 Sanjay Kharte1†
Sanjay Kharte1† Ashish Kumar1*†
Ashish Kumar1*† Priyamvada Mishra2
Priyamvada Mishra2 R. S. Ramakrishnan3
R. S. Ramakrishnan3 Stuti Sharma4
Stuti Sharma4 Nishi Mishra5
Nishi Mishra5 Puneet Singh Chauhan2
Puneet Singh Chauhan2 Radheshyam Sharma6
Radheshyam Sharma6 Vedant Gautam1
Vedant Gautam1 Shweta Tiwari5
Shweta Tiwari5 Vinod Goyal5Sonu Sharma5G. K. Koutu5
Vinod Goyal5Sonu Sharma5G. K. Koutu5 N. K. Joshi7
N. K. Joshi7- 1Department of Plant Pathology, Jawaharlal Nehru Agricultural University, Jabalpur, Madhya Pradesh, India
- 2CSIR-National Botanical Research Institute, Lucknow, Uttar Pradesh, India
- 3Department of Plant Physiology, Jawaharlal Nehru Agricultural University, Jabalpur, Madhya Pradesh, India
- 4Department of Genetics and Plant Breeding, Jawaharlal Nehru Agricultural University, Jabalpur, Madhya Pradesh, India
- 5Seed Technology Research Centre, Jawaharlal Nehru Agricultural University, Jabalpur, Madhya Pradesh, India
- 6Biotechnology Centre, Jawaharlal Nehru Agricultural University, Jabalpur, Madhya Pradesh, India
- 7Department of Entomology and Plant Pathology, University of Arkansas, Fayetteville, AR, United States
Background: Fusarium oxysporum f. sp. lentis is a major fungal pathogen that causes vascular wilt in lentil crops, leading to significant reductions in yield. Despite its importance, the genetic underpinnings of this pathogen remain poorly understood.
Methods: We performed whole-genome sequencing of F. oxysporum f. sp. lentis using the Illumina Shotgun Sequencing platform. The resulting high-quality genome assembly consisted of 12,366 contigs with a total length of 124.48 Mb. Genome completeness was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis, and functional annotation was performed through comparisons with several public databases, including Uniprot, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Pfam, and Clusters of Orthologous Groups (COG). Pathogenicity-related genes were identified using the PHI-base database, and secondary metabolite biosynthesis was analyzed with AntiSMASH.
Results: The genome assembly achieved 99% completeness, identifying 116,998 protein-coding genes. A total of 16,779 carbohydrate-active enzymes (CAZymes) could be detected, highlighting the pathogen’s potential for plant cell wall degradation. Pathogenicity analysis revealed genes linked with moderate virulence. AntiSMASH detected 77 biosynthetic gene clusters (BGCs), including those encoding Type I polyketide synthases (T1PKS) and non-ribosomal peptide synthetases (NRPS), which may contribute to pathogenicity.
Discussion:: The comprehensive genomic analysis of F. oxysporum f. sp. lentis offers valuable insights into its pathogenic mechanisms, including plant cell wall degradation and secondary metabolite production. These findings pave the way for future research on host-pathogen interactions and the development of targeted disease management strategies.
1 Introduction
Lentil (Lens culinaris (L.) Medik.) is a nutritionally significant pulse crop cultivated across various regions, including the Indian subcontinent, West Asia, Ethiopia, and North Africa. In India, chickpea, pigeon pea, and lentil are the predominant pulse crops, collectively accounting for over 60% of the nation’s total pulse production. Globally, lentil production reached approximately 5.73 million tons (FAO, 2021). Lentils are an annual, self-pollinating diploid crop characterized by a genome size of approximately 4 Gb (2n = 14) (Arumuganathan and Earle, 1991). Lentil growers worldwide face similar biotic and abiotic stresses which significantly reduce productivity, particularly due to soil and seed-borne diseases, that cause high mortality (Bayaa and Erskine, 1998; Infantino et al., 2006). Notably, strains within the Fusarium genus impose substantial constraints on global grain and forage legume production (Rubiales et al., 2015). Fusarium oxysporum is a widely distributed soilborne fungal pathogen responsible for vascular wilt diseases in numerous plant species. Its management remains challenging, despite attempts through chemicals (Gordon, 2017; Kharte et al., 2022) and biological control (Garkoti et al., 2013). Among the various biotic stresses affecting lentil yield, wilt disease, caused by F. oxysporum f. sp. lentis, presents a significant challenge. Addressing this issue is crucial for enhancing lentil production and ensuring higher yields (Choudhary and Mohanka, 2012). In India, wilt is a major concern in the states of Uttar Pradesh, Madhya Pradesh, Bihar, West Bengal, and other lentil growing regions, with reported infections ranging from 25% to 95% (Khare, 1980). Despite its agricultural significance, the molecular basis of pathogenicity, host specificity, and adaptation mechanisms in F. oxysporum f. sp. lentis remains largely unexplored. Resistant cultivars remain a key strategy for managing lentil wilt, with resistant germplasm serving as valuable input for molecular breeding programs (Kumar et al., 2021).
Several molecular markers have been employed for the characterization and classification of Fusarium species (Baayen et al., 1997; Lievens et al., 2008; Baysal et al., 2010; Sharma et al., 2014; van Dam et al., 2016). Additionally, genes associated with virulence have been targeted for the molecular discrimination of fungal strains. (Lievens et al., 2008; van Dam et al., 2016). Many studies have reported that secreted-in xylem (SIX) genes, primarily located on a single chromosome, are linked to the pathogenicity of F. oxysporum (Rep et al., 2004; Houterman et al., 2007; Kashiwa et al., 2013; Carvalhais et al., 2019). Strains of F. oxysporum exhibit variations in gene content, sequences, and chromosome numbers due to rearrangements and the mobility of lineage-specific chromosomes (Davière et al., 2001; Ma et al., 2010; Schmidt et al., 2013; Vlaardingerbroek et al., 2016; Nelson et al., 1983). Analysis of publicly available F. oxysporum genome assemblies reveal that genome sizes ranges from approximately 50–70 Mb, varying among various formats. (data of the NCBI Genome database, https://www.ncbi.nlm.nih.gov/genome/browse/#!/eukaryotes/707/). The genome of F. oxysporum exhibits high plasticity, comprising a conserved core genome essential for survival and an accessory genome enriched with lineage-specific virulence factors (Ma et al., 2010). The accessory genome contains key pathogenicity-related elements, including SIX effectors, transposable elements, and secondary metabolite gene clusters, all of which contribute to host specificity and adaptation (van Dam et al., 2016). Comparative genomic analyses across different F. oxysporum formae speciales have revealed substantial variation in effector repertoires, highlighting the role of horizontal gene transfer in evolution of pathogenicity (Schmidt et al., 2013). Advances in high-throughput sequencing technologies have significantly expanded fungal genomics, enabling a deeper understanding of pathogen diversity, virulence mechanisms, and intricate host-pathogen interactions at the genomic level (Kersey, 2019). Whole-genome sequencing (WGS) offers valuable insights into genetic variations, virulence-associated genes, and the biosynthetic pathways of secondary metabolites that enhance fungal pathogenicity (Schardl et al., 2013). Among sequencing platforms, Illumina sequencing technology has gained widespread acceptance due to its high accuracy and cost-effectiveness, making it suitable for fungal genome sequencing. This technology significantly facilitates de novo genome assembly and comparative genomic studies, thereby advancing our understanding of fungal biology (Goodwin et al., 2016). In this study, we employed Illumina-based whole-genome sequencing to generate a high-quality draft genome assembly of F. oxysporum f. sp. lentis. Genome annotation was performed to identify candidate pathogenicity gene clusters, carbohydrate-active enzymes (CAZymes), and secondary metabolite biosynthetic gene clusters associated with lentil infection. Our findings provide a genomic framework that elucidates the molecular mechanisms underlying F. oxysporum pathogenicity and contribute to the development of genome-informed strategies for disease resistance in lentil breeding programs.
2 Materials and methods
2.1 Fungal strain culture and isolation
The F. oxysporum f. sp. lentis isolate used in this study was collected from the infected lentil plant showing typical Fusarium wilt symptoms from the wilt infected fields of Damoh, Madhya Pradesh, India. The isolate was cultured on potato dextrose agar (PDA) and incubated at 27°C ± 2°C for 7 days. Fungal mycelia were picked from liquid potato dextrose broth (PDB) cultures grown under shaking conditions (150 rpm) at 25°C for 5 days (Sicard et al., 1997; Nelson et al., 1983).
2.2 DNA extraction and purification
The high-quality DNA was extracted from a seven-day-old using the cetyltrimethylammonium bromide (CTAB) method with slight modifications (Doyle and Doyle, 1987). Briefly, mycelia were collected by vacuum filtration, and ground to a fine powder in liquid nitrogen using a mortar and pestle. The fine powdered samples were incubated in DNA extraction buffer (2% CTAB, 1.4 M NaCl, 100 mM Tris-HCl pH 8.0, 20 mM EDTA, and 0.2% β-mercaptoethanol) at 65°C for 45 min. DNA was purified with phenol:chloroform: isoamyl alcohol (25:24:1) solution and precipitated with cold isopropanol. The DNA pellet was washed with 70% ethanol, air-dried, and resuspended in 1X TE buffer. The quantification of extracted DNA was calculated using a Nanodrop spectrophotometer (Thermo Fisher Scientific, United States) and a Qubit fluorometer (Invitrogen, United States). DNA integrity was evaluated by agarose gel electrophoresis (0.8% w/v). Only high-quality DNA (A260/A280 ratio ∼1.8–2.0, and no visible degradation) was preferred for sequencing.
2.3 DNA library preparation and sequencing
A single isolate of Fusarium was used for Whole Genome Sequencing by Illumina (Illumina Novaseq with 150 × 2 chemistry). The sequencing library was constructed using isolated DNA according to the manufacturer’s instructions in NEBNext® Ultra™ II FS DNA Library Prep Kit for Illumina (https://international.neb.com/protocols/2017/10/25/protocol-for-fs-dna-library-prep-kit-e7805-e6177-with-inputs-greater-than-or-equal--to-100-ng). The quality control of prepared library was executed using Agilent tape station. Subsequently sequenced 150 bp reads at each end using Illumina.
2.4 Genome assembly and quality assessment
With 121 k-mer lengths, the Spades assembler version 3.15.5 (Bankevich et al., 2012) performed the de novo genome assembly of produced sequence data. The outcomes for reliable assembly were gained after eliminating contigs whose length was less than 200 bp. Using Illumina reads, the complete genome sequence was assembled and then Quake (Kelley et al., 2010) was used for error correction before scaffolding with the help of SOAPdenovo v2.04 (Luo et al., 2012). GapCloser v1.12 (Luo et al., 2012) was laboring to close gaps within the scaffolds. The quality and the completeness of the assembled genome were evaluated through the use of BUSCO v5.3.2 (Benchmarking Universal Single-Copy Orthologs) with the Ascomycota_odb10 database (Simão et al., 2015).
2.5 Genome prediction and annotation
Protein coding sequences were predicted via the automated pipeline MAKER2 (v2.31.9) (Holt and Yandell, 2011). The first reiteration of MAKER2 combined evidence from known mRNAs, proteins, and ab initio predictions with SNAP (Korf, 2004) and GeneMark-ES v2.3a (Cantarel et al., 2008; Ter-Hovhannisyan et al., 2008). The SNAP hidden Markov models (HMM) were adjusted using the Core Eukaryotic Genes Mapping Approach (CEGMA) output, while GeneMark-ES parameters were self-trained using genome scaffolds >90 bp. The result was then used to train the evidence-based predictor, Augustus (Stanke and Morgenstern, 2005). The resultant gene sets were combined to acquire the most inclusive set of non-redundant reference genes.
2.6 Functional annotation
Functional annotation of protein-coding genes was executed based on sequence homology searches counter to major biological databases. The eggNOG-mapper tool (Huerta-Cepas et al., 2019) was utilized to assign Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (https://www.genome.jp/kegg/). In addition, protein models were aligned using BLASTP (Altschul et al., 1997) with an e-value threshold of ≤1e-5, Non-Redundant (NR) database, protein families (Pfam), and SwissProt (BLASTP cut-off e-value≤1e-5), and then classified according to Clusters of Orthologous Groups of proteins (COG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000). KOBAS software was used to test the statistical enrichment of differentially expressed genes in KEGG pathways (Xie et al., 2011). The Gene Ontology (GO) terms were used for tree hierarchical classification. KEGG pathway analysis assisted metabolic functions, and PHI-base (Pathogen-Host Interaction Database) (https://www.phi-base.org/) was used to identify putative pathogenicity-related genes (Urban et al., 2020). Repetitive sequences were soft-masked using the Tantan tool integrated into the Funannotate pipeline to retain genome integrity during gene prediction (Palmer and Stajich, 2017). Initially, raw genomic sequences were preprocessed using the funannotate clean command to remove contaminant or low-quality sequences. Tantan was then employed to identify and soft-mask simple repeats by converting them to lowercase letters, thereby preserving sequence length and positional information without altering the underlying sequence data. The masked genome was subsequently used as input for funannotate predict, ensuring accurate gene annotation while accounting for repetitive regions. Further, tRNA genes were predicted using tRNAscan-SE, integrated within the Funannotate pipeline, to support comprehensive genome annotation. The analysis began with genome preparation, followed by execution of the funannotate predict module, which incorporates tRNAscan-SE for the identification of tRNA genes. The resulting annotations were reviewed to confirm the presence and classification of tRNA loci, contributing to the overall gene annotation profile of the genome.
2.7 Identification of carbohydrate-active enzymes
Carbohydrate-active enzymes (CAZymes) in the F. oxysporum f. sp. lentis genome were recognized using the CAZy database (Lombard et al., 2014). Protein sequences were interrogated against the CAZy database using BLASTP with DIAMOND and HMMscan, applying an e-value threshold of e-value ≤1e−5. Predicted CAZymes were classified into glycoside hydrolases (GHs), glycosyltransferases (GTs), carbohydrate-binding modules (CBMs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), and auxiliary activities (AAs) based on their catalytic functions.
2.8 Identification of secondary metabolite gene clusters
Biosynthetic gene clusters (BGCs) related to secondary metabolite (SM) production in F. oxysporum f. sp. lentis were recognized using the fungal antiSMASH v7.0 beta pipeline (Blin et al., 2023). Default parameters were selected for the analysis. AntiSMASH employs profile hidden Markov models (HMMs) to precisely detect all known secondary metabolite gene clusters.
To further illustrate the identified gene clusters, BLASTP searches were executed against the NCBI Genome Portal Software Platform (NCBI, 2023), and functional annotations were assigned. The known gene clusters were classified into polyketide synthases (PKSs), non-ribosomal peptide synthetases (NRPSs), hybrid PKS-NRPSs, terpene synthases, and other known biosynthetic gene clusters.
2.9 Synteny Analysis
Whole-genome protein sequences and gene positions for F. oxysporum f. sp. coagulans (reference strain) were retrieved from the NCBI Genome Database [Accession number: GCA_014154955.1, GCA_014154955.1_SMRT_HiC_Fo5176_genomic] (NCBI, 2023). If a gene had numerous transcripts, only the first transcript was taken for analysis. Comparative genomic assessment was examined to evaluate gene synteny between F. oxysporum F. sp. lentis and the reference genome. Protein-coding genes were compared using BLASTP, retentive the top five non-self-hits per target genome with an e-value threshold of e-value≤1e−5. Synteny analysis was performed by MCScanX package (Wang et al., 2012), aiding the documentation of conserved gene blocks and genome rearrangements.
3 Result
The genome of F. oxysporum f. sp. lentis was sequenced using a whole-genome shotgun strategy on the Illumina platform. A total of 157,942,234 reads (Figure 1) were generated, comprising 78,971,117 paired-end reads (150 bp each). Subsequently quality control and preprocessing, 77,890,107 clean reads (ranging from 36 to 150 bp) were reserved, with a GC content of 48% in raw reads and 47% in clean reads (Table: 1). De novo assembly of the processed reads resulted in 12,366 contigs. The majority of contigs (65,981) ranged from 1 to 1,000 bp in length, followed by 7,992 contigs in the 1,001–5,000 bp range (Figures 2A–D). The total assembly spanned 114,006,452 bp, with the largest contig measuring 1,197,421 bp. The highest total contig length (124,483,966 bp) was observed in the 1–1,000 bp range. The assembly yielded an N50 value of 51,992 bp and an L50 of 353 (Table 2), indicating a high-quality genome assembly.
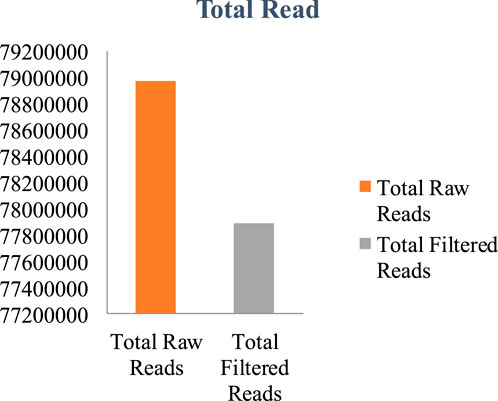
Figure 1. Whole genome assembly- Total raw and filtered reads.

Table 1. Quality control attributes of reads of sequence generated.
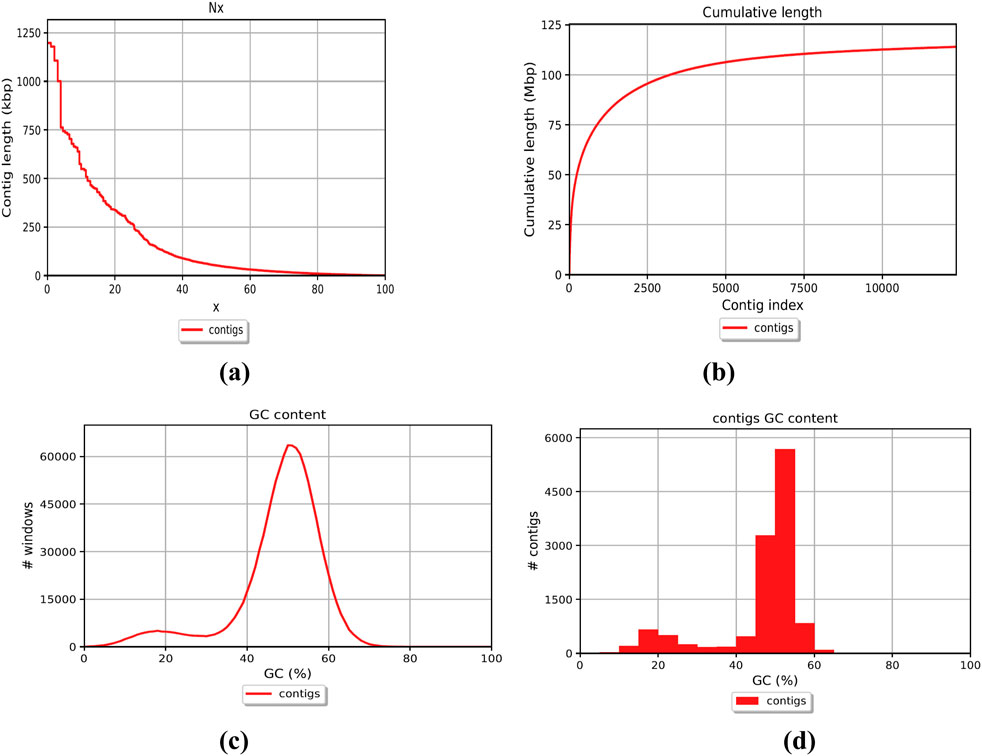
Figure 2. Whole genome assembly; Contigs representation (a) Contig length (b) Contig’s cumulative length (c) Contig GC percent (d) Contig’s coverage histogram.
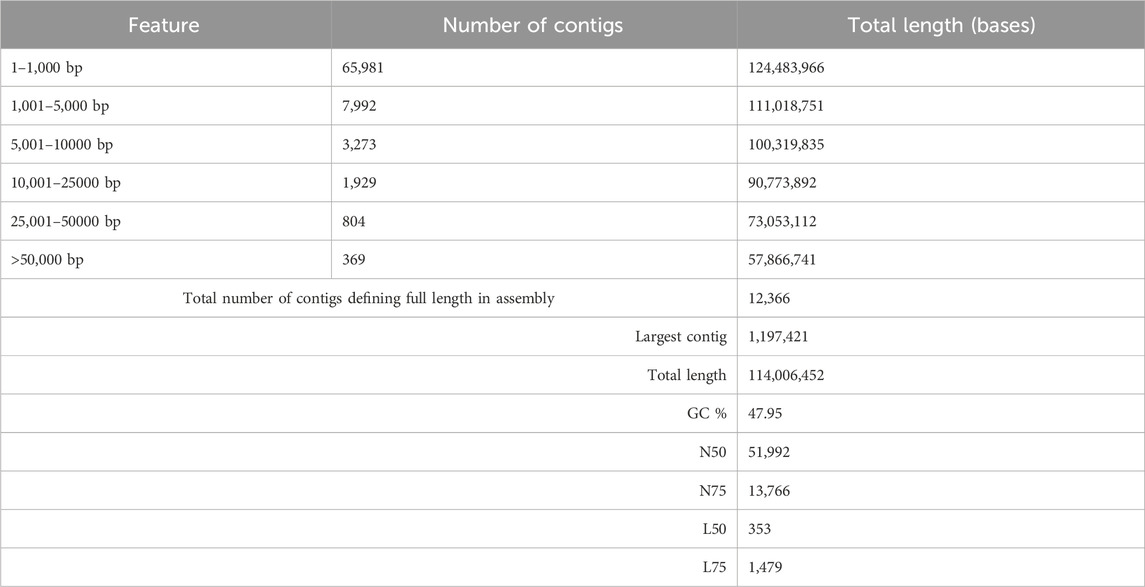
Table 2. Whole genome sequencing assembly statistics.
3.1 Gene prediction and annotation
The completeness of the assembled genome was estimated via Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis, which specified a 99% completeness score. Of the 758 BUSCO groups analyzed, 58 were recognized as complete single-copy BUSCOs, 694 as complete duplicated BUSCOs, 1 as a fragmented BUSCO, and 5 as missing BUSCOs (Figure 3; Table 3). A total of 116,998 protein-coding genes were predicted in the F. oxysporum genome. Functional annotation of the predicted genes identified homologies with known databases such as UniProt; 30,579 genes (26.14%), Gene Ontology (GO); 44,376 genes (37.93%), KEGG Pathways; 433 genes (0.37%), Pfam (Protein Families Database); 209,329 genes, Clusters of Orthologous Groups (COG); 21,001 genes (17.95%), Carbohydrate-Active Enzymes (CAZymes); 16,779 genes (14.34%), PHI-base (Pathogen-Host Interactions Database); 2,937 genes (2.51%), and antiSMASH (Secondary Metabolite Gene Clusters Analysis); 76 genes, which identified genes associated with secondary metabolism. The results highlighted a well-annotated genome enriched with miscellaneous functional elements related to metabolic processes, pathogenicity, and secondary metabolite biosynthesis (Figure 4; Table 4).

Figure 3. Whole genome assembly analysis: Completeness of genome–BUSCO (Benchmarking Universal Single-Copy Orthologs) analysis.
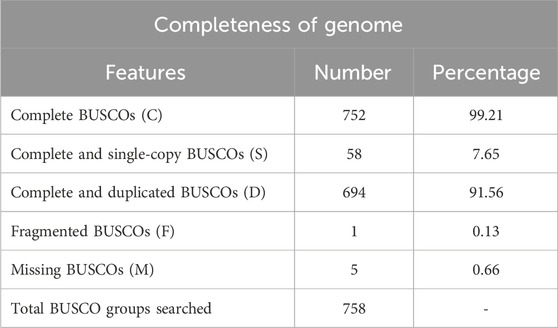
Table 3. BUSCO (Benchmarking Universal Single-Copy Orthologs) analysis for completeness of genome.
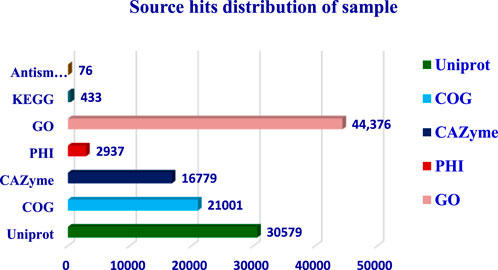
Figure 4. Whole genome assembly analysis: Gene prediction and Functional annotation.
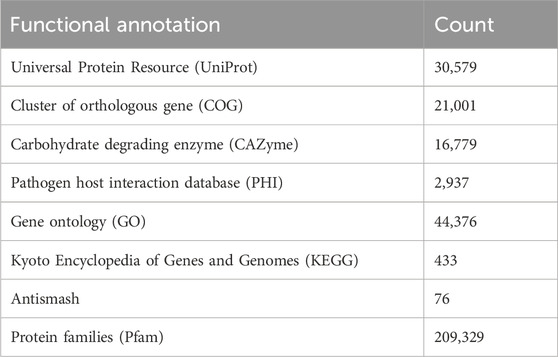
Table 4. Gene prediction and functional annotation.
3.2 Functional analysis
The functional annotation of the F. oxysporum f. sp. lentis genome revealed that the highest number of hits were detected against the Pfam database, followed by Gene Ontology (GO) and UniProt databases (Figure 4). To further examine the functional classification of genes, 35,458 unigenes were exposed to GO analysis. The unigenes were organized into 44,376 functional groups across three main GO categories, i.e., cellular component, molecular function, and biological process (Figure 5).
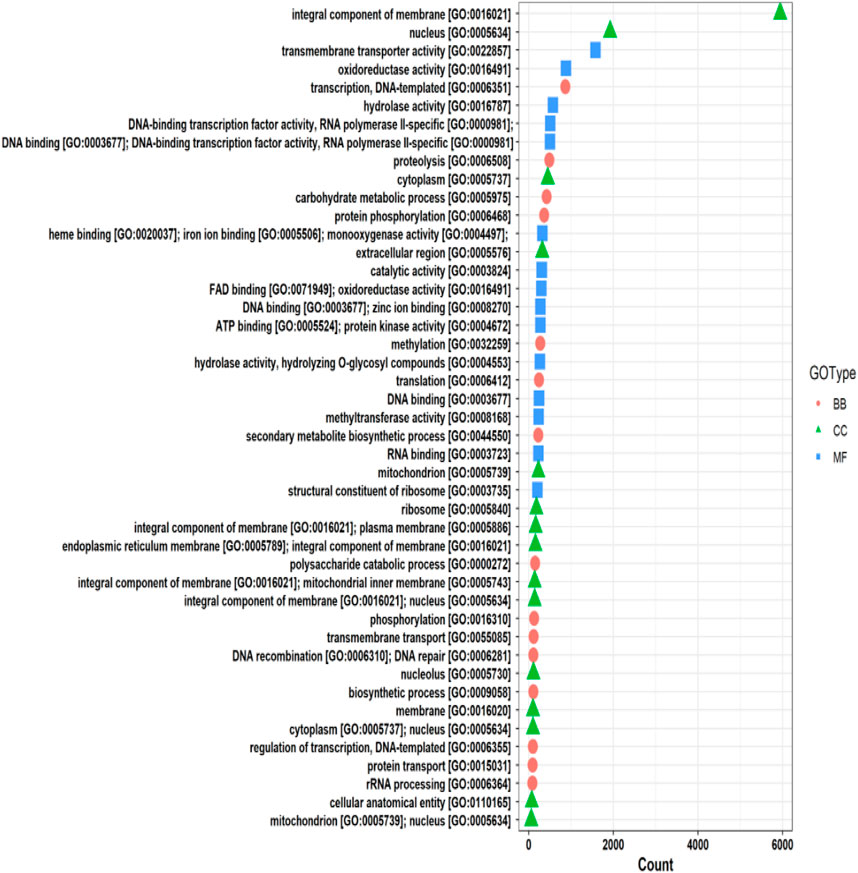
Figure 5. Gene Ontology (GO) classifications of unigenes.
The maximum number of hits was examined against the Pfam database, followed by the GO and UniProt database (Figure 4). To analyze the roles of all unigenes above 35,458 unigenes were used for GO analysis. These unigenes were classified into 44,376 functional groups in three main classes (cellular component, molecular function, and biological process) (Figure 5). The GO annotation results revealed that terms of integral component of membrane, nucleus cytoplasm, and extracellular region were dominant in the cellular component, those of transmembrane transporter activity oxidoreductase activity, binding and catalytic activity were key in molecular function, and those of transcription, proteolysis, carbohydrate metabolic process, protein phosphorylation, and methylation were foremost in biological process (Figure 5). To gain deeper insights into physiological processes, the KOBAS software was utilized to analyze the statistical enrichment of DEGs (Differentially Expressed Genes) within KEGG pathways. This analysis revealed that 433 unigenes were mapped to 107 KEGG pathways. Most of the correlative genes were differently expressed for protein modification and ubiquitination, glycan metabolism (pectin degradation), amino acid degradation (L-valine degradation), lipid metabolism (sphingolipids metabolism), ubiquinone, other amino acid biosynthesis, carbohydrate biosynthesis and metabolism and glycolipid biosynthesis and nucleotide-sugar biosynthesis and sulfur metabolism. Among 107 pathways, the largest pathway was protein modification (178), amino acid degradation (91) and glycan metabolism (112 genes), followed by glycolysis/gluconeogenesis (33 genes), lipid metabolism (71 genes) and purine metabolism (82 genes) (Figure 6). The results of this study showed that all unigenes were involved in development and metabolism indicating that these unigenes much more likely play important roles for fungal developmental stage. The COG functional classification showed that 21,001 unigene were distributed across 25 COG categories (Figure 7). It also showed that general function prediction only accounts for the largest percentage followed by translation, ribosomal structure, and biogenesis, amino acid transport and metabolism, and coenzyme transport and metabolism. Many unigenes were also involved in cluster transcription and signal transduction mechanisms. These results indicated that genes in the above categories play important roles in development. The Fusarium genome sequence was analyzed to determine the presence of Carbohydrate-Active enzymes (CAZymes). The analysis revealed 16,779 putative genes encoding CAZymes, which were distributed across 91 CAZyme protein families. CAZyme are proteins with polysaccharide-degrading enzymatic activities on polysaccharides. We identified 16,779 putative genes that encode CAZyme in the genome of Fusarium. Of the six CAZyme classes, Carbohydrate Esterases (CE), Glycoside Hydrolases (GH), and Polysaccharide Lyases (PL) are Plant Cell Wall Degrading Enzymes (PCWDEs). There were with 306, 274, 518, 82, 1,300, and 139 genes identified in the Auxiliary Activities (AA), Carbohydrate-Binding Molecules (CBM), Glycoside Hydrolases (GT), Polysaccharide Lyases (PL), Glycoside Hydrolases (GH) and Carbohydrate Esterases (CE) classes, respectively (Figure 8) (Table 5).
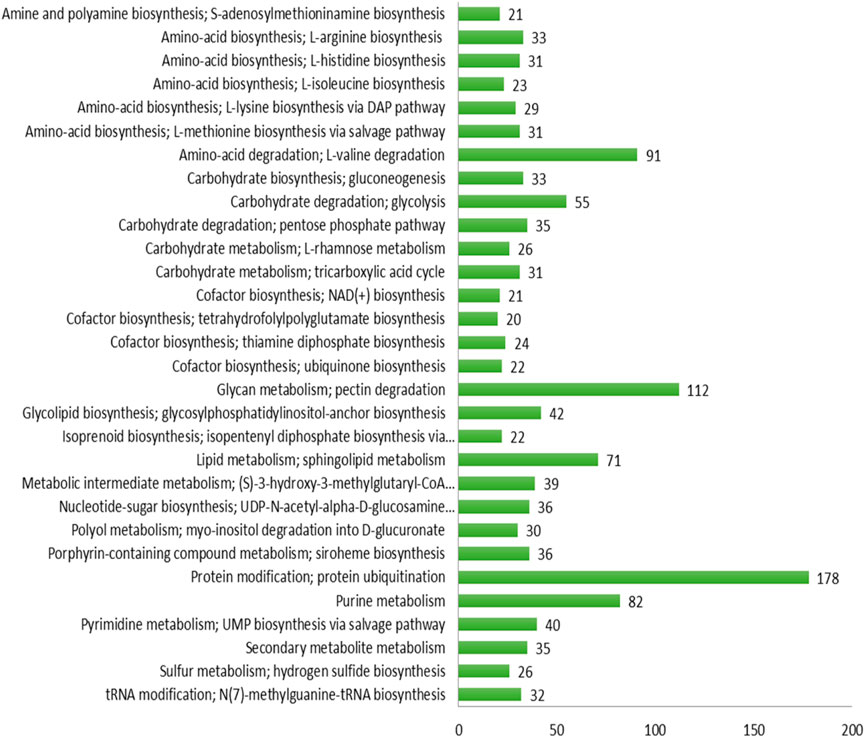
Figure 6. KEGG: Pathway analysis.
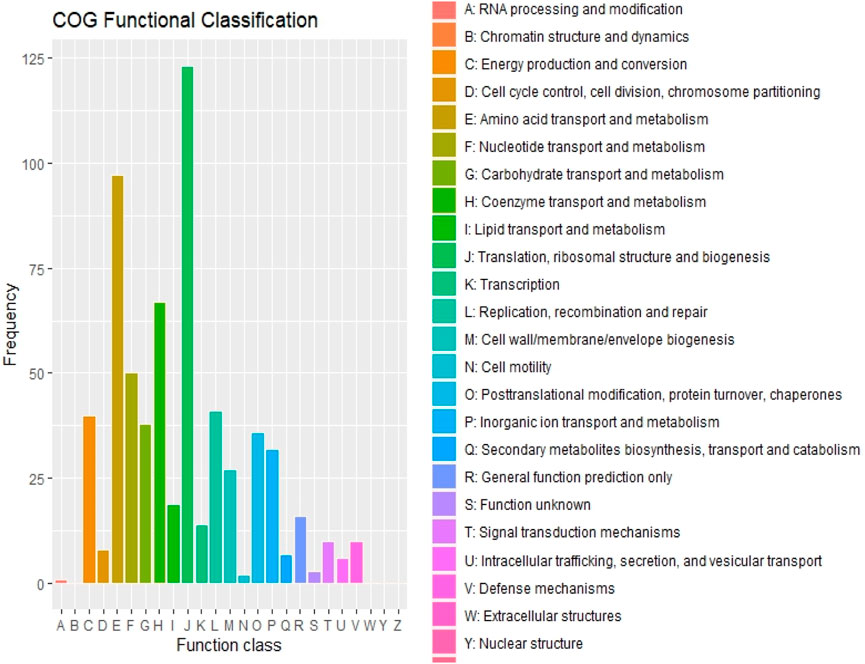
Figure 7. Cluster of Orthologous Genes and their Functional Classification.
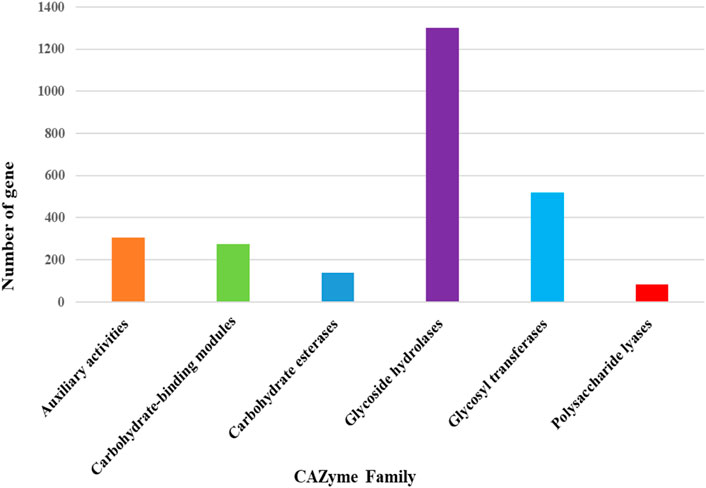
Figure 8. Distribution of CAZyme family putative genes in different classes.
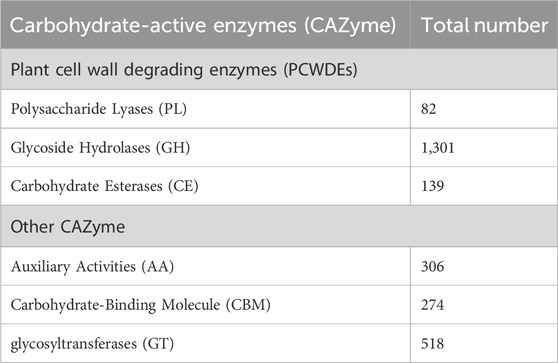
Table 5. Distribution of different classes of Carbohydrate-Active Enzymes (CAZyme).
This includes 306 Auxiliary Activities (AAs) [Figure 9a; Supplementary Tables S1a–f], 274 Carbohydrate-Binding Modules (CBMs) [Figure 9b; Supplementary Tables S1a–f], 139 Carbohydrate Esterases (CEs) [Figure 9c; Supplementary Tables S1a–f], 1,301 Glycoside Hydrolases (GHs) [Figure 9d; Supplementary Tables S1a–f], 518 Glycosyltransferases (GTs) [Figure 9e; Supplementary Tables S1a–f] and 82 Polysaccharide Lyases (PLs) [Figure 9f; Supplementary Tables S1a–f]. The predictions suggested more GHs than GTs in the Fusarium genome. GHs (glycosidases or glycosyl hydrolases, EC 3.2.1.) are enzymes that catalyze the hydrolysis of glycosidic bonds of complex carbohydrates and key enzymes involved in carbohydrate metabolism. In addition, GHs are common enzymes in nature that degrade the most abundant biomasses, such as cellulose, hemicellulose, and starch.
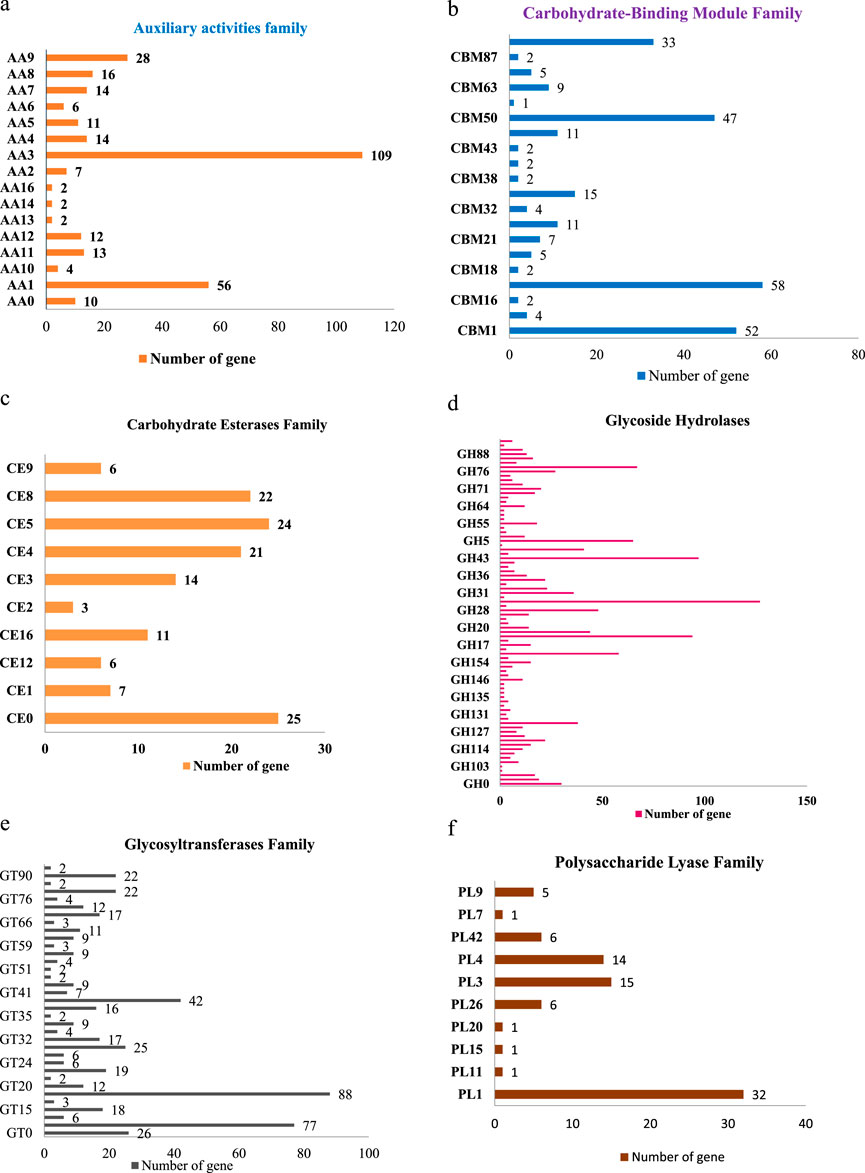
Figure 9. (a) Distribution of CAZyme family putative genes in Auxiliary activities (AA). (b) Distribution of CAZyme family putative genes in Carbohydrate-Binding Module (CBM) Family. (c) Distribution of CAZyme family putative genes in Carbohydrate Esterases (CE) Family. (d) Distribution of CAZyme family putative genes in Glycoside Hydrolases (GH) Family. (e) Distribution of CAZyme family putative genes in Glycosyltransferases (GT) Family. (f) Distribution of CAZyme family putative genes in Polysaccharide Lyase (PL) Family.
The genome assembly comprised 12,366 scaffolds, totaling approximately 114 million base pairs (Mb). Using Tantan within the Funannotate pipeline, 4.53% of the genome was identified as repetitive elements and soft-masked accordingly. These repetitive sequences, which include various transposable elements, were preserved in lowercase format to maintain sequence integrity for downstream analyses. The soft-masking approach facilitated accurate gene annotation by preventing misannotation within repetitive regions. Additionally, the identification of these elements provides insights into genome evolution and stability, offering a foundation for comparative genomics and further functional analysis aimed at improving genome annotation accuracy.
Genome annotation using tRNAscan-SE within the Funannotate pipeline identified 598 valid tRNA genes. All predicted tRNAs were non-overlapping, supporting the accuracy of gene model predictions and minimizing the risk of annotation artifacts. These tRNA genes, essential for the translational machinery, contribute to the fidelity of protein synthesis by delivering specific amino acids to ribosomes. Their distinct organization likely reflects a stable genome architecture and functional conservation. The relatively high number of tRNA genes suggests a well-structured genome with potential implications for codon usage bias, gene regulation, and evolutionary adaptation.
3.3 Pathogen host interactions
The Pathogen Host Interactions database (PHI-base) has manually curated experimentally verified pathogenicity, virulence, and effector genes from fungal, bacterial, and protist pathogens. The amino acid sequence of the target species of Fusarium isolate was compared with the PHI database by using the BLAST software, and the gene of the target species was combined with the functional annotation information to obtain an annotation result (Figure 10). Fusarium strain harbors abundant PHI-base genes, including reduced virulence (1,196), increased virulence (hypervirulence) (20), loss of pathogenicity (106), lethal (175), unaffected pathogenicity (1,433), chemical target (5), effector (plant avirulence determinant) (5) and. Reduced virulence and unaffected pathogenicity are the major annotation genes, suggesting that Fusarium strain is not a highly pathogenic strain (Figure 10).
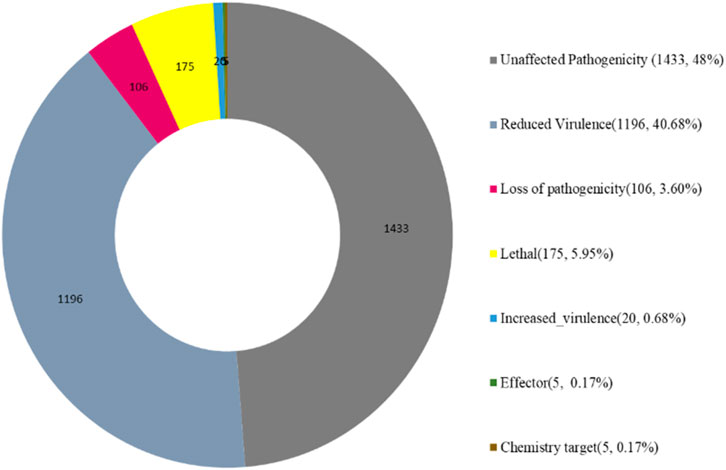
Figure 10. Functions of PHI proteins.
3.4 Analysis of secondary metabolite biosynthetic gene clusters
AntiSMASH analysis suggested that Fusarium strain possesses 77 Secondary Metabolite (SM) biosynthetic gene clusters (BGCs), including 23 T1PKS, 23 NRPS, 13 NRPS-like, 5 indole, 12 terpene, 4 fungal-RiPP like, 2 phosphonate, 1 CDPS, 2 NRP-metallophore biosynthetic genes (Figure 11) (Table 6). Only 30% of these BGCs showed gene homologies with known clusters in the MIBiG database. By further comparison with the gene sequences of other reference strains, several BGCs of the Fusarium strain with high similarity were identified and predicted to be responsible for the biosynthesis of ACT-Toxin II (Polyketide) in region 24.1, choline (NRPS-like) in region 26.1, α-acorenol (terpene) in region 280.1, Sansalvamide (NRPS) in region 362.1, Prolipyrone B (T1PKS) in region 363.1, Ochratoxin A (NRP + Polyketide) in region 655.1, Karaiol (Terpene) in region 873.1(Figures 12a–g).

Figure 11. Graphical representation of Fusarium fungal genomes revealing putative natural products of many different types.
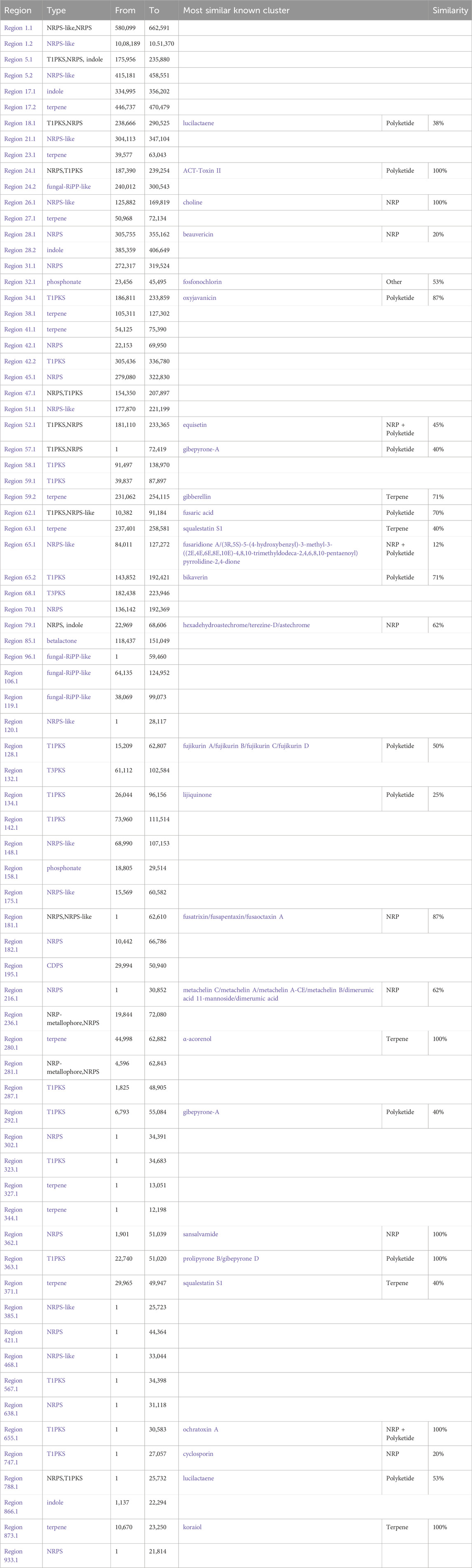
Table 6. Identification of Secondary metabolite (SM) biosynthetic gene clusters (BGCs) using AntiSMASH.

Figure 12. (a) AntiSMASH Report; ACT-toxin II: Region 24.1. (b) AntiSMASH Report; choline: Region 26.1. (c) AntiSMASH Report; α-acorenol; Region 280.1. (d) AntiSMASH Report; sansalvamide: Region 362.1. (e) AntiSMASH Report; Prolipyrone B: Region 363.1. (f) AntiSMASH Report; Ochratoxin A: Region 655.1. (g) AntiSMASH Report; Koraiol: Region 873.1.
AntiSMASH analysis showed that genes within the region 26.1 had a significant BLAST hit with the choline BGC (GenBank: NW_022194793) from Fusarium proliferatum ET1 genome assembly (3400500–3441734). BGC region 280.1 of Fusarium strain displayed significant similarity with that of α-acorenol (GenBank: CM000599 & NC_030996) from F. oxysporum f. sp. lycopersici (127,387–148652). Sansalvamide, a cyclic pentadepsipeptide with a potent anticancer effect, was originally isolated from one marine Fusarium species. The chemical structure of sansalvamide, with four proteogenic amino acids and one hydroxyl acid, suggested that it could be synthesized by a five-module NRPS where each of the modules would be responsible for incorporating one of the amino acids. BGC region 873.1 of Fusarium strain displayed significant similarity with that of Koraiol (GenBank: CM000600 & NC_030997) from F. oxysporum f. sp. lycopersici (1,264,684–1286383). BGC region 24.1 of the Fusarium strain displayed significant similarity with that of ACT-Toxin II (GenBank: NW_022194804) from F. proliferatum ET1 genome (252,933–304792).
Based on the antiSMASH analysis, compounds a were putatively biosynthesized by various NRPS & PKSs, compound b was putatively synthesized from NRPS-like while compounds c was plausible products of the NRPS only, compound d was a plausible product of the Terpene only (Figure 13). BGC region 24.1 possesses several additional enzymes including ilicicolin H biosynthetic gene cluster from Talaromyces variabilis (GenBank: MK539848.1) and ilicicolin J and 8-epi-ilicicolin H Neonectria sp. DH2 (GenBank: RQWH01000002.1). BGC region 24.1 possesses several additional enzymes including UNII-YC2Q1O94PT and gibe-pyrone D biosynthetic gene cluster from Alternaria alternata (GenBank: AB725683.1).

Figure 13. AntiSMASH analysis showing identification of putative compounds biosynthesized by different gene clusters (a, b1, b2, c) NRPs and PKSs, NRPS-like, NRPs, Terpene only.
3.5 Synteny analysis
The synteny analysis was performed to compare the genome sequence of F. oxysporum f. sp. conglutinans causing Fusarium wilt of cabbage with the draft genome sequence of Fusarium assembled in this study. This comparative genomic approach revealed notable collinearity between specific scaffolds in the draft genome and chromosomes in the reference genome. Specifically, scaffold 2 (scf2) and scaffold 10 (scf10) exhibited strong collinear relationships with chromosome 9 (chr9) of F. oxysporum f. sp. conglutinans, indicating a high level of conserved gene order and orientation across these regions. In addition, scaffold 1 (scf1) and scaffold 8 (scf8) showed substantial collinear blocks with chromosome 3 (chr3) of the reference genome, suggesting evolutionary conservation and potential functional similarities in these genomic regions. Moreover, larger segments of scaffold 3 (scf3), scaffold 4 (scf4), and scaffold 6 (scf6) displayed extensive syntenic blocks with chromosome 2 (chr2) of the reference genome, further supporting the presence of conserved genome architecture between the studied isolate and F. oxysporum f. sp. conglutinans. These patterns of synteny not only validate the structural integrity of the draft genome but also provide insights into the evolutionary conservation and potential gene function relationships across different Fusarium isolates (Figures 14a-d).
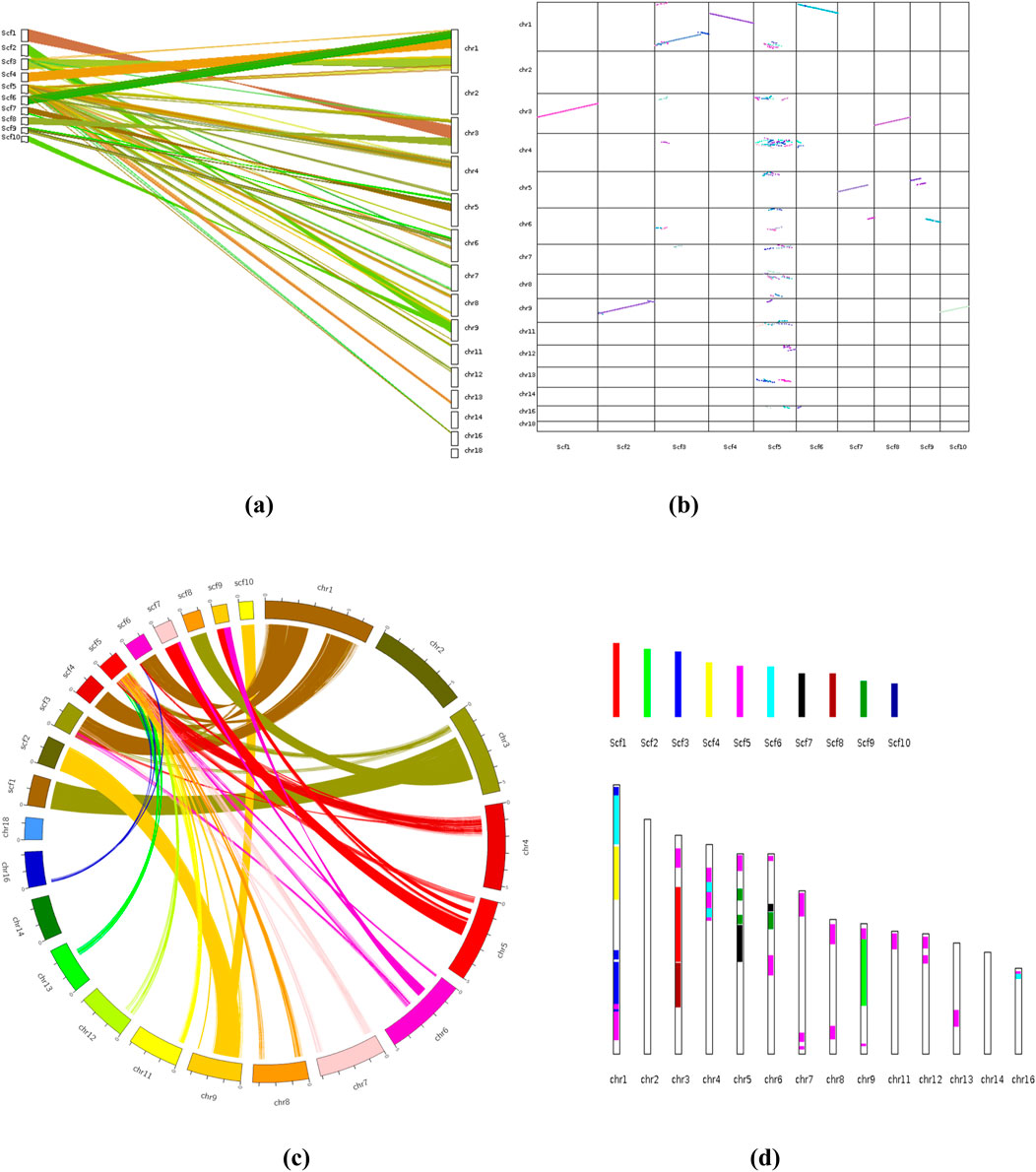
Figure 14. (a) Synteny Analysis: Different types of plots showing patterns of synteny and collinearity; [(a, b) Dual synteny plot& Dot plot]. (b) Chromosomes are labeled “species abbreviation” + ”chromosome ID” in the format. (c) Circle plot, (d) Bar plot. Sc, Fusarium oxysporum strain; chr, Fusarium oxysporum f. sp. coaglutinans (Reference species: GCA_014154955.1_SMRT_HiC_Fo5176_genomic).
4 Discussion
In this study, we present the first whole-genome sequencing of F. oxysporum f. sp. lentis, pathogen responsible for vascular wilt in lentils, using the Illumina Shotgun Sequencing platform. The development of genomic resources is crucial for understanding the genetic basis of pathogenicity and other biological processes in F. oxysporum f. sp. lentis. The F. oxysporum species complex exhibits extensive genetic and functional diversity. Pathogenic strains demonstrate host specificity, whereas non-pathogenic strains do not induce disease symptoms, despite the absence of morphological differences (Nelson et al., 1981; Steinberg et al., 2016). Molecular genetic studies are essential for deciphering pathogenicity mechanisms, which, in turn, are crucial for devicing effective disease management strategies. Our sequencing efforts resulted in a high-quality genome assembly comprising 12,366 contigs with a total assembly length of 124,483,966 bases. BUSCO analysis confirmed 99% completeness, indicating a robust and reliable assembly. The N50 and L50 values were 51,992 and 353, respectively, further indicating a high-quality well-assembled genome.
Functional annotation of the genome revealed 116,998 protein-coding genes, with substantial homology identified across multiple databases, including Uniprot, GO, KEGG, Pfam, COG, CAZyme, PHI, and AntiSMASH. Gene ontology (GO) analysis classified 35,458 unigenes into three major categories: cellular components, molecular functions, and biological processes. Key enriched GO terms included transmembrane transporter activity, oxidoreductase activity, binding, and catalytic activity, suggesting an active role in metabolic and cellular processes. Additionally, 433 unigenes were mapped to 107 KEGG pathways, with significant representation in protein modification and ubiquitination, glycan metabolism, amino acid degradation, lipid metabolism, and carbohydrate biosynthesis.
Carbohydrate-active enzymes (CAZymes) play a pivotal role in fungal pathogenicity by facilitating plant cell wall degradation. We identified 16,779 CAZyme-encoding genes distributed across 91 protein families, with glycoside hydrolases (GH) and glycosyltransferases (GT) being the most abundant classes. The presence of hemicellulose-degrading enzymes (GH10, GH11, GH115) and pectin-degrading enzymes (GH43, GH51, GH78, GH93, PL1, PL3, PL4) underscores the ability of F. oxysporum f. sp. lentis to efficiently degrade plant cell walls. Interestingly, an expansion of fungal cell wall biosynthesis-related CAZymes was also observed, emphasizing the importance of chitin and β-glucan remodeling in fungal development and host colonization.
Pathogenicity-related gene analysis using the PHI-base database identified multiple genes associated with different pathogenicity levels: reduced virulence (1,196), hypervirulence (20), loss of pathogenicity (106), lethal effects (175), and unaffected pathogenicity (1,433). The predominance of reduced virulence and unaffected pathogenicity genes suggests that while this strain possesses virulence factors, it may not be highly aggressive compared to other pathogenic F. oxysporum strains.
The identification of secondary metabolite biosynthetic gene clusters (BGCs) is critical for understanding fungal virulence and toxin production. AntiSMASH analysis revealed 77 secondary metabolite clusters, including 23 Type I polyketide synthase (T1PKS), 23 non-ribosomal peptide synthetases (NRPS), 13 NRPS-like, 5 indole, 12 terpene, 4 fungal RiPP-like, 2 phosphonate, 1 CDPS, and 2 NRP-metallophore biosynthetic genes. These findings lay the groundwork for further investigations into the role of secondary metabolites in pathogenicity and host interactions.
Advancements in sequencing technologies have significantly improved genome assembly quality. While early genome sequencing of F. oxysporum relied on Sanger sequencing (Broad Institute, 2007; Ma et al., 2010). However, next-generation sequencing (NGS) platforms such as Illumina and BGI have accelerated genome assembly at reduced cost. Third-generation sequencing technologies, such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), offer longer read lengths, with superior genome assemblies (van Dijk et al., 2018; Amarasinghe et al., 2020; Giani et al., 2020).
5 Conclusion
This study presents the first whole-genome sequencing and assembly of F. oxysporum f. sp. lentis using Illumina short-read sequencing technology. The high-quality genome assembly, with 99% completeness based on BUSCO analysis, provides critical insights into the genetic basis of pathogenicity, carbohydrate-active enzymes (CAZymes), and secondary metabolite biosynthetic gene clusters (BGCs). Functional annotation revealed key gene families associated with virulence, metabolic pathways, and host adaptation, enhancing our understanding of F. oxysporum evolution and pathogenic mechanisms. Although this strain harbors significant virulence genes, PHI-base analysis advocates moderate pathogenic potential, underscoring the need for further investigation into host-pathogen interactions. This study offers novel insights into the genetic makeup, virulence factors, and metabolic potential of F. oxysporum f. sp. lentis, contributing to future research on host-pathogen interactions and disease management strategies and lentil breeding strategies. Future research should prioritize comparative genomics of F. oxysporum f. sp. lentis isolates to identify race-specific virulence factors and host resistance mechanisms. The integration of long-read sequencing technologies, such as PacBio or Oxford Nanopore, will enhance genome assembly, facilitating the identification of structural variations and transposable elements linked to pathogenicity. Additionally, transcriptomic and proteomic studies will provide functional validation of key effector genes and metabolic pathways involved in lentil infection. Understanding the molecular basis of virulence and secondary metabolism in F. oxysporum f. sp. lentis will facilitate the development of disease-resistant lentil cultivars and targeted biocontrol strategies to mitigate Fusarium wilt.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/s and accession number(s) can be found at online repositories at NCBI https://www.ncbi.nlm.nih.gov/sra/?term=SRR25383692.
Author contributions
SK: Conceptualization, Writing – original draft. AK: Conceptualization, Methodology, Software, Supervision, Writing – original draft, Writing – review and editing. PM: Data curation, Software, Writing – original draft. RR: Methodology, Writing – review and editing. SS: Methodology, Resources, Writing – review and editing. NM: Writing – original draft. PC: Formal Analysis, Writing – original draft. RS: Investigation, Methodology, Software, Writing – original draft. VeG: Formal Analysis, Methodology, Software, Validation, Visualization, Writing – original draft. ST: Visualization, Writing – review and editing. ViG: Visualization, Writing - review and editing. SS: Software, Validation, Writing – review and editing. GK: Funding acquisition, Project administration, Writing – review and editing. NJ: Data curation, Methodology, Software, Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors acknowledge JNKVV, Jabalpur for providing facilities for conducting the work. AK gratefully acknowledges the ICAR National Agricultural Higher Education Project–Centre of Advanced Agricultural Science and Technology (NAHEP-CAAST) for supporting the faculty exchange program, and the Department of Entomology and Plant Pathology at the University of Arkansas, Fayetteville, AR, for hosting the exchange visit, which was instrumental in learning some of the data analysis approaches used in this study. AK also sincerely thanks Dr. R. Adams (University of Arkansas) for valuable advice on data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1585510/full#supplementary-material
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25 (17), 3389–3402. doi:10.1093/nar/25.17.3389
Amarasinghe, S. L., Su, S., Dong, X., Zappia, L., Ritchie, M. E., and Gouil, Q. (2020). Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 21 (1), 30. doi:10.1186/s13059-020-1935-5
Arumuganathan, K., and Earle, E. D. (1991). Nuclear DNA content of some important plant species. Plant Mol. Biol. Report. 9, 208–218. doi:10.1007/bf02672069
Baayen, R. P., Van Dreven, F., Krijger, M. C., and Waalwijk, C. (1997). Genetic diversity in Fusarium oxysporum f. sp. dianthi and Fusarium redolens f. sp. dianthi. Eur. J. Plant Pathology 103, 395–408. doi:10.1023/A:1008604416107
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi:10.1089/cmb.2012.0021
Baysal, Ö., Siragusa, M., and Carimi, F. (2010). Molecular characterization of Fusarium oxysporum isolates from different hosts in Turkey. J. Phytopathology 158 (8), 534–541. doi:10.1111/j.1439-0434.2009.01652.x
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M., et al. (2023). antiSMASH 7.0: new and improved predictions for genomic analysis of secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 51 (W1), W463–W469. doi:10.1093/nar/gkad344
Broad Institute (2007). Fusarium comparative sequencing Project. Available online at: http://www.broadinstitute.org.
Cantarel, B. L., Korf, I., Robb, S. M. C., Parra, G., Ross, E., Moore, B., et al. (2008). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18 (1), 188–196. doi:10.1101/gr.6743907
Carvalhais, L. C., Henderson, J., Rincon-Florez, V. A., O’Dwyer, C., Czislowski, E., Aitken, E. A. B., et al. (2019). Molecular diagnostics of banana Fusarium wilt targeting Secreted-in-Xylem genes. Front. Plant Sci. 10, 547–612. doi:10.3389/fpls.2019.00547
Choudhary, S., and Mohanka, R. (2012). In vitro antagonism of indigenous Trichoderma isolates against phytopathogen causing wilt of lentil. Int. J. Life Sci. & Pharma Res. 2, 1–5.
Davière, J. M., Langin, T., and Daboussi, M. J. (2001). Potential role of transposable elements in the genomic plasticity of Fusarium oxysporum. Nucleic Acids Res. 29 (13), 2894–2901. doi:10.1093/nar/29.13.2894
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19 (1), 11–15.
FAO (Food and Agriculture Organization) (2021). Crop production and trade data. Available online at: http://www.fao.org/faostat/en/#home (Accessed September 18, 2021).
Garkoti, A., Kumar, A., and Tripathi, H. (2013). Management of Fusarium wilt of lentil through fungicides. J. Mycol. Plant Pathology 43, 333–335.
Giani, A. M., Gallo, G. R., Gianfranceschi, L., and Formenti, G. (2020). Long walk to genomics: history and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J. 18, 9–19. doi:10.1016/j.csbj.2019.11.002
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17 (6), 333–351. doi:10.1038/nrg.2016.49
Gordon, T. R. (2017). Fusarium oxysporum and the Fusarium wilt syndrome. Annu. Rev. Phytopathology 55, 23–39. doi:10.1146/annurev-phyto-080615-095919
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinforma. 12, 491. doi:10.1186/1471-2105-12-491
Houterman, P. M., Cornelissen, B. J. C., Rep, M., Manuyakorn, A., Puttikhunt, C., Kasinrerk, W., et al. (2007). Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog. 3 (5), e183. doi:10.1371/journal.ppat.0030183
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S.K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47 (D1), D309–D314. doi:10.1093/nar/gky1085
Infantino, A., Kharrat, M., Riccioni, L., Coyne, C. J., McPhee, K. E., and Grunwald, N. J. (2006). Screening techniques and sources of resistance to root diseases in cool season food legumes. Euphytica 147, 201–221. doi:10.1007/s10681-006-6963-z
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi:10.1093/nar/28.1.27
Kashiwa, T., Inami, K., Fujinaga, M., Ogiso, H., Yoshida, T., Teraoka, T., et al. (2013). An avirulence gene homologue in the tomato wilt fungus Fusarium oxysporum f. sp. lycopersici race 1 functions as a virulence gene in the cabbage yellows fungus F. oxysporum f. sp. conglutinans. J. General Plant Pathology 79, 412–421. doi:10.1007/s10327-013-0471-5
Kelley, D. R., Schatz, M. C., and Salzberg, S. L. (2010). Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 11 (11), R116. doi:10.1186/gb-2010-11-11-r116
Kersey, P. J. (2019). Plant genome sequences: past, present, future. Curr. Opin. Plant Biol. 48, 1–8. doi:10.1016/j.pbi.2018.11.001
Kharte, S., Kumar, A., Sharma, S., Ramakrishnan, R. S., Kumar, S., Malvi, S., et al. (2022). In vitro evaluation of fungicides and bio-agents for the management of lentil wilt caused by Fusarium oxysporum f. sp. lentis. Biol. Forum – An Int. J. 14 (4), 489–495. Available online at: https://www.researchgate.net/publication/367598549_In_vitro_Evaluation_of_Fungicides_and_Bio-agents_for_the_Management_of_Lentil_Wilt_caused_by_Fusarium_oxysporum_f_sp_lentis.
Kumar, A., Bohra, A., Mir, R. R., Sharma, R., Tiwari, A., Khan, M. W., et al. (2021). Next generation breeding in pulses: present status and future directions. Crop Breed. Appl. Biotechnol. 21 (Special Issue), e394221S13. doi:10.1590/1984-70332021v21sa26
Lievens, B., Rep, M., and Thomma, B. P. H. J. (2008). Recent developments in the molecular discrimination of formae speciales of Fusarium oxysporum. Pest Manag. Sci. 64, 781–788. doi:10.1002/ps.1564
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42 (D1), D490–D495. doi:10.1093/nar/gkt1178
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1 (1), 18. doi:10.1186/2047-217X-1-18
Ma, L. J., van der Does, H. C., Borkovich, K. A., Coleman, J. J., Daboussi, M. J., Di Pietro, A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium oxysporum. Nature 464 (7287), 367–373. doi:10.1038/nature08850
NCBI (2023). NCBI genome database. Bethesda, MD: National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/genome/.
Nelson, P. E., Toussoun, T. A., and Cook, R. J. (1981). Fusarium: diseases, biology, and taxonomy. Pennsylvania State University Press.
Nelson, P. E., Toussoun, T. A., and Marasas, W. F. O. (1983). Fusarium species: an illustrated manual for identification. University Park, PA: Pennsylvania State University Press.
Rep, M., Van Der Does, H. C., Meijer, M., Van Wijk, R., Houterman, P. M., Dekker, H. L., et al. (2004). A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 53, 1373–1383. doi:10.1111/j.1365-2958.2004.04177.x
Rubiales, D., Fondevilla, S., Chen, W., Gentzbittel, L., Higgins, T. J., Castillejo, M. A., et al. (2015). Achievements and challenges in legume breeding for pest and disease resistance. Crit. Rev. Plant Sci. 34 (1–3), 195–236. doi:10.1080/07352689.2014.898445
Schardl, C. L., Young, C. A., Hesse, U., Amyotte, S. G., Andreeva, K., Calie, P. J., et al. (2013). Plant-symbiotic fungi as chemical engineers: multi-genome analysis of the Clavicipitaceae reveals dynamics of alkaloid loci. BMC Genomics 14, e1003323–23. doi:10.1371/journal.pgen.1003323
Schmidt, S. M., Lukasiewicz, J., and Farrer, R. A. (2013). Comparative genomics of plant fungal pathogens. Fungal Genet. Biol. 61, 48–60.
Sharma, M., Nagavardhini, A., Mahendar, T., Raju Ghosh, S., Rajeev, K. V., and Varshney, R. K. (2014). Development of DArT markers and assessment of diversity in Fusarium oxysporum f. sp. ciceris, wilt pathogen of chickpea (Cicer arietinum L.). BMC Genomics 15, 454. doi:10.1186/1471-2164-15-454
Sicard, D., Michalakis, Y., Dron, M., and Neema, C. (1997). Genetic diversity and pathogenic variation of colletotrichum lindemuthianum in the three centers of diversity of its host, Phaseolus vulgaris. Phytopathology 87 (8), 807–813. doi:10.1094/PHYTO.1997.87.8.807
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33 (Suppl. 2), W465–W467. doi:10.1093/nar/gki458
Steinberg, G., Perez-Martin, J., and Snetselaar, K. M. (2016). Fungal morphogenesis and host invasion. Curr. Opin. Microbiol. 34, 1–8. doi:10.1016/j.mib.2016.07.002
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). GeneMark-ES: ab initio gene prediction supported by a statistical model of transcriptome structure. Genome Biol. 9 (1), R7.
Urban, M., Cuzick, A., Seager, J., Wood, V., Rutherford, K., Venkatesh, S. Y., et al. (2020). PHI-base: the pathogen–host interactions database. Nucleic Acids Res. 48 (D1), D613–D620. doi:10.1093/nar/gkz904
van Dam, P., Fokkens, L., Schmidt, S. M., Linmans, J. H. J., Kistler, H. C., Ma, L. J., et al. (2016). Effector profiles distinguish formae speciales of Fusarium oxysporum. Environ. Microbiol. 18 (11), 4087–4102. doi:10.1111/1462-2920.13445
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D., and Thermes, C. (2018). The third revolution in sequencing technology. Trends Genet. 34 (9), 666–681. doi:10.1016/j.tig.2018.05.008
Vlaardingerbroek, I., Beerens, B., Rose, L., Fokkens, L., Cornelissen, B. J. C., and Rep, M. (2016). Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum. Environ. Microbiol. 18 (11), 3702–3713. doi:10.1111/1462-2920.13281
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi:10.1093/nar/gkr1293
Keywords: Fusarium oxysporum f. sp. lentis, illumina shotgun sequencing, pathogenicity, CAZymes, secondary metabolites, virulence factors, host-pathogen interactions, gene annotation
Citation: Kharte S, Kumar A, Mishra P, Ramakrishnan RS, Sharma S, Mishra N, Chauhan PS, Sharma R, Gautam V, Tiwari S, Goyal V, Sharma S, Koutu GK and Joshi NK (2025) Whole genome sequencing and functional annotation of Fusarium oxysporum f. sp. lentis to unravel virulence and secondary metabolite biosynthesis gene clusters. Front. Genet. 16:1585510. doi: 10.3389/fgene.2025.1585510
Received: 28 February 2025; Accepted: 06 May 2025;
Published: 18 June 2025.
Edited by:
Jeff Bennetzen, University of Georgia, United StatesReviewed by:
Asif Bashir Shikari, Sher-e-Kashmir University of Agricultural Sciences and Technology of Kashmir, IndiaLeonardo Alfredo Ornella, Cubiqfoods SL, Spain
Copyright © 2025 Kharte, Kumar, Mishra, Ramakrishnan, Sharma, Mishra, Chauhan, Sharma, Gautam, Tiwari, Goyal, Sharma, Koutu and Joshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashish Kumar, YXNoaXNoa3VtYXJAam5rdnYub3Jn
†These authors have contributed equally to this work and share first authorship