 Yefeng Wang
Yefeng Wang Xinghan Wu2†
Xinghan Wu2† Ningan Xu
Ningan Xu- 1Department of Cardiology, The Affiliated Children’s Hospital of Xiangya School of Medicine, Central South University (Hunan Children’s Hospital), Changsha, Hunan, China
- 2Medical Genetics Department, The Affiliated Children’s Hospital of Xiangya School of Medicine, Central South University (Hunan Children’s Hospital), Changsha, Hunan, China
- 3Healthcare Center, The Affiliated Children’s Hospital of Xiangya School of Medicine, Central South University (Hunan Children’s Hospital), Changsha, Hunan, China
Introduction: Rubinstein-Taybi syndrome is an extremely rare autosomal dominant genetic disease. The incidence of RSTS ranges from 1/100 000 to 125 000.
Methods: We retrospectively reviewed the phenotype and genotype of two children who were diagnosed with RSTS in Hunan Province Children’s Hospital from January 2022 to December 2023. Clinical data of the children were collected. Whole-exome sequencing was performed on the children. The candidate variants were verified by Sanger sequencing in the pedigree, followed by pathogenicity analysis.
Results: The main clinical presentations of the two cases were growth retardation, special facial features, and mild intellectual disability. Three mutations were detected by exome sequencing, all of which were sporadic mutations verified by Sanger sequencing. In case 1, pathological mutations were detected in EP300 gene and NSD1 gene. A heterozygous mutation c. 3934C>T (p. Arg1312Ter) was detected in exon 24 of EP300 gene. A heterozygous mutation c. 5843G>A (p. Arg1948 His) was detected in exon 18 of NSD1 gene. In case 2, a heterozygous mutation (c.2749C>T) (p. Gln917 *) was detected in exon 14 of EP300 gene, which has not been reported in the literature so far. According to ACMG guidelines, this mutation was preliminarily determined to be pathogenic. Comparative analysis of phenotypic differences between the Chinese cohort and the Cohen JL and Fergelot P. cohorts revealed that arched eyebrows, downslanting palpebral fissures, and low-set ears were significantly more common in the Chinese population.
Discussion: EP300 gene c.2749C>T heterozygous mutation may be the genetic cause of Rubinstein Taybi syndrome. EP300 gene combined with NSD1 gene mutation may lead to atypical clinical presentations. These findings further enrich the variation spectrum of EP300 gene.
1 Introduction
Rubinstein-Taybi syndrome (RSTS) is an extremely rare autosomal dominant genetic disease, first reported by Rubinstein and Taybi in 1963. The incidence of RSTS ranges from 1/100,000 to 125,000. It was reported mostly in Caucasians, and there was no difference between male and female populations. The main features of RSTS are growth retardation, microcephaly, facial deformities (e.g., lateral canthal slope, wide nasal bridge, and rostral nose), wide thumb and big toe, and mental and motor retardation (Milani et al., 2015). cAMP response element-binding protein (CREB) is encoded by chromosome 16p13.3 (OMIM #600140). CREBBP gene (Petrij et al., 1995)and chromosome 22q13.2 encodes E1A binding protein p300 (EP300) (OMIM #602700) EP300 gene (Roelfsema et al., 2005). The variants of CREBBP gene account for 50%∼60%, which is called RSTS1 type; EP300 gene mutations account for 8%∼10%, which is called RSTS type 2, but about 30% of patients have no pathogenic genes (Yang and Chen, 2023; Sun et al., 2021). This study reported the clinical phenotype and EP300 gene variation of two children with RSTS, and explored the relationship between clinical phenotype and genotype of RSTS.
2 Study objects and methods
2.1 Research object
We retrospectively reviewed the phenotype and genotype of two children who were diagnosed with RSTS in Hunan Province Children’s Hospital from January 2022 to December 2023. The diagnosis of RSTS is established in a proband with characteristic clinical features (growth retardation, microcephaly, facial deformities wide thumb and big toe, and mental and motor retardation),with a heterozygous pathogenic variant (or likely pathogenic) or a chromosomal deletion involving one or more exons of the EP300 gene. Children without genetic results and imaging data were not included in this study. This study was approved by the Ethics Committee of Hunan Province Children’s Hospital (HCHLL-2023-16), and the parents of the two children signed informed consent forms.
2.2 Research methods
2.2.1 Clinical data collection
The basic information of the patients was collected through the electronic medical record system, including age at first diagnosis, clinical manifestations, growth and development history, maternal history and other general conditions, physical examination, laboratory examination results, gene sequencing results, treatment and prognosis, etc.
2.2.2 Whole-exome sequencing (WES) and sanger validation
Whole-exome sequencing and Sanger sequencing validation were performed on the patients (conducted by Maijinuo Technology Co., Ltd.). Venous blood samples (2 mL each) were collected from the patients and their parents using EDTA anticoagulation. Genetic testing was carried out by Maijinuo Technology Co., Ltd. Exome capture was performed using the Agilent SureSelect method, followed by high-throughput sequencing on the Illumina platform. Sequencing data were analyzed using the GATK software, and variants were filtered using the TGex software. WES allowed the identification of putative causative mutations in EP300 and NSD1, which were confirmed by Sanger sequencing. Primers were designed using the online primer design tool NCBI Primer-BLAST. PCR amplification products were purified with the Tiangen purification kit and sequenced on an ABI 3500DX Genetic Analyzer (Applied Biosystems, United States of America). Sequence alignment and analysis of the sequencing data were performed using SeqMan software.
The primer sequences of Sanger sequencing were as follows.
EP300- c.3934C>T-F1: GATTAGCATGTTCCCTGCACTC
EP300- c.3934C>T-R1: TTCTGCCATCTCTCCACTGTC
EP300- c.2749C>T-F1: GCTCTTCATCAGAATTCACCC
EP300- c.2749C>T-R1: GGATTGTGTCCCCTTGTCG
NSD1- c.5843G>A-F1: TGGCAGGGTACAGATCTTCA
NSD1- c.5843G>A-R1: ACCTCCTACACAGTGACCATG
2.2.3 Bioinformatics analysis
The sequencing data were aligned to the human genome reference hg19 (GRCh37), and exome coverage and sequencing quality regions were evaluated. The raw Fastq data from whole-exome sequencing were analyzed using the ISoGenetic genetic variant interpretation system (integrating GATK, BWA mem, samtools, and Picard) and the CNVexon™ algorithm to identify copy number variations (CNVs), single nucleotide variants (SNVs), and small insertions/deletions (Indels). Detected genetic variants were annotated using databases including OMIM (http://www.omim.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), HGMD (http://www.hgmd.cf.ac.uk/ac/validate.php), gnomAD (http://gnomad.broadinstitute.org/about), and Ensembl (http://grch37.ensembl.org/index.html). Pathogenicity predictions for the variants were performed using SIFT and PROVEAN (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org/), and CADD (https://cadd.gs.washington.edu/). Variants were classified into five categories based on the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines and the Clinical Genome Resource (ClinGen) expert recommendations (Richards et al., 2015): ① pathogenic, ② likely pathogenic, ③ uncertain significance (VOUS), ④ likely benign, and ⑤ benign.
Protein sequences were retrieved from the UniProt database (https://www.uniprot.org), and wild-type and mutant protein structural models were generated using the SWISS-MODEL database (https://swissmodel.expasy.org/). Structural alterations in mutant proteins were further analyzed using PyMOL software.
2.2.4 Literature search methods
A literature search was conducted using the keywords “Rubinstein-Taybi syndrome” and “EP300 gene” in the CNKI, VIP, and Wanfang databases. Additionally, the keywords “Chinese Rubinstein-Taybi Syndrome” and “EP300 gene” were used to search the PubMed database. The goal was to identify articles reporting the clinical characteristics of children with Rubinstein-Taybi syndrome (RSTS) caused by EP300 gene mutations, with duplicate cases removed. Phenotypic differences between Chinese populations and cohorts of other ethnicities were compared. The search period spanned from the inception of each database to December 2024.
2.2.5 Statistical methods
SPSS 20.0 software was used to analyze the data. Chi-square test or Fisher’s exact test was used to compare the clinical phenotypes. P < 0.05 was statistical difference.
3 Results
3.1 Clinical data
3.1.1 Case 1
A 10-year and 7-month-old boy presented with growth retardation. He was the first child, born full-term with a birth weight of 3.0 kg (birth length unknown). He experienced hypoxia and asphyxia at birth. Developmental milestones: began conscious vocalizations (e.g., calling family members) at 1 year and 6 months, walked independently at 1 year and 8 months. Currently, his language expression lags behind peers, with poor motor coordination. He is in the third grade but performs poorly academically. Growth retardation in both height and weight was observed after birth, though the exact growth velocity remains unclear. No headaches, dizziness, visual field defects, polydipsia, or polyuria. VSD repair was performed at 1 year 3 months, followed by bilateral orchiopexy at age seven due to cryptorchidism. Family history: father’s height is 165 cm; mother’s height is 157 cm; mother has epilepsy managed with oxcarbazepine, with no seizures during pregnancy. Physical examination: weight is 24.2 kg (−1∼-2 SD), height is 128.6 cm (<-2 SD), head circumference is 48.5 cm (<-2 SD); proportionate build. Dysmorphic features: microcephaly, long eyelashes, downslanting palpebral fissures, small auricles, broad nasal bridge, prominent nasal ridge, micrognathia, thick eyebrows, exotropia (Figures 1a,b), webbed neck, hirsutism, low posterior hairline (Figure 1c). No broad thumbs (Figure 1e), slightly broadened great toes (Figure 1d). Tanner stage I genitalia. Investigations: blood/urine tests, biochemistry, and thyroid function are normal. Wechsler Intelligence Scale: IQ 65. Bone age (TW3 method): 11 years. Brain MRI: mildly widened left cerebellar sulci, significant right deviation of the nasal septum, and hypertrophic bilateral inferior turbinates (Figure 2).
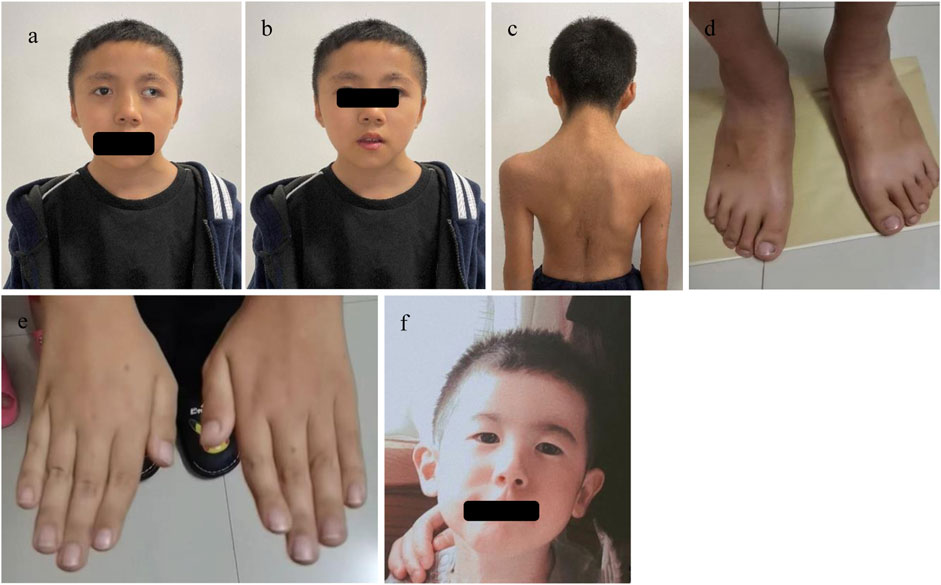
Figure 1. The facial features and other phenotypes of the cases: In Case 1, (a) shows microtia, broad nasal bridge, prominent nasal ridge, deviated nasal septum, down-slanting outer canthus, and exotropia; (b) shows thick eyebrows and micrognathia; (c) shows webbed neck, hypertrichosis, and low posterior hairline; (d) shows a slightly broad big toe; (e) shows no obvious broad thumb or fifth finger flexion deformity. In Case 2, (f) shows microcephaly, down-slanting outer canthus, and prominent nasal ridge.
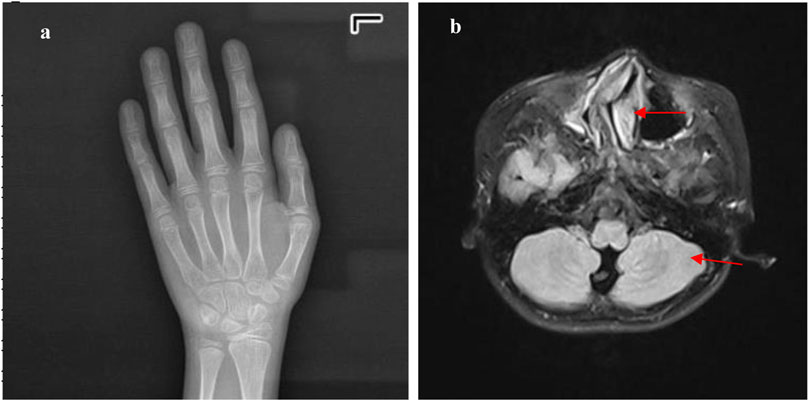
Figure 2. The metacarpal and phalangeal bones and cranial MRI of Case 1: (a) shows a bone age of 11 years, with an actual age of 10 years and 9 months, indicating the bone age is neither advanced nor delayed. (b) shows that the cranial MRI indicates slightly widened cerebellar sulci on the left side (red arrow), a markedly rightward deviation of the nasal septum in the middle (red arrow), and hypertrophy of the bilateral inferior nasal turbinates.
3.1.2 Case 2
A 2-year and 3-month-old boy presented with growth retardation. He was the first child, born preterm at 35 weeks via vaginal delivery with a birth weight of 2.13 kg (birth length unknown). Hospitalized in the neonatal unit due to preterm low birth weight; no intrauterine growth restriction noted prenatally. Family history: father’s height is 170 cm; mother’s height is 157 cm; unremarkable prenatal history. All developmental milestones are delayed compared to age-matched peers. Currently, the patient exhibits only minimal vocalizations, lacks meaningful words, and is unable to walk independently. Physical examination: height is 80 cm (<-2 SD), weight is 9.8 kg (<-2 SD), head circumference is 46.0 cm (<-2 SD); proportionate build. Dysmorphic features: microcephaly, downslanting palpebral fissures, micrognathia, broad nasal bridge (Figure 1f). No broad thumbs or great toes. Tanner stage I genitalia. Investigations: blood/urine tests, biochemistry, and thyroid function are normal. Gesell Developmental Schedules: Adaptive 62.7, Gross Motor 39.8, Fine Motor 54.2, Language 44.2, Personal-Social 41.8 (mild developmental delay). Brain MRI: No significant abnormalities.
3.2 Genetic test results
3.2.1 Case 1
A heterozygous nonsense mutation in exon 24 of the EP300 gene, c.3934C>T (p. Arg1312Ter), was identified. This mutation introduces a premature stop codon, resulting in a truncated protein. Both parents of the proband were tested and found to have no variation at this site, confirming it as a de novo mutation (Figure 3). This variant has been previously reported in the literature (Negri et al., 2015). According to the ACMG guidelines, this variant is preliminarily classified as pathogenic (PVS1 + PM2_Supporting + PS2 + PS4_Supporting).
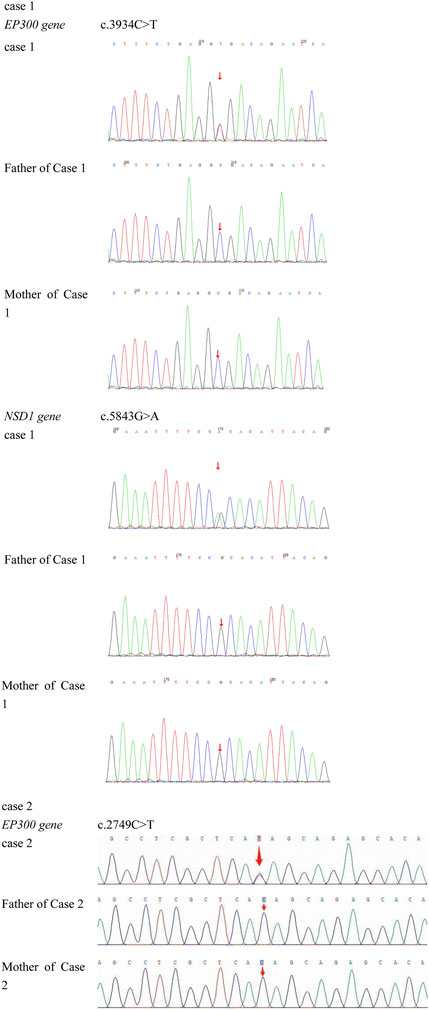
Figure 3. Case 1 carries a heterozygous mutation in exon 24 of the EP300 gene: c.3934C>T (p.Arg1312Ter). Genetic testing of the parents showed no variation at this locus, confirming it as a de novo mutation. Additionally, Case 1 exhibits a heterozygous mutation in exon 18 of the NSD1 gene: c.5843G>A (p.Arg1948His). Parental testing at this locus similarly revealed no variations, indicating another de novo mutation. Case 2 has a heterozygous mutation in exon 14 of the EP300 gene: c.2749C>T (p.Gln917*). Testing of both parents demonstrated no genetic variation at this site, confirming it as a de novo mutation.
3.2.2 Case 2
A heterozygous mutation in exon 14 of the EP300 gene, *c.2749C>T (p. Gln917) **, was identified. This mutation results in a premature stop codon at amino acid position 917, leading to a truncated protein and potential loss of function. This variant has not been reported in the literature or large-scale population frequency databases. Parental testing confirmed it as a de novo mutation (Figure 3). According to the ACMG guidelines, this variant is preliminarily classified as pathogenic (PVS1 + PM2_Supporting + PS2).
Structural analysis using SWISS- MODEL (https://swissmodel.expasy.org/) online database and PyMOL software revealed that both mutations in the EP300 gene result in truncated proteins, leading to loss of function (Figures 4b,c). Additionally, copy number variation (CNV) analysis was performed for Case 2, and no pathogenic CNVs were detected.
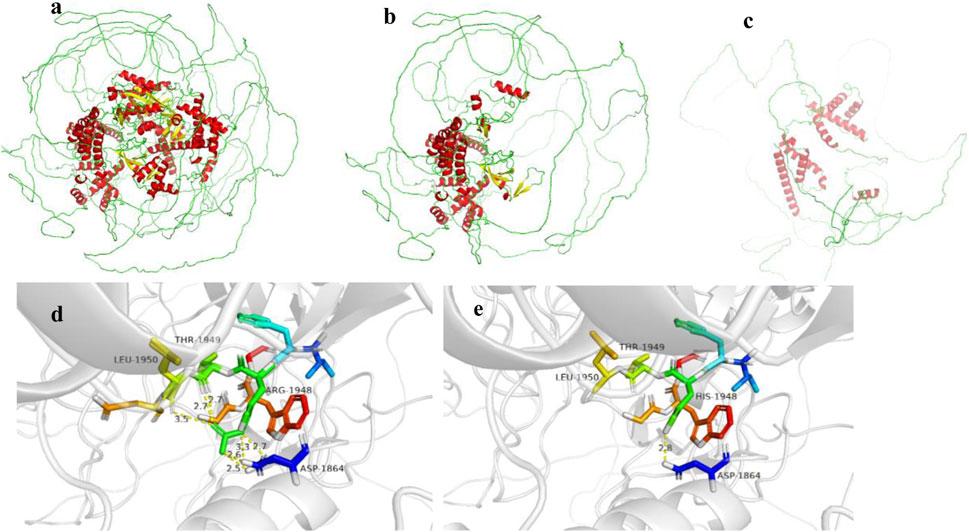
Figure 4. (a–c) show the EP300 gene wild type, c.3934C>T (p.Arg1312Ter) mutation, and c.2749C>T (p.Gln917*) mutation, respectively. (d,e) show the NSD1 gene wild type and c.5843G>A (p.Arg1948His) mutation, respectively.
Case 1 also harbored a heterozygous mutation in exon 18 of the NSD1 gene, c.5843G>A (p. Arg1948His), which is a missense mutation located in a mutation hotspot region (Figure 3). While mutations at the same position have been reported in the literature (Pohjola et al., 2012), the specific amino acid change (p. Arg1948His) has not been documented. Bioinformatics predictions using REVEL (score: 0.831), SIFT, PolyPhen-2, MutationTaster, and GERP + all indicated that this variant is deleterious. Parental testing confirmed it as a de novo mutation. This variant has not been reported in the literature, and its pathogenicity in the ClinVar database is classified as uncertain significance. Its frequency in the general population databases is 0.0001087. According to the ACMG guidelines, this variant is preliminarily classified as pathogenic (PM1 + PM5 + PP3_Moderate + PS2_P + PM2_P). Structural analysis using PyMOL software revealed that the 1948th amino acid of the NSD1 protein is located in a β-sheet secondary structural domain. The substitution of arginine with histidine disrupts hydrogen bonds with aspartic acid at position 1864, threonine at position 1949, and leucine at position 1950, potentially leading to local structural instability and impaired protein function (Figure 4e).
3.3 Results of literature review
A total of 4 English articles and 4 Chinese articles meeting the criteria were identified. After removing duplicates and cases with incomplete information, six patients with clinical phenotypes and genotypes were collected. Including the two newly reported cases in this study, a total of 8 Chinese patients with Rubinstein-Taybi syndrome (RSTS) caused by EP300 gene variants were included (Yang and Chen, 2023; Sun et al., 2021; Huang et al., 2023; Du et al., 2023; Bai et al., 2023; Yang et al., 2024) (details in Table 1). Among the eight patients, 1 case (12.5%) had a missense mutation, 3 cases (37.5%) had frameshift mutations, and 4 cases (50%) had nonsense mutations.
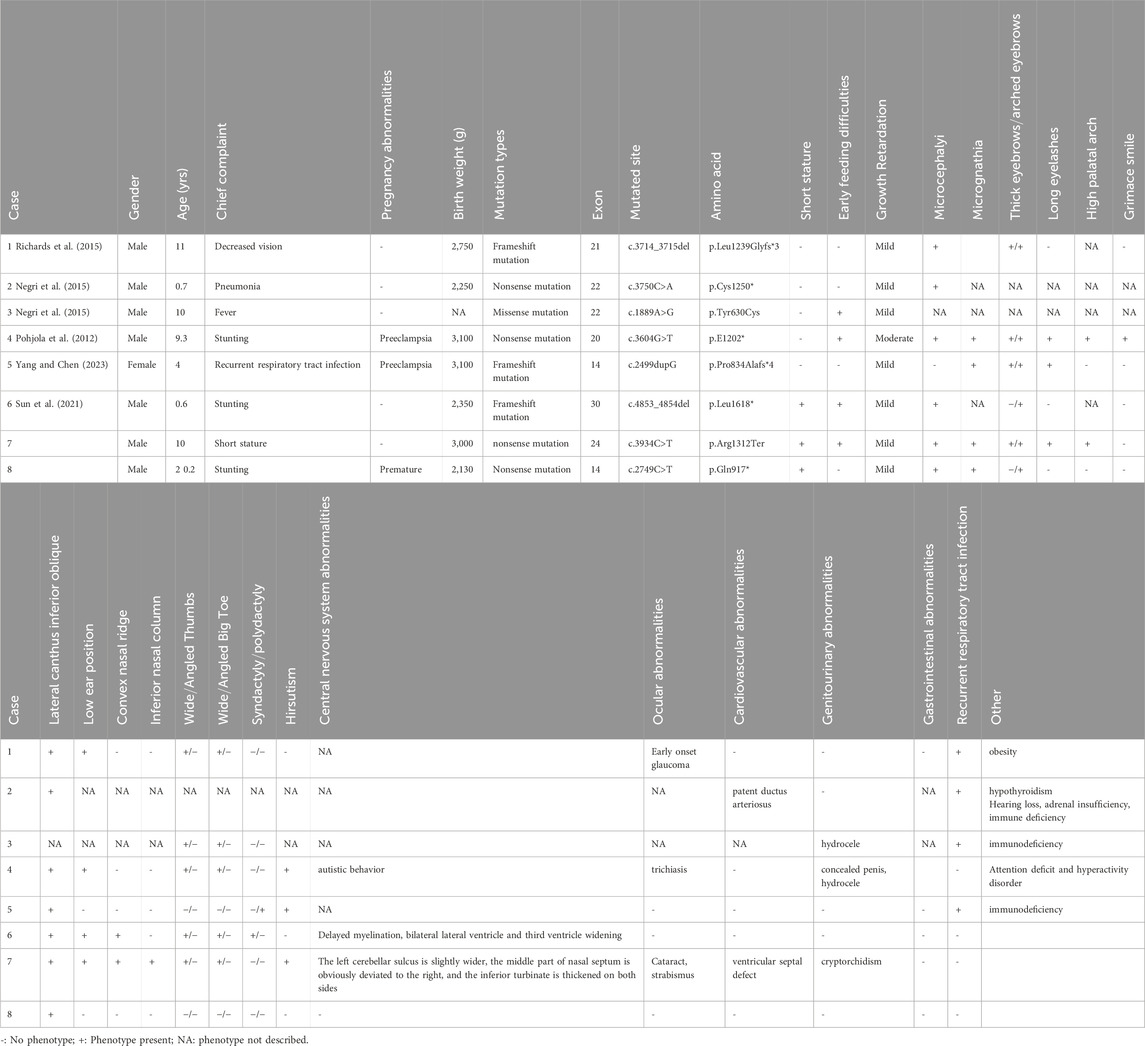
Table 1. Clinical phenotypes of Chinese patients with EP300 gene mutation RSTS.
Comparative analysis of phenotypic differences between the Chinese cohort and the Cohen JL and Fergelot P. cohorts revealed that arched eyebrows, downslanting palpebral fissures, and low-set ears were significantly more common in the Chinese population, with statistical significance. Phenotypes such as angulated thumbs, severe intellectual disability, epilepsy, scoliosis, and keloid formation were not reported in the Chinese population (details in Figure 5; Table 2).
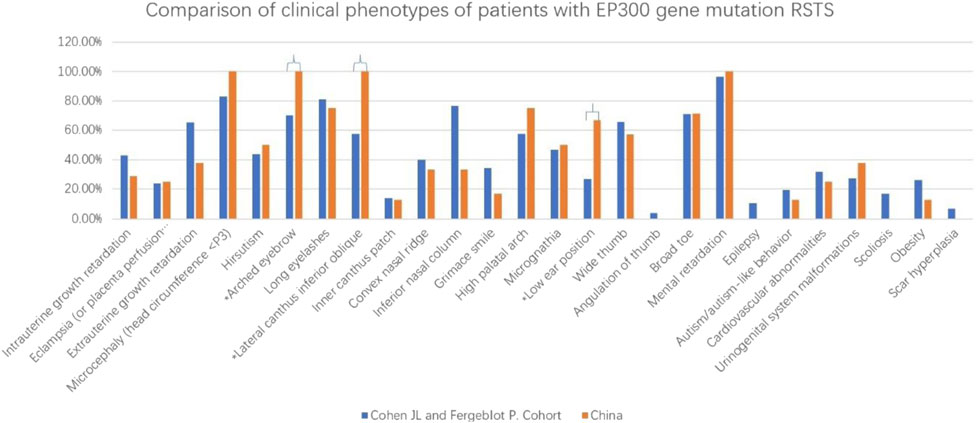
Figure 5. This figure revealed that arched eyebrows, downslanting palpebral fissures, and low-set ears were significantly more common in the Chinese population. *p < 0.05.
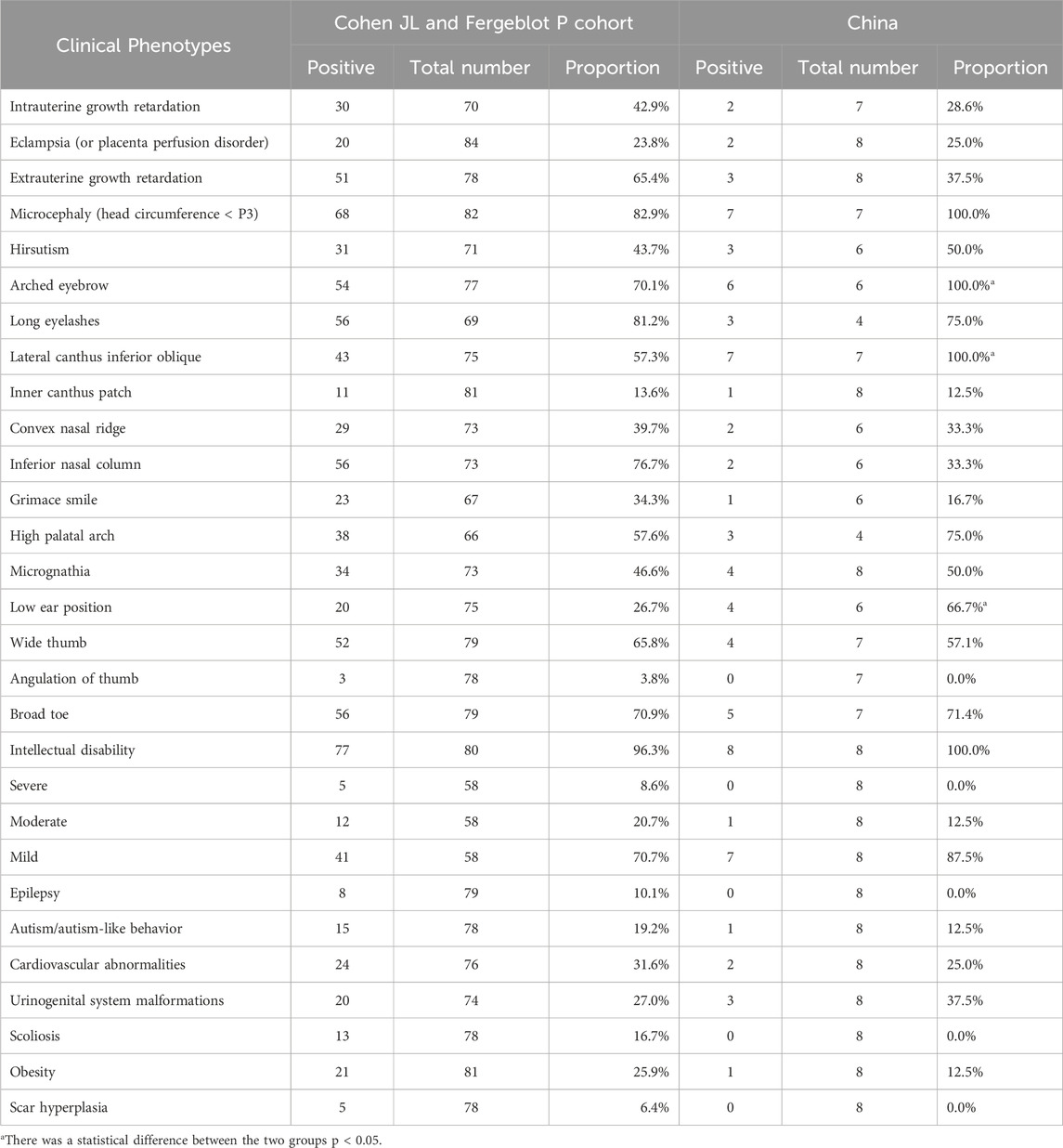
Table 2. Comparison of clinical phenotypes of patients with EP300 gene mutation RSTS.
4 Discussion
Rubinstein-Taybi Syndrome (RSTS) is a genetic disorder affecting multiple systems. Clinically, it is also referred to as “Broad Thumb-Hallux Syndrome”. Its main features include craniofacial abnormalities (e.g., microcephaly, thick hair, low hairline, arched/thick eyebrows, downslanting palpebral fissures, epicanthal folds, high-arched palate, micrognathia), skeletal abnormalities (e.g., broad thumbs/halluces, clinodactyly, scoliosis), short stature, and intellectual disability. Additionally, it may involve abnormalities in the respiratory, gastrointestinal, cardiovascular, nervous, and genitourinary systems (Van Gils et al., 2021).
In this study, both patients with EP300 gene mutations exhibited short stature and dysmorphic features (e.g., microcephaly, downslanting palpebral fissures, micrognathia, broad nasal bridge), but no typical “grimacing smile” or broad/angulated thumbs. Their intellectual development was mildly delayed, consistent with previous reports. Case 1 had additional features such as webbed neck, hirsutism, and broad halluces, along with structural anomalies like cryptorchidism, ventricular septal defect, strabismus, and deviated nasal septum, while Case 2 lacked these phenotypes.
Pathogenic mutations in the CREBBP and EP300 genes can cause RSTS. The encoded proteins, EP300 and CREBBP, share high sequence similarity in the bromodomain, cysteine-histidine-rich region, and histone acetyltransferase (HAT) domain. Both play critical roles in embryonic development, growth control, and homeostasis by coupling chromatin remodeling with transcription factor recognition. Mutation types include intragenic deletions, whole-gene deletions, frameshift mutations, nonsense mutations, missense mutations, splice-site mutations, and duplications. Currently, most studies suggest no clear correlation between the severity or specificity of clinical phenotypes and the type of single-gene variant (Roelfsema and Peters, 2007; Kuai, 2023; Cohen et al., 2020).
EP300 gene mutations account for 8%–11% of RSTS cases. The EP300 protein, encoded by the EP300 gene, functions as a histone acetyltransferase involved in chromatin remodeling and transcriptional regulation, which are essential for cell proliferation and differentiation. Additionally, EP300 mediates cAMP gene regulation by specifically binding to phosphorylated CREB protein (Dyson and Wright, 2016). Previous reports indicate that patients with EP300 variants often exhibit milder clinical manifestations compared to those with CREBBP variants or chromosome 16p13.3 abnormalities. The characteristic “grimacing smile” is less common in EP300 variant patients, and features such as broad thumbs and angulated thumbs are rare. Intellectual impairment in EP300 variant patients is typically milder, often presenting as mild to moderate intellectual disability or normal intelligence with learning difficulties (Fergelot et al., 2016).
Analysis of ClinVar database entries for pathogenic or likely pathogenic variants revealed that missense mutations in the EP300 gene account for 18.8% (35/186), while loss-of-function variants (nonsense, frameshift, splice-site, and start codon loss) account for 76.3% (142/186). This distribution is similar to that observed in Chinese patients. The two newly reported EP300 mutations in this study are truncating variants, which alter protein length, leading to loss of function, impaired transcription, and disrupted cell proliferation and differentiation, resulting in a range of clinical manifestations.
Case 1 also harbored a pathogenic variant in the NSD1 gene, which is rare given that both variants were de novo. The parents were not of advanced age, and the mother took folic acid before and during pregnancy. The mutations may be associated with the mother’s long-term use of antiepileptic drugs. NSD1 gene mutations cause Sotos syndrome (OMIM 117550), a rare autosomal dominant disorder characterized by four main features: (1) dysmorphic facial features (e.g., macrocephaly, prominent forehead, high hairline, downslanting palpebral fissures, long narrow face, pointed chin, sparse frontotemporal hair); (2) overgrowth (prenatal and postnatal accelerated growth, height and/or head circumference ≥2 SD); (3) advanced bone age; and (4) developmental delay (learning difficulties, mild to severe intellectual disability) (Luo, 2020). Case 1 exhibited high-arched palate, strabismus, micrognathia, and learning difficulties but lacked typical facial features and overgrowth, with no advanced bone age. This atypical presentation may be due to overlapping effects of the EP300 gene.
Both cases also required differentiation from Menke-Hennekam syndrome. Menke and Hennekam analyzed 11 patients with CREBBP mutations and 2 with EP300 homologous region variants, who lacked typical RSTS features but exhibited developmental delay, autism-like behaviors, short stature, and microcephaly (DDD study, 2018). Additional features included feeding difficulties, vision and hearing impairments, recurrent upper respiratory infections, and epilepsy. The two patients in this study lacked autism-like behaviors, significant feeding difficulties, recurrent infections, or epilepsy, supporting a diagnosis of RSTS based on clinical phenotypes and genetic testing.
With the widespread clinical application of new genetic technologies, an increasing number of genes have been linked to short stature. Both patients in this study presented to our pediatric clinic for growth retardation. Case 1 was initially diagnosed with Noonan syndrome (NS) due to features like downslanting palpebral fissures, webbed neck, and low hairline, along with short stature (<10th percentile), ventricular septal defect, intellectual disability, and cryptorchidism. However, genetic testing did not identify NS-associated genes but revealed pathogenic mutations in EP300 and NSD1. This highlights the importance of genetic testing for accurate diagnosis and treatment in children with short stature and dysmorphic features, as well as for assessing potential disease risks. Both parents initially requested growth hormone (GH) therapy. While no literature supports GH therapy for RSTS, studies suggest a link between EP300 mutations and gastrointestinal tumors (Wang et al., 2023). Considering potential risks, neither patient received GH therapy. Therefore, genetic testing is essential for children with short stature and dysmorphic features to clarify the etiology and guide appropriate GH therapy.
Additionally, this study compared phenotypic differences between Chinese RSTS patients with EP300 mutations and European/American cohorts (Cohen et al., 2020), finding that arched eyebrows, downslanting palpebral fissures, and low-set ears were more common in the Chinese population. However, these features are not specific, and the statistical differences may be due to small sample sizes.
4.1 Study limitations
Due to the low incidence of EP300 variants and limited reported cases, the relationship between genotype and phenotype requires further validation with larger sample sizes. In the future, we look forward to international multicenter studies and more case reports to make the phenotypic spectrum of EP300-related RSTS more comprehensive.
5 Conclusion
In summary, this study reports the clinical and genetic characteristics of two RSTS patients with EP300 mutations, including one with dual mutations in EP300 and NSD1, enriching our understanding of RSTS. Further research into the role of EP300 gene in neuronal and skeletal cell development and the exploration of new treatments for RSTS are needed to improve early diagnosis, treatment, and prognosis.
Data availability statement
The data presented in the study are deposited in ClinVar, accession numbers SUB15286200 and SUB15286227.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Hunan Province Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
YW: Formal Analysis, Methodology, Writing – original draft. XW: Methodology, Software, Visualization, Writing – review and editing. SZ: Data curation, Investigation, Methodology, Writing – review and editing. NX: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the grants from the Hunan Provincial Science and Technology Department Project (2024JJ5219), the Research project of Hunan Provincial Health Project (202206045182).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bai, Z., Li, G., and Kong, X. (2023). Case report: a Chinese girl like atypical Rubinstein-Taybi syndrome caused by a novel heterozygous mutation of the EP300 gene. BMC Med. Genomics 16 (1), 24. doi:10.1186/s12920-022-01424-4
Cohen, J. L., Schrier Vergano, S. A., Mazzola, S., Strong, A., Keena, B., McDougall, C., et al. (2020). EP300-related Rubinstein-Taybi syndrome: highlighted rare phenotypic findings and a genotype-phenotype meta-analysis of 74 patients. Am. J. Med. Genet. A 182 (12), 2926–2938. doi:10.1002/ajmg.a.61883
DDD study, Menke, L. A., Gardeitchik, T., Hammond, P., Heimdal, K. R., Houge, G., et al. (2018). Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am. J. Med. Genet. A 176 (4), 862–876. doi:10.1002/ajmg.a.38626
Du, C., Li, Z., Zou, B., Li, X., Chen, F., Liang, Y., et al. (2023). Novel heterozygous variants in the EP300 gene cause Rubinstein-Taybi syndrome 2: reports from two Chinese children. Mol. Genet. Genomic Med. 11 (9), e2192. doi:10.1002/mgg3.2192
Dyson, H. J., and Wright, P. E. (2016). Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding protein (CBP) and p300. J. Biol. Chem. 291 (13), 6714–6722. doi:10.1074/jbc.R115.692020
Fergelot, P., Van Belzen, M., Van Gils, J., Afenjar, A., Armour, C. M., Arveiler, B., et al. (2016). Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by EP300 mutations. Am. J. Med. Genet. A 170 (12), 3069–3082. doi:10.1002/ajmg.a.37940
Huang, X., Rui, X., Zhang, S., Qi, X., Rong, W., and Sheng, X. (2023). De novo variation in EP300 gene cause Rubinstein-Taybi syndrome 2 in a Chinese family with severe early-onset high myopia. BMC Med. Genomics 16 (1), 84. doi:10.1186/s12920-023-01516-9
Kuai, Y. (2023). Rubinstein-Taybi syndrome: a case report and literature review. Chin. J. Reprod. Health 34 (6), 581–585. doi:10.3969/j.issn.1671-878X.2023.06.019
Luo, T. (2020). Progress in diagnosis and treatment of Sotos syndrome. Chin. J. Med. Genet. 37 (02), 536–539. doi:10.19538/j.ek2023070612
Milani, D., Manzoni, F. M. P., Pezzani, L., Ajmone, P., Gervasini, C., Menni, F., et al. (2015). Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Ital. J. Pediatr. 41, 4. doi:10.1186/s13052-015-0110-1
Negri, G., Milani, D., Colapietro, P., Forzano, F., Della Monica, M., Rusconi, D., et al. (2015). Clinical and molecular characterization of Rubinstein-Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin. Genet. 87 (2), 148–154. doi:10.1111/cge.12348
Petrij, F., Giles, R. H., Dauwerse, H. G., Saris, J. J., Hennekam, R. C., Masuno, M., et al. (1995). Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376 (6538), 348–351. doi:10.1038/376348a0
Pohjola, P., Peippo, M., Penttinen, M. T., Elenius, K., and Kääriäinen, H. (2012). Translation of a research-based genetic test on a rare syndrome into clinical service testing, with sotos syndrome as an example. Genet. Test. Mol. Biomarkers 16 (10), 1188–1194. doi:10.1089/gtmb.2012.0153
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Roelfsema, J. H., and Peters, D. J. (2007). Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev. Mol. Med. 9 (23), 1–16. doi:10.1017/S1462399407000415
Roelfsema, J. H., White, S. J., Ariyürek, Y., Bartholdi, D., Niedrist, D., Papadia, F., et al. (2005). Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 76 (4), 572–580. doi:10.1086/429130
Sun, J., Wu, Q., and Zhang, T. (2021). A case of Rubinstein-Taybi syndrome caused by EP300 gene mutation and literature review. Chin. J. Child. Health Care 29 (12), 1387–1389. doi:10.11852/zgetbjzz2020-1215
Van Gils, J., Magdinier, F., Fergelot, P., and Lacombe, D. (2021). Rubinstein-taybi syndrome: a model of epigenetic disorder. Genes (Basel) 12 (7), 968. doi:10.3390/genes12070968
Wang, C., Lin, H., Zhao, W., Liang, Y., Chen, Y., and Wang, C. (2023). MiR-26a-5p exerts its influence by targeting EP300, a molecule known for its role in activating the PI3K/AKT/mTOR signaling pathway in CD8+tumor-infiltrating lymphocytes of colorectal cancer. Cell Mol. Biol. (Noisy-le-grand) 69 (12), 232–241. doi:10.14715/cmb/2023.69.12.37
Yang, B. T., and Chen, T. T. (2023). A case of Rubinstein-Taybi syndrome caused by EP300 gene mutation. Chin. J. Med. Genet. 40 (3), 360–363. doi:10.3760/cma.j.cn511374-20220726-00493
Keywords: Rubinstein-Taybi syndrome, EP300 gene, NSD1 gene, growth retardation, children
Citation: Wang Y, Wu X, Zhao S and Xu N (2025) Rubinstein Taybi syndrome caused by EP300 gene mutation: what we learned from two cases and literature review. Front. Genet. 16:1588657. doi: 10.3389/fgene.2025.1588657
Received: 06 March 2025; Accepted: 04 June 2025;
Published: 02 July 2025.
Edited by:
Mara Marongiu, National Research Council (CNR), ItalyReviewed by:
Cristina Skrypnyk, Arabian Gulf University, BahrainYong Wu, Shenzhen Baoan Women’s and Children’s Hospital, China
Copyright © 2025 Wang, Wu, Zhao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ningan Xu, NDk0Mzc5OUBxcS5jb20=
†These authors have contributed equally to this work and share first authorship