 Leonardo Affronte1,2
Leonardo Affronte1,2 Antonella Pini2*Claudia Pizzoli3Emanuele Coccia4
Antonella Pini2*Claudia Pizzoli3Emanuele Coccia4 Serena Mazzone3Arber Golemi5Melania Giannotta2
Serena Mazzone3Arber Golemi5Melania Giannotta2 Duccio Maria Cordelli3,6
Duccio Maria Cordelli3,6 Valerio Carelli6,7
Valerio Carelli6,7 Alessandro Vaisfeld1,4
Alessandro Vaisfeld1,4 Flavia Palombo7
Flavia Palombo7- 1Department of Medical and Surgical Sciences (DIMEC), Alma Mater Studiorum, University of Bologna, Bologna, Italy
- 2IRCCS Istituto delle Scienze Neurologiche di Bologna, Pediatric Neuromuscular Unit, UO Neuropsichiatria dell’Età Pediatrica, Bologna, Italy
- 3IRCCS Istituto Delle Scienze Neurologiche di Bologna, UOC Neuropsichiatria dell'Età Pediatrica, Bologna, Italy
- 4Medical Genetics Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy
- 5Medicina Nucleare, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy
- 6Department of Biomedical and Neuromotor Sciences (DIBINEM), University of Bologna, Bologna, Italy
- 7IRCCS Istituto delle Scienze Neurologiche di Bologna, Programma di Neurogenetica, Bologna, Italy
ATP13A2 is a gene localized on chromosome 1p36.13 and coding for a transmembrane protein found in the lysosomes and late endosomes, which is involved in many cellular metabolic activities. Pathogenetic variants of ATP13A2 are associated with a wide range of neurodegenerative disorder including Kufor Rakeb syndrome (KRS), a rare autosomal recessive form of levodopa responsive juvenile onset parkinsonism (MxMD-ATP13A2), characterized by rapidly progressive muscular stiffness, bradykinesia, spasticity, pyramidal findings, dementia and supranuclear gaze palsy. The aim of this study is to provide detailed clinical descriptions of two siblings, carriers of novel biallelic ATP13A2 variants. One of them showed KRS levodopa-responsive motor dystonic features at the age of 10 years preceded by moderate cognitive impairment, while the other only showed mild cognitive impairment at our last evaluation at 11 years of age. Additionally, we reviewed the previously published cases, focusing on early signs and symptoms, clinical evolution and response to therapy. To our knowledge, this is the only work that groups all reported KRS patients and compares their clinical and molecular features.
Introduction
ATP13A2 is a gene localized on chromosome 1p36.13 and coding for a transmembrane protein found in the lysosomes and late endosomes. The protein belongs to a P-type ATPases family whose role is to transport substrates through membranes by ATP hydrolysis and whose alterations underlie impairment in the metal/cation complex and mitochondrial homeostasis as well as lysosomal function (Martin et al., 2015; Sørensen et al., 2018; Van Veen et al., 2014). ATP13A2 variants are associated with a wide range of neurodevelopment and neurodegenerative disorders including Kufor Rakeb syndrome (KRS), early-onset parkinsonism (EOPD), neuronal ceroid lipofuscinosis (NCL), hereditary spastic paraplegia (HSP) and amyotrophic lateral sclerosis-like form (Estrada-Cuzcano et al., 2017; Yahya et al., 2023). KRS is a rare autosomal recessive form of levodopa-responsive juvenile onset parkinsonism, previously known as PARK9 and now as MxMD-ATP13A2 (Lange et al., 2022), that was initially described in 1994 in five members of a large family who lived in the Jordanian town of Kufor Rakeb, after which the disease was named (Najim al-Din et al., 1994; Brüggemann et al., 2010). In 2006 a large Chilean family with early-onset parkinsonism, whose characteristics resembled those of the Jordanian family, was reported and the ATP13A2 gene was firstly associated to the syndrome (Ramirez et al., 2006). To confirm this association, ATP13A2 pathogenic variants were investigated and found in the members of the original family (Ramirez et al., 2006).
KRS is characterized by early onset parkinsonism, usually between 12 and 16 years, and a rapid progression of clinical signs and symptoms. Motor manifestations include extrapyramidal findings such as rigidity, tremor, bradykinesia, postural instability with festinating gait, pyramidal findings such as spasticity with muscular stiffness, myoclonus, hyperreflexia and in some cases positive Babinski’s sign (Najim al-Din et al., 1994; Williams et al., 2005; Ramirez et al., 2006; Brüggemann et al., 2010). Additional motor signs include dystonia, ataxia, dyskinesia like facial-faucial-finger mini-myoclonus, dysarthria, dysphagia, slowed vertical and/or horizontal saccade eye movement and supranuclear upgaze palsy. Non-motor findings include cognitive decline that leads in some cases to dementia, visual hallucination and autonomic disorders such as urinary and fecal incontinence (Schneider et al., 2010; Yang and Xu, 2014). Only in a few cases the motor symptoms are reported to be preceded by cognitive impairment (Schneider et al., 2010; Crosiers et al., 2011; Niemann and Jankovic, 2019). In children, features such as psychomotor delay, early cognitive decline, pyramidal signs, ataxia and atypical movement disorders such as dystonia or choreoathetosis could make the diagnosis even more complicated (Paviour et al., 2004; Schrag and Schott, 2006; Garcia-Cazorla and Duarte, 2014; Niemann and Jankovic, 2019). This wide variety of signs and symptoms is probably due to a widespread brain involvement. KRS patients were examined with magnetic resonance imaging (MRI): common findings include diffuse brain atrophy and bilateral abnormal hypodensity on the T2 images in the putamen and caudate nuclei, due to iron accumulation (Brüggemann et al., 2010; Schneider et al., 2010; Yang and Xu, 2014). Moreover, in some patients the dopamine transporter (DAT) images revealed striatal tracer uptake under physiological levels (Brüggemann et al., 2010).
At present, there is no cure for KRS and the treatment is symptomatic. The major symptoms are sufficiently well controlled for some years by levodopa and carbidopa in most cases, while other medications such as dopamine agonists or trihexyphenidyl are reported as beneficial in some patients (Kola et al., 2022). Psychiatric symptoms, if they occur, can be controlled with antipsychotic drugs (McNeil-Gauthier et al., 2019). Second generation antipsychotic drugs should be used in order to reduce the occurrence of extrapyramidal side effects. Finally, deep brain stimulation can be considered as a treatment in the advanced cases (Kola et al., 2022).
The purpose of this work is to describe the phenotype of two siblings carrying the same novel pathogenic variants: a boy presenting with the typical phenotype at the age of 10 years, and his younger sister with only neuropsychological impairment. Furthermore, we provide a review of all cases published to date, with a focus on molecular data and phenotypic implications.
Case 1 description
Patient 1 is a 14-year-old Italian boy, born at term by a normal delivery after a non-complicated pregnancy. His parents are not consanguineous and there is no history of neurological or psychiatric pathologies in the family. He had a mild motor milestone delay, starting to walk independently at around 2 years of age. He began to use single words and then to talk at standard age, having some phonological issues for which he was treated by a speech therapist for a few years, with a good response. During primary school he showed learning difficulties and at 7 years the Wechsler Intelligence Scale for Children–Fourth (WISC IV) was performed, showing a mild to moderate cognitive deficit (VCI 64, PRI 69, WMI 58, PSI 65, Full Scale IQ 53). At the age of 8 years he came for the first time to our attention. Neurological examination was normal, he was able to walk and run fast with no coordination problems and he was independent in everyday activities. A wakeful and sleep EEG showed occasional sharp waves in the fronto-central regions of the left hemisphere, not increased with sleep. EEG was repeated, but the previously observed abnormalities were no longer detected.
At about 10 years of age he became slower and showed an overall flexed posture. Neurological examination revealed a dystonic and flexed posture of the left arm and bilateral tremor of the hands. Urinary incontinence was reported. Strength and reflexes were normal. In a few months his condition worsened and amimia, facial-faucial-finger mini-myoclonus, reluctant speech, slowed vertical and horizontal saccade eye movement appeared. Moreover, worsening of cognitive ability was observed: WISC IV scale was performed at 11 years old (VCI 64, PRI 65, WMI 46, PSI 53, Full Scale IQ 44). This new result showed that those tasks evaluating the efficiency of cognitive processes were mainly involved, whereas pure verbal and non-verbal reasoning skills remained stable. Therefore, it seems that the functions worsening were verbal working memory (VMI) and executive speed tasks (PSI).
Routine blood tests and motor and sensory nerve conduction, showed no alteration. Brain and spinal cord MRI at the age of 11 was normal.
Whole exome sequencing (WES) was performed, showing compound heterozygosity for ATP13A2 (MxMD-ATP13A2) variants (NM_022089.4): the c.2425dup variant, never reported and maternally inherited, is classified as likely pathogenic (PVS1, PM2_SUP), causing the misalignment of the reading frame with production of a truncated p. Ala809GlysTer49 protein; the c.3153dup variant, rare (2/1,614,040 alleles in gnomAD) and paternally inherited, is classified as pathogenic (PVS1, PM2_SUP), also causing a misalignment of the reading frame with production of a truncated protein p. Ser1052LeufsTer62. Moreover, the c.3153dup is already reported in ClinVar as a Pathogenic variant (VCV001968613.4). Pathogenic and likely pathogenic variants in other genes related to parkinsonism, to childhood onset dystonia, chorea or related movement disorder and to intellectual disability were excluded. Both ATP13A2 variants were validated through Sanger sequencing. Parents did not show learning disability and were intellectually normal.
After the diagnosis a dopamine transporter scan (DaTscan) was performed, revealing bilaterally reduced availability of the presynaptic dopamine transporters in the putamen and a mild bilateral uptake reduction in the caudate nucleus compared with normal controls. These findings represent a bilateral and symmetric impairment of the nigro-striatal system (Figure 1).
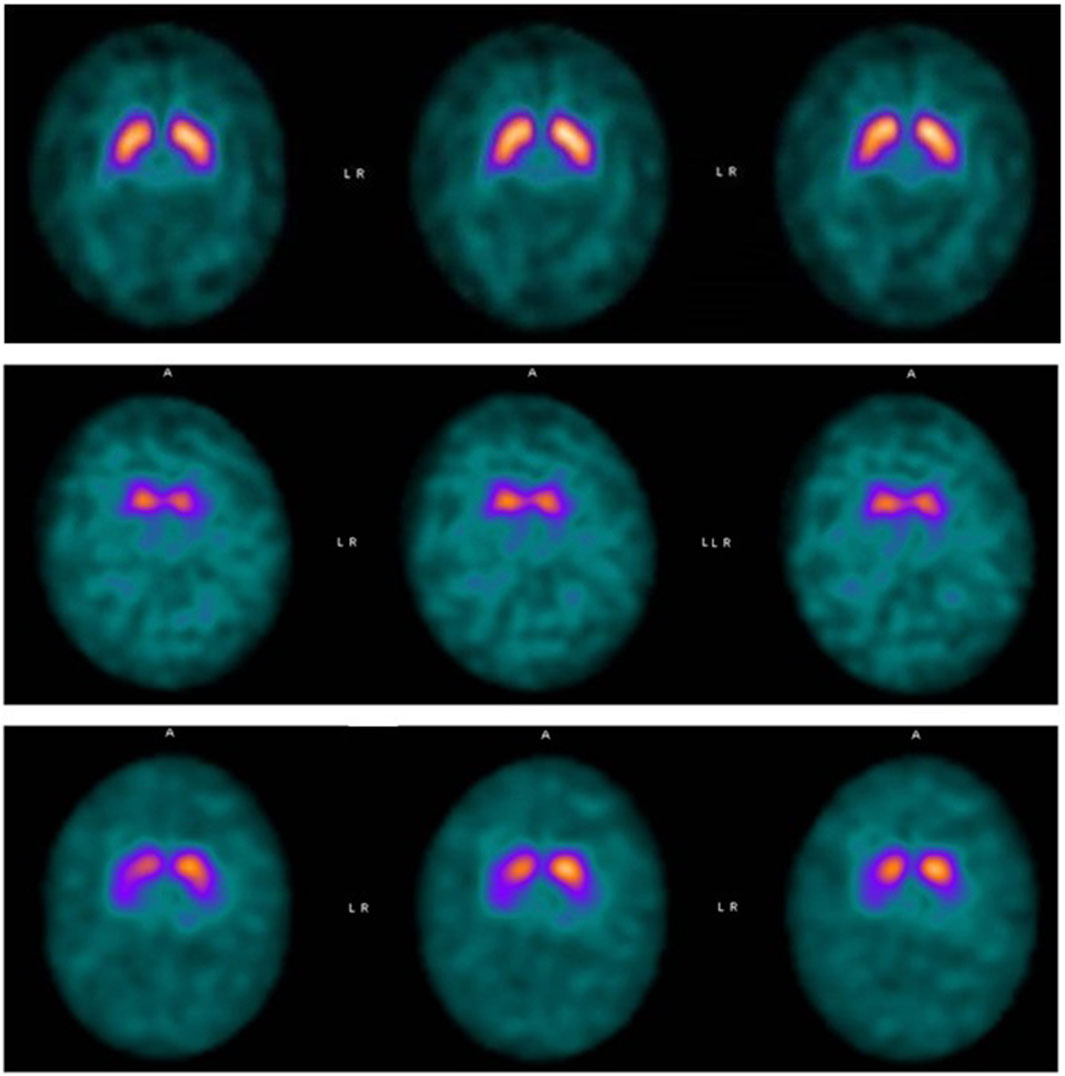
Figure 1. Top panel: DaTSCAN SPECT normal. Middle panel: Patient 1: ligand binding in both putamina was almost absent. The uptake was also markedly and symmetrically reduced in the caudate nucleus bilaterally. Bottom panel: Patient 2: DaTSCAN SPECT showed considerably decreased ligand binding in both putamina. Initial uptake reduction was observed also in the right caudate.
The autonomic control of the cardiovascular system was studied by deep-breathing test and tilt test, both of which were normal for age.
The child was treated from the age of 12 years with levodopa, with benefits. Clinically, movements became more fluid and rapid, gait skills improved, the face was more expressive, he regained sphincteric control and his speech was also more comprehensible. Moreover, a scopolamine patch was used to control sialorrhea. No collateral effects related to levodopa have been observed so far, at 14 years of age. The cognitive profile on the WISC IV scale at the age of 13 resulted in VCI 58, PRI 56, WMI 46, PSI 47, and Full Scale IQ 40; a reevaluation at age 14 years and 8 months showed improvement in visual logical reasoning skills and poor development in processing speed and verbal memory (VCI 52, PRI 63, WMI 46, PSI 47 Full scale IQ 40). Auditory attention in a signal detection task also proved to be in the expected average for age (subtest Auditory Attention Nepsy 2 50th for age). This task was not administered the previous year due to difficulty in maintaining delivery and number of false recognitions exceeding correct responses.
Case 2 description
The sister of patient 1 came to our attention at the age of 11 years following her brother’s diagnosis and carried the same genetic diagnosis.
Her neurological examination was normal. She appeared as a quiet girl, her father reported adequate social and relational skills. The neuropsychological evaluation showed general cognitive skills below average, assessed by the WISC-IV test (VCI 80, PRI 78, WMI 76, PSI 65, Full Scale IQ 67). The profile was homogeneous, apart from the visuo-graphic processing speed index. This score is influenced by the specific subitem “Coding” of WISC-IV, the graphic speed test, which shows a lower result than the other tests, as if to indicate a greater involvement of fine motor skills. Planning and visuomotor integration skills were also investigated by VMI test, showing a lower score than the average for age (VMI, 8th percentile). It therefore seemed that motor action planning was the first and most compromised domain. Awake EEG was normal. Brain DaTSCAN SPECT showed considerably decreased ligand binding in both putamina (Figure 1). Initial uptake reduction was observed also in the right caudate. Vegetative system function was also studied in this patient. The autonomic control of the cardiovascular system was assessed by performing deep-breathing test, tilt test, and Valsalva maneuver, which were normal for age.
The girl is currently not treated with any medication and attends school with support.
Discussion
Parkinsonism is a frequent condition characterized by bradykinesia, rigidity, tremor and postural instability, most often affecting patients over 60 years of age. Only around 5% of the patients are less than 50 years old (Niemann and Jankovic, 2019). The population with early-onset parkinsonism (EOP) is further arbitrarily subdivided by age of onset. Individuals whose symptoms begin between the ages of 21 and 50 are categorized under the term “young onset parkinsonism” (YOP). However, if the neurological symptoms start before 20 years of age the syndrome is called “juvenile parkinsonism” (JP) (Paviour et al., 2004; Garcia-Cazorla and Duarte, 2014; Niemann and Jankovic, 2019). From an etiological point of view, JP may be the results of acquired or genetic causes. The first group includes some of the most common causes including drug side effects, structural brain lesions, such as hypoxic-ischemic encephalopathy or basal ganglia tumors, encephalitis and immunomediated disease. The second group could be split up into inborn errors of metabolism (IEM) or other genetic causes different from IEM (Garcia-Cazorla and Duarte, 2014). KRS is classified in the JP group and is known for being an autosomal recessive disease that causes iron accumulation in the brain, thus also classified as IEM (Schneider et al., 2010; Garcia-Cazorla and Duarte, 2014; Estrada-Cuzcano et al., 2017).
KRS is an extremely rare disease and this emerges from the small number of cases reported in the literature. Possibly, this is also due to the recent identification of the genetic basis for this disorder. Thus, we reviewed all published cases and to our knowledge, this is the first analysis that groups all reported KRS patients comparing their clinical and molecular features (Table 1). Variants are listed according to the position on the of ATP13A2 protein (Figure 2). We also summarize 15 patients harboring heterozygous pathogenic ATP13A2 variants, who have received a clinical diagnosis of EOPD (Supplementary Table S1) (Di Fonzo et al., 2007; Lin et al., 2008; Djarmati et al., 2009; Chen et al., 2011; Fong et al., 2011; Wang et al., 2020). Clinical and neuroimaging data of KRS patients are summarized in Table 2. Our patients presented with symptoms that are partly characteristic of KRS and partly distinctive.
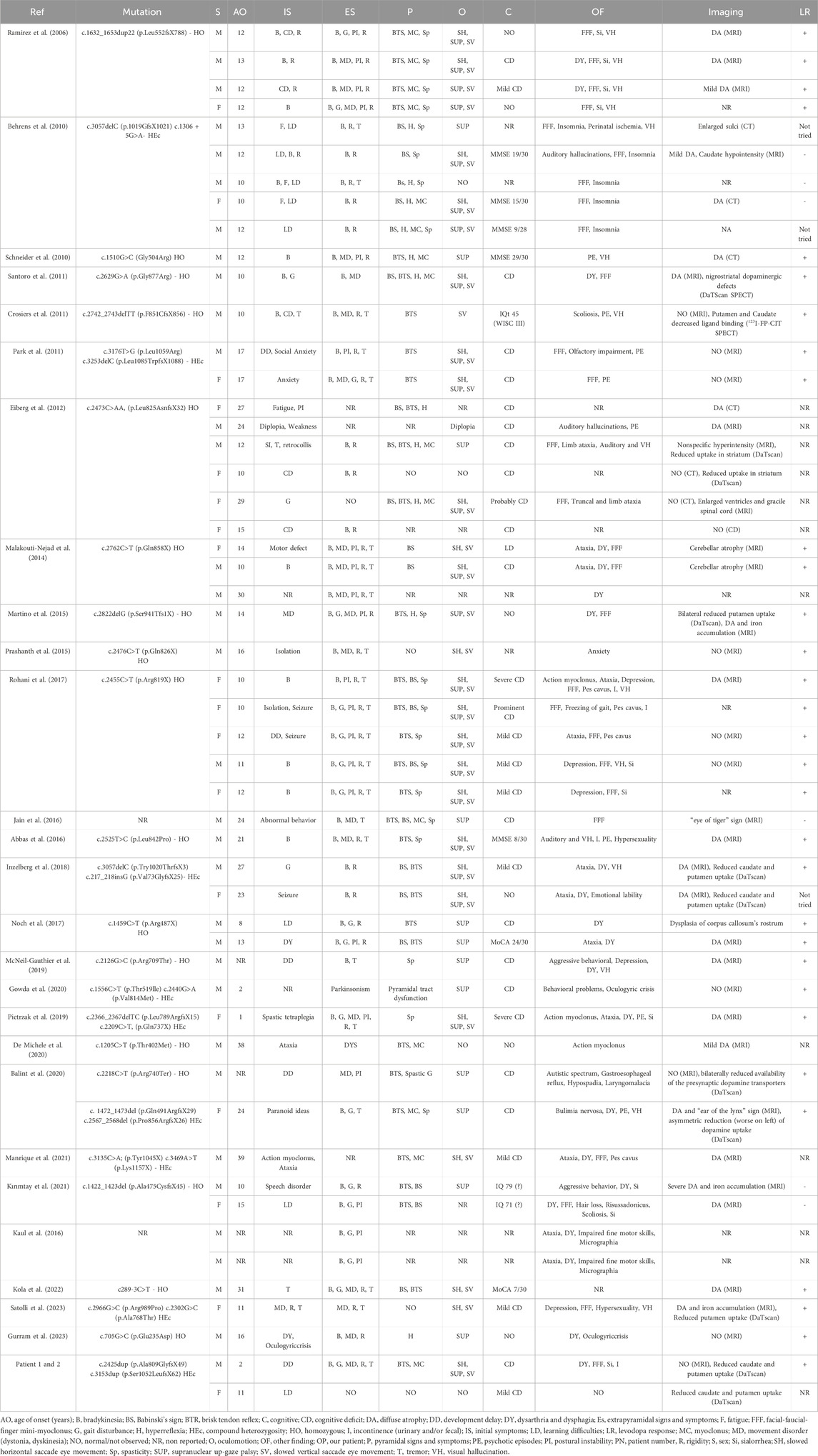
Table 1. Patients with a clinical diagnosis of KRS.
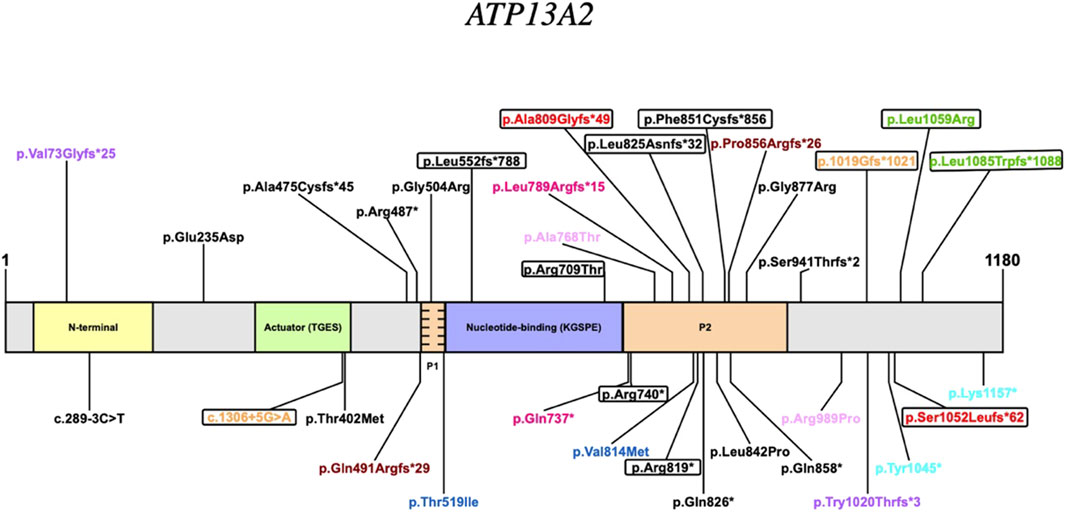
Figure 2. Pathogenic variants of the ATP13A2 gene. Homozygous variants are depicted in black; compound heterozygous variants share the same colour (variants of our cases are in red); encircled variants are reported in patients who presented with “DD developmental delay” or “CD cognitive deficit” as initial symptoms.
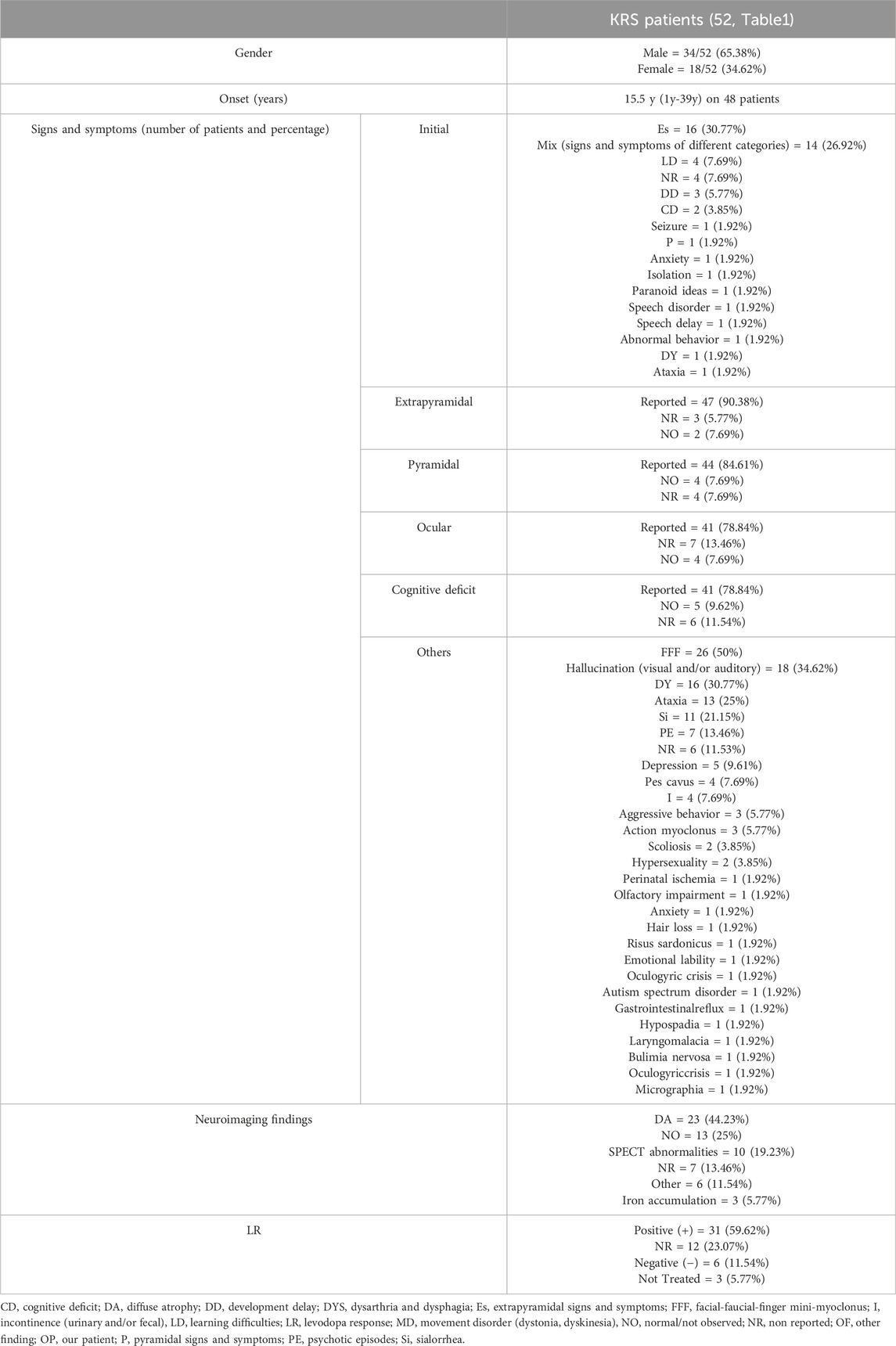
Table 2. Summary of clinical and neuroimaging data of KRS.
A gender bias is observed in KRS patients, being the male-to-female ratio almost 2:1 (Table 2). Concerning the age of onset, mean age is 15.5 years, with a wide range, spanning from a minimum of 1 year to a maximum of 39 years (Table 2), (Ramirez et al., 2006; Rohani et al., 2017; Niemann and Jankovich.,2019). According with literature data, our patients present an early onset at 2 and 11 years, respectively. However, the initial symptoms observed in our patients are remarkable compared with the reported KRS population: the developmental delay observed in patient 1 has been described in only 2 other cases (5.77%) and the learning difficulties observed in patient 2 have been reported in 3 other cases (7.69%). Patient 1 showed a rapidly progressive disease course as most reported patients (Ramirez et al., 2006; Kirimtay et al., 2021; Satolli et al., 2023). The presence of extrapyramidal signs and symptoms, observed in 90.38% of the KRS population, and pyramidal signs and symptoms, described in 84.61% of the KRS population, are observed in patient 1 but not yet in patient 2, at the age of 11 years. Ocular movements impairment, present in patient 1, is reported in the majority of KRS cases (78.84%). The cognitive decline shown by our patient 1 is a very frequent symptom among KRS patients (78.84%). Many other signs and symptoms can be found in KRS: amongst them, facial-finger mini myoclonus, dysphagia, sialorrhea and urinary incontinence. Those are all present in our patient 1. They are some of the most frequent additional findings in KRS, with a recurrence rate of 50%, 30.77%, 21.15% and 7.69%, respectively.
MRI images showed no abnormality in patient 1, corresponding to the second most observed finding in KRS (25%). The young age of patient 1 and the early execution of the MRI are the factors that probably determined the normal result of the examination, which is generally altered in patients with a longer disease course. Moreover, as for some other patients, [123I]FP-CIT–SPECT showed in both our siblings a decrease of dopamine transporter (DaT) density in the striatum that corresponds to a neuronal loss in this brain area. This finding has been observed in other 8 KRS patients (19.23% of the total). A good response to the symptomatic treatment by levodopa is an important element to highlight as the 59.62% of KRS patients respond positively, like in our patient 1. However, a high rate (23.07%) of “not reported” response must be considered.
It is tempting to speculate that the observed clinical differences in KRS patients could be explained by the type and position of ATP13A2 variants. In general, there seems to be a predominance of Loss of Function (LoF) variants (24/35) (Figure 2). Although, it cannot be ruled out that missense variants may also have a LoF effect. Literature data from reported KRS patients suggests that the P2 domain could be a hotspot (Table 1). In fact, variants affecting this domain are numerous accounting for 40% of the total variants (14/35) with a total of 24 patients having at least one allele with a variant in this region. Some phenotypic similarities are observed among the patients in this group such as the age of the onset less than or equal to 11 years of age. Another common feature of the group is the cognitive deficit of variable severity appearing, as in Patient 1 and 2, at the onset of the disease in 5 other patients. From a therapeutic perspective, all patients in this group who were administered levodopa presented a positive response.
However, the two patients described in the present study display also peculiar features that allow some speculations about the brain involvement in KRS due to ATP13A2 gene variants. Patient 1 presented a main involvement of the cognitive efficiency systems and a more severe intellectual disability than those reported in the literature. In patient 2 neuropsychological examination showed only movement planning and visuo-motor integration skills impairment. Therefore, both patients show cognitive impairment with different severity at onset, despite the absence of possible pathogenic variants in DD/ID genes that may act as modifiers. Given the functions involved, although different, there seems to be a primary involvement of the parietal-frontal areas.
In the families reported to date, the phenomenon of “incomplete penetrance”, i.e., cases with pathogenic variants and absence of symptoms, has never been reported. Patients with a late onset and/or slow progression, which can therefore be considered as paucisymptomatic, at least at the onset, are instead described (De Michele et al., 2020; Manrique et al., 2021). However, since they have no siblings, no comparisons could be made. In multi-member families the onset and clinical presentation are homogeneous, except for two families with a certain variability of symptom onset (e.g., from 10 to 29 years) (Eiberg et al., 2012; Malakouti-Nejad et al., 2014). However, once the symptoms have arisen, they rapidly evolved to a similar severity in all members. We also explored a gender-related difference of clinical expressivity and/or age of onset, which did not emerge in the families reported. Undoubtedly, the major bias in reporting a variable expressivity in siblings symptomatology is due to early diagnosis in younger one, as in our family, and this could explain the absence, so far, of the movement disorder phenotype in patient 2 which may eventually develop in the future.
The reasons behind the two unusual presentations are unclear, however we hypothesize that this may depend on a more extensive involvement of the brain, and not only of the basal ganglia. Remaining on the speculative ground also the motor functions of nigro-striatal circuits are affected later than cognitive functions in the presence of an ATP13A2-related cellular energy deficit. Further, we studied in both patients the vegetative system, which had never been done in ATP13A2 patients, finding that was essentially unimpaired based on the tests performed.
Another possible cause of phenotypic variability could be the modifying effect of specific variants within the ATP13A2 protein, according to variant’s type and position. The P2 domain seems to be a hotspot, with a developmental delay severity that is apparently linked to specific variants. However, the small number of cases does not allow definitive conclusions. In addition, the fact that patients with the same ATP13A2 variants experiences different disease onset and progression suggests that other unknown genetic factors may modify the overall effect.
Conclusion
We report here novel pathogenic variants in the ATP13A2 gene causing early onset Kufor Rabek Syndrome presenting with early onset cognitive impairment. To our knowledge patient 2 is the first case with genetic diagnosis characterized by cognitive impairment preceding the motor presentation characteristic of KRS displaying concomitant cognitive motor action planning impairment and learning disability. In this specific case, the diagnosis was due to the previously diagnosed brother. Due to the potential therapeutic approach and to better understand the disease course, we suggest that KRS should be considered even in atypical presentations with only cognitive impairment and ATP13A2 should be included in the genes panel for cognitive delay.
Data availability statement
The data presented in the study are deposited in the BioProject database, accession number BioProject ID: PRJNA1214690.
Ethics statement
The studies involving humans were approved by Ethics Committee of Area Vasta Emilia Romagna (CE-AVEC -17151-17152). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participant’s legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
LA: Writing – original draft, Writing – review and editing. AP: Conceptualization, Supervision, Writing – original draft, Writing – review and editing. CP: Data curation, Formal Analysis, Writing – review and editing. EC: Data curation, Formal Analysis, Writing – review and editing. SM: Data curation, Formal Analysis, Writing – review and editing. AG: Data curation, Formal Analysis, Writing – review and editing. MG: Data curation, Formal Analysis, Writing – review and editing. DMC: Supervision, Writing – review and editing. VC: Supervision, Writing – review and editing. AV: Data curation, Formal Analysis, Writing – review and editing. FP: Conceptualization, Data curation, Formal Analysis, Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
AP, MG and VC are part of the European Reference Network for neuromuscular diseases. The authors would like to thank Prof. Pietro Cortelli for performing the vegetative test on our patients and for the clinical discussion of this paper. Other thanks go to Cecilia Baroncini for technical support. Finally, we would also like to thank the family of our patients for sharing the clinical information and for giving their consent for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1588812/full#supplementary-material
References
Abbas, M. M., Govindappa, S. T., Sheerin, U. M., Bhatia, K. P., and Muthane, U. B. (2016). Exome sequencing identifies a novel homozygous missense ATP13A2 mutation. Mov. Disord. Clin. Pract. 4 (1), 132–135. doi:10.1002/mdc3.12353
Balint, B., Damasio, J., Magrinelli, F., Guerreiro, R., Bras, J., and Bhatia, K. P. (2020). Psychiatric manifestations of ATP13A2 mutations. Mov. Disord. Clin. Pract. 7 (7), 838–841. doi:10.1002/mdc3.13034
Behrens, M. I., Brüggemann, N., Chana, P., Venegas, P., Kägi, M., Parrao, T., et al. (2010). Clinical spectrum of kufor-rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov. Disord. 25, 1929–1937. doi:10.1002/mds.22996
Brüggemann, N., Hagenah, J., Reetz, K., Schmidt, A., Kasten, M., Buchmann, I., et al. (2010). Recessively inherited parkinsonism: effect of ATP13A2 mutations on the clinical and neuroimaging phenotype. Arch. Neurol. 67 (11), 1357–1363. doi:10.1001/archneurol.2010.281
Chen, C. M., Lin, C. H., Juan, H. F., Hu, F. J., Hsiao, Y. C., Chang, H. Y., et al. (2011). ATP13A2 variability in Taiwanese parkinson's disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B (6), 720–729. doi:10.1002/ajmg.b.31214
Crosiers, D., Ceulemans, B., Meeus, B., Nuytemans, K., Pals, P., Van Broeckhoven, C., et al. (2011). Juvenile dystonia-parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Park. Relat. Disord. 17, 135–138. doi:10.1016/j.parkreldis.2010.10.011
De Michele, G., Galatolo, D., Lieto, M., Fico, T., Saccà, F., Santorelli, F. M., et al. (2020). Ataxia-myoclonus syndrome due to a novel homozygous ATP13A2 mutation. Park. Relat. Disord. 76, 42–43. doi:10.1016/j.parkreldis.2020.06.001
Di Fonzo, A., Chien, H. F., Socal, M., Giraudo, S., Tassorelli, C., Iliceto, G., et al. (2007). ATP13A2 missense mutations in juvenile parkinsonism and young onset parkinson disease. Neurology 68 (19), 1557–1562. doi:10.1212/01.wnl.0000260963.08711.08
Djarmati, A., Hagenah, J., Reetz, K., Winkler, S., Behrens, M. I., Pawlack, H., et al. (2009). ATP13A2 variants in early-onset parkinson's disease patients and controls. Mov. Disord. 24 (14), 2104–2111. doi:10.1002/mds.22728
Eiberg, H., Hansen, L., Korbo, L., Nielsen, I. M., Svenstrup, K., Bech, S., et al. (2012). Novel mutation in ATP13A2 widens the spectrum of kufor-rakeb syndrome (PARK9). Clin. Genet. 82 (3), 256–263. doi:10.1111/j.1399-0004.2011.01745.x
Estrada-Cuzcano, A., Martin, S., Chamova, T., Synofzik, M., Timmann, D., Holemans, T., et al. (2017). Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 140 (2), 287–305. doi:10.1093/brain/aww307
Fong, C. Y., Rolfs, A., Schwarzbraun, T., Klein, C., and O'Callaghan, F. J. (2011). Juvenile parkinsonism associated with heterozygous frameshift ATP13A2 gene mutation. Eur. J. Paediatr. Neurol. 15 (3), 271–275. doi:10.1016/j.ejpn.2011.01.001
Garcia-Cazorla, A., and Duarte, S. T. (2014). Parkinsonism and inborn errors of metabolism. J. Inherit. Metab. Dis. 37 (4), 627–642. doi:10.1007/s10545-014-9723-6
Gowda, V. K., Srinivasan, V. M., and Shivappa, S. K. (2020). Kufor-rakeb syndrome/parkinson disease type 9. Indian J. Pediatr. 87 (3), 231–232. doi:10.1007/s12098-019-03167-0
Gurram, S., Holla, V. V., Kumari, R., Dhar, D., Kamble, N., Yadav, R., et al. (2023). Dystonic opisthotonus in kufor-rakeb syndrome: expanding the phenotypic and genotypic spectrum. J. Mov. Disord. 16 (3), 343–346. doi:10.14802/jmd.23098
Inzelberg, R., Estrada-Cuzcano, A., Laitman, Y., De Vriendt, E., Friedman, E., and Jordanova, A. (2018). Kufor-rakeb Syndrome/PARK9: one novel and one possible recurring Ashkenazi ATP13A2 mutation. J. Park. Dis. 8 (3), 399–403. doi:10.3233/JPD-181360
Jain, R. S., Khan, I., and Sisodiya, M. (2016). Kufor–rakeb syndrome (KRS): clinico-radiological phenotype of a probable sporadic case with “eye of tiger” sign. Indian J. Med. Specialities 7, 127–129. doi:10.1016/j.injms.2016.09.002
Kaul, A., Franzese, K., and Cohen, J. (2016). Poster 298 ataxia and dysarthria in two siblings with kufor rakeb syndrome: a case report. PM R. 8 (9S), S257. doi:10.1016/j.pmrj.2016.07.470
Kırımtay, K., Temizci, B., Gültekin, M., Yapıcı, Z., and Karabay, A. (2021). Novel mutations in ATP13A2 associated with mixed neurological presentations and iron toxicity due to nonsense-mediated decay. Brain Res. 1750, 147167. doi:10.1016/j.brainres.2020.147167
Kola, S., Meka, S. S. L., Syed, T. F., Kandadai, R. M., Alugolu, R., and Borgohain, R. (2022). Kufor rakeb syndrome with novel mutation and the role of deep brain stimulation. Mov. Disord. Clin. Pract. 9 (7), 1003–1007. doi:10.1002/mdc3.13518
Lange, L. M., Gonzalez-Latapi, P., Rajalingam, R., Tijssen, M. A. J., and Ebrahimi-Fakhari, D. (2022). On behalf of the Task Force on Genetic Nomenclature in Movement Disorders. Nomenclature of genetic movement disorders: recommendations of the international parkinson and movement disorder society task force - an update. Mov. Disord. 37 (5), 905–935. doi:10.1002/mds.28982
Lin, C. H., Tan, E. K., Chen, M. L., Tan, L. C., Lim, H. Q., Chen, G. S., et al. (2008). Novel ATP13A2 variant associated with parkinson disease in Taiwan and Singapore. Neurology 71 (21), 1727–1732. doi:10.1212/01.wnl.0000335167.72412.68
Malakouti-Nejad, M., Shahidi, G. A., Rohani, M., Shojaee, S. M., Hashemi, M., Klotzle, B., et al. (2014). Identification of p.Gln858* in ATP13A2 in two EOPD patients and presentation of their clinical features. Neurosci. Lett. 577, 106–111. doi:10.1016/j.neulet.2014.06.023
Manrique, L., Sánchez-Rodríguez, A., Pelayo-Negro, A. L., Corral-Juan, M., Matilla-Dueñas, A., and Infante, J. (2021). Ataxia and action myoclonus related to novel mutations in ATP13A2 gene. Mov. Disord. Clin. Pract. 8 (6), 969–971. doi:10.1002/mdc3.13260
Martin, S., Holemans, T., and Vangheluwe, P. (2015). Unlocking ATP13A2/PARK9 activity. Cell Cycle 14 (21), 3341–3342. doi:10.1080/15384101.2015.1093420
Martino, D., Melzi, V., Franco, G., Kandasamy, N., Monfrini, E., and Di Fonzo, A. (2015). Juvenile dystonia-parkinsonism syndrome caused by a novel p.S941Tfs1X ATP13A2 (PARK9) mutation. Park. Relat. Disord. 21 (11), 1378–1380. doi:10.1016/j.parkreldis.2015.09.036
McNeil-Gauthier, A. L., Brais, B., Rouleau, G., Anoja, N., and Ducharme, S. (2019). Successful treatment of psychosis in a patient with kufor-rakeb syndrome with low dose aripiprazole: a case report. Neurocase 25 (3-4), 133–137. doi:10.1080/13554794.2019.1625928
Najim al-Din, A. S., Wriekat, A., Mubaidin, A., Dasouki, M., and Hiari, M. (1994). Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: kufor-rakeb syndrome. Acta Neurol. Scand. 89 (5), 347–352. doi:10.1111/j.1600-0404.1994.tb02645.x
Niemann, N., and Jankovic, J. (2019). Juvenile parkinsonism: differential diagnosis, genetics, and treatment. Park. Relat. Disord. 67, 74–89. doi:10.1016/j.parkreldis.2019.06.025
Noch, E., Henchcliffe, C., Hellmers, N., Chu, M. L., Pappas, J., Moran, E., et al. (2017). Kufor-rakeb syndrome due to a novel ATP13A2 mutation in 2 Chinese-American brothers. Mov. Disord. Clin. Pract. 5 (1), 92–95. doi:10.1002/mdc3.12567
Park, J. S., Mehta, P., Cooper, A. A., Veivers, D., Heimbach, A., Stiller, B., et al. (2011). Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing kufor-rakeb syndrome, a form of early-onset parkinsonism. Hum. Mutat. 32 (8), 956–964. doi:10.1002/humu.21527
Paviour, D. C., Surtees, R. A. H., and Lees, A. J. (2004). Diagnostic considerations in juvenile parkinsonism. Mov. Disord. 19, 123–135. doi:10.1002/mds.10644
Pietrzak, A., Badura-Stronka, M., Kangas-Kontio, T., Felczak, P., Kozubski, W., Latos-Bielenska, A., et al. (2019). Clinical and ultrastructural findings in an ataxic variant of kufor-rakeb syndrome. Folia Neuropathol. 57 (3), 285–294. doi:10.5114/fn.2019.88459
Prashanth, L. K., Murugan, S., Kamath, V., Gupta, R., Jadav, R., Sreekantaswamy, S., et al. (2015). First report of kufor-rakeb syndrome (PARK 9) from India, and a novel nonsense mutation in ATP13A2 gene. Mov. Disord. Clin. Pract. 2 (3), 326–327. doi:10.1002/mdc3.12175
Ramirez, A., Heimbach, A., Gründemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi:10.1038/ng1884
Rohani, M., Lang, A. E., Sina, F., Elahi, E., Fasano, A., Hardy, J., et al. (2017). Action myoclonus and seizure in kufor-rakeb syndrome. Mov. Disord. Clin. Pract. 5 (2), 195–199. doi:10.1002/mdc3.12570
Santoro, L., Breedveld, G. J., Manganelli, F., Iodice, R., Pisciotta, C., Nolano, M., et al. (2011). Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics 12 (1), 33–39. doi:10.1007/s10048-010-0259-0
Satolli, S., Di Fonzo, A., Zanobio, M., Pezzullo, G., and De Micco, R. (2023). Kufor rakeb syndrome without gaze palsy and pyramidal signs due to novel ATP13A2 mutations. Neurol. Sci. 44 (10), 3723–3725. doi:10.1007/s10072-023-06899-2
Schneider, S. A., Paisan-Ruiz, C., Quinn, N. P., Lees, A. J., Houlden, H., Hardy, J., et al. (2010). ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov. Disord. 25 (8), 979–984. doi:10.1002/mds.22947
Schrag, A., and Schott, J. M. (2006). Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet. Neurol. 5, 355–363. doi:10.1016/S1474-4422(06)70411-2
Sørensen, D. M., Holemans, T., Van Veen, S., Martin, S., Arslan, T., Haagendahl, I. W., et al. (2018). Parkinson disease related ATP13A2 evolved early in animal evolution. PLoS One 13 (3), e0193228. doi:10.1371/journal.pone.0193228
Van Veen, S., Sørensen, D. M., Holemans, T., Holen, H. W., Palmgren, M. G., and Vangheluwe, P. (2014). Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front. Mol. Neurosci. 7, 48. doi:10.3389/fnmol.2014.00048
Wang, D., Gao, H., Li, Y., Jiang, S., and Yang, X. (2020). ATP13A2 gene variants in patients with Parkinson’s disease in Xinjiang. Biomed. Res. Int. 2020, 6954820. doi:10.1155/2020/6954820
Williams, D. R., Hadeed, A., al-Din, A. S., Wreikat, A. L., and Lees, A. J. (2005). Kufor rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov. Disord. 20 (10), 1264–1271. doi:10.1002/mds.20511
Yahya, V., Di Fonzo, A., and Monfrini, E. (2023). Genetic evidence for endolysosomal dysfunction in parkinson’s disease: a critical overview. Int. J. Mol. Sci. 24 (7), 6338. doi:10.3390/ijms24076338
Keywords: ATP13A2, WES, juvenile onset parkinsonism, kufor rakeb syndrome, KRS cases revision
Citation: Affronte L, Pini A, Pizzoli C, Coccia E, Mazzone S, Golemi A, Giannotta M, Cordelli DM, Carelli V, Vaisfeld A and Palombo F (2025) Case Report: Novel ATP13A2 pathogenic variants associated with early-onset parkinsonism and a mini-review. Front. Genet. 16:1588812. doi: 10.3389/fgene.2025.1588812
Received: 06 March 2025; Accepted: 10 July 2025;
Published: 29 July 2025.
Edited by:
Hong-Fu Li, Zhejiang University, ChinaReviewed by:
Jorge Diogo Da Silva, University of Minho, PortugalMartje Gesine Pauly, Luebeck University of Applied Sciences, Germany
Copyright © 2025 Affronte, Pini, Pizzoli, Coccia, Mazzone, Golemi, Giannotta, Cordelli, Carelli, Vaisfeld and Palombo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonella Pini, YW50b25lbGxhLnBpbmlAaXNuYi5pdA==