M. T., Rovo, A., et al. (2022). Male-specific late effects in adult hematopoietic cell transplantation recipients: a systematic review from the late effects and quality of life working committee of the center for international blood and marrow transplant research and transplant complications working party of the European society of blood and marrow transplantation. Bone Marrow Transpl. 57, 1150–1163. doi:10.1038/s41409-022-01591-z
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Rossi, M. L., Ghosh, A. K., and Bohr, V. A. (2010). Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 9, 331–344. doi:10.1016/j.dnarep.2009.12.011
Schmidt, L., Heyes, E., and Grebien, F. (2020). Gain-of-Function effects of N-terminal CEBPA mutations in acute myeloid leukemia. Bioessays 42, e1900178. doi:10.1002/bies.201900178
Smith, M. L., Cavenagh, J. D., Lister, T. A., and Fitzgibbon, J. (2004). Mutation of CEBPA in familial acute myeloid leukemia. N. Engl. J. Med. 351, 2403–2407. doi:10.1056/NEJMoa041331
Tawana, K., and Fitzgibbon, J. (1993). “CEBPA-associated familial acute myeloid leukemia (AML),” in GeneReviews®. Editors M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and A. Amemiya (Seattle (WA): University of Washington, Seattle).
Tawana, K., Wang, J., Renneville, A., Bödör, C., Hills, R., Loveday, C., et al. (2015). Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126, 1214–1223. doi:10.1182/blood-2015-05-647172
Keywords: whole exome sequencing, personalized medicine, tumor sequencing, case report, phenocopy
Citation: Demaret T, Feret D, Lambert B, Pranger D, Dargent J-L, Martin Martinez MD, Renda A and Maystadt I (2025) CEBPA-associated familial acute myeloid leukemia mimicking Werner syndrome: a case report. Front. Genet. 16:1575651. doi: 10.3389/fgene.2025.1575651
Received: 13 February 2025; Accepted: 02 April 2025;
Published: 17 April 2025.
Edited by:
Enrico Attardi, St. Jude Children’s Research Hospital, United StatesReviewed by:
Giacomo Coltro, University of Florence, ItalyLuca Guarnera, Policlinico Tor Vergata, Italy
Copyright © 2025 Demaret, Feret, Lambert, Pranger, Dargent, Martin Martinez, Renda and Maystadt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanguy Demaret, dGFuZ3V5LmRlbWFyZXRAaXBnLmJl
†ORCID: Tanguy Demaret, orcid.org/0000-0001-7605-9031; Barbara Lambert, orcid.org/0000-0002-9458-8751; Jean-Louis Dargent, orcid.org/0000-0002-1950-0289; Isabelle Maystadt, orcid.org/0000-0001-7170-8186