 Hongwei Li1†
Hongwei Li1† Liming Zhao
Liming Zhao- 1Department of Cardiology, Hospital of Chengdu Office of People’s Government of Xizang Autonomous Region (Hospital.C.X.), Chengdu, China
- 2School of Medicine, Xizang Minzu University, Xianyang, Shaanxi, China
- 3Biobank, Hospital of Chengdu Office of People’s Government of Xizang Autonomous Region (Hospital.C.X.), Chengdu, China
Objective: Patent foramen ovale (PFO), a prevalent congenital cardiac defect, is linked to clinical conditions such as cryptogenic stroke and migraine. The genetic underpinnings of PFO remain poorly elucidated, particularly in Tibet. This study aimed to identify potential pathogenic mutations in Tibetan PFO patients via whole exome sequencing (WES) to clarify its genetic basis.
Methods: Eighteen Tibetan PFO patients diagnosed by echocardiography were enrolled. Peripheral blood samples underwent WES using Illumina HiSeq platform, followed by bioinformatics analysis to filter rare variants. Pathogenicity was assessed using predictive tools (SIFT, PolyPhen V2, and MutationTaster) and cardiac development-related gene databases (OMIM, HPO, HGMD, and MGI).
Results: In this study, we identified four novel pathogenetic mutations in Tibetan PFO patients, including GABRP rs201584759 (c.421C>T: p. R141C), GJB4 rs200602523 (c.292C>T: p. R98C), RTTN rs199568901 (c.5410G>A: E1804K), and USH2A rs144768593 (c.5608C>T: p. R1870W). Further analysis indicated that GABRP, GJB4, and RTTN were significantly associated with the occurrence of congenital heart disease.
Conclusion: This study first reveals genetic characteristics of Tibetan PFO patients, implicating GABRP, GJB4, RTTN, and USH2A mutations in disrupting cardiac developmental pathways, potentially contributing to the occurrence of PFO. Findings underscore genetic factors regarding PFO prevalence in populations living in high-altitude and provide insights for molecular research and precision medicine.
Introduction
Patent foramen ovale (PFO), a congenital cardiac structural defect resulting from incomplete embryonic closure of the foramen ovale, is pathologically characterized by a persistent interatrial channel between the left and right atria (Cho et al., 2021). This condition affects approximately 25% of the general population, though the majority remain clinically asymptomatic (Sposato et al., 2024). Interestingly, recent studies have reported higher PFO prevalence in specific populations. For instance, Möller et al. (2024) observed a PFO prevalence of 47% among Tibetans and Han Chinese living at high altitude (2,275 m), suggesting potential environmental or genetic influences on PFO occurrence (Möller et al., 2024). Notably, PFO has been implicated in diverse clinical sequelae through paradoxical embolism and right-to-left shunt mechanisms. Epidemiological evidence indicates that 40% of cryptogenic stroke patients exhibit concurrent PFO, underscoring its significant association with paradoxical embolic stroke (Mac Grory et al., 2022; Baik et al., 2022). Furthermore, this anatomical defect demonstrates clinical correlations with migraine and hypoxemia syndromes, and emerging research suggests potential involvement in atrial fibrillation and heart failure pathogenesis (Nguyen et al., 2024; Yan and Li, 2021; Mohan and Litwin, 2023). Regarding genetic mechanisms, genome-wide association studies (GWAS) have identified critical genes involved in angiogenesis and cardiac septation, including TBX5 and GATA4, where loss-of-function mutations may disrupt endocardial cushion remodeling and impair foramen ovale closure (Apostolos et al., 2024). However, substantial heterogeneity persists in current evidence for pathogenic PFO-associated variants, with an incomplete understanding of population-specific penetrance differences and phenotype-genotype correlations (Paolucci et al., 2021). Systematic elucidation of PFO’s genetic architecture holds translational significance, not only for constructing risk stratification models but also for identifying molecular targets for precision interventions (Yeh and DeFaria Yeh, 2021).
The Tibetan population is predominantly distributed across the Tibetan Plateau and adjacent high-altitude regions in China, characterized by unique environmental adaptations and distinct genetic profiles. Studies show a significant link between high altitude and higher rates of congenital heart disease (CHD), with incidence rising as elevation increases (González-Andrade, 2020). Due to their exposure to relatively isolated environmental stressors and specific physiological traits, cardiovascular health issues in Tibetans—particularly those associated with PFO—remain understudied. Investigating the genetic determinants of cardiovascular diseases in this population holds significant scientific value as it provides critical insights into the interplay between environmental and hereditary factors shaping disease susceptibility within this ethnically distinct group.
Whole exome sequencing (WES), a high-throughput genomic technology, enables comprehensive analysis of protein-coding exonic regions in the genome. Compared to whole genome sequencing (WGS), this method demonstrates superior cost-efficiency for detecting coding sequence variations due to its targeted design, making it particularly valuable for identifying disease-associated genetic alterations (Morton et al., 2022). Consequently, WES has emerged as a critical tool in genetic research. Recent advancements in WES applications have significantly enhanced CHD investigations. This technology has facilitated the identification of monogenic pathogenic variants underlying various CHD subtypes, including hypoplastic left heart syndrome (Gordon et al., 2022), heterotaxy (Bolkier et al., 2022), and patent ductus arteriosus (Gao et al., 2022). Additionally, Li et al. (2024) demonstrated potential cardiovascular risk associations in PFO-related variants (Li et al., 2024). Notably, current genetic findings are predominantly derived from Han Chinese populations. The scarcity of WES data from Tibetan populations in high-altitude regions with elevated CHD prevalence creates substantial knowledge gaps, hindering cross-ethnic investigations into disease mechanisms and PFO-related genetic architecture.
This study employs WES technology to investigate genetic characteristics of PFO in Tibetan populations, focusing on identifying disease-associated mutations. The research seeks to provide novel insights into the genetic mechanisms underlying PFO development while establishing critical references for optimizing cardiovascular health strategies among ethnic Tibetan communities. Through comprehensive genomic analysis, we aim to reveal population-specific molecular markers that could enhance diagnostic precision and inform personalized prevention approaches for this cardiac defect.
Materials and methods
Study participants
This study enrolled 18 Tibetan patients with echocardiographically confirmed PFO from the Second People’s Hospital of Tibet Autonomous Region. Inclusion criteria comprised: 1) Diagnosis of PFO through transesophageal echocardiography (TEE) or contrast-enhanced transcranial Doppler (c-TCD); 2) Exclusion of other CHDs including atrial septal defect (ASD), ventricular septal defect (VSD) and patent ductus arteriosus; 3) No family history of inherited cardiovascular diseases in three-generation of direct relatives. Exclusion criteria were: 1) Comorbid acquired cardiac conditions (such as rheumatic heart disease or infective endocarditis); 2) Pulmonary hypertension (mean pulmonary artery pressure ≥25 mmHg as measured by right heart catheterization); 3) Cardiac structural/functional alterations secondary to systemic disorders (such as connective tissue diseases, thyroid dysfunction, chronic anemia); 4) Receiving catheter-based interventions within the last 3 months. Basic clinical data of the patients were collected from the hospital’s electronic medical records, as shown in Table 1. The research protocol received ethical approval from the Institutional Review Board of the Hospital of Chengdu Office of People’s Government of Xizang Autonomous Region (Hospital.C.X.) (Approval No.: Med-Eth-Re [2022] 77). Written informed consent was obtained from the legal guardians of all participants, and this study adhered to the ethical principles outlined in the Declaration of Helsinki throughout its process.
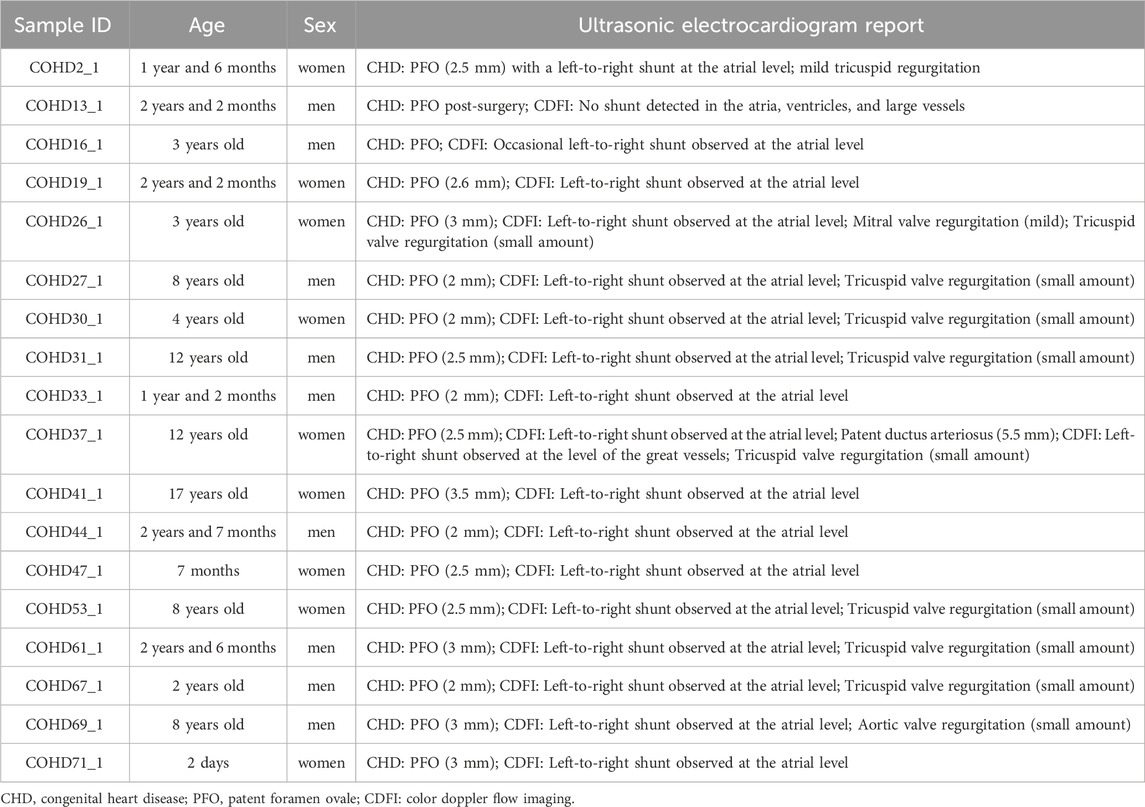
Table 1. Demographic and clinical data of PFO cases.
WES
Genomic DNA was extracted from venous blood samples of PFO patients using the Gentra Puregene Blood Kit (QIAGEN, USA). The DNA quality was further evaluated by agarose gel electrophoresis, with the requirement for a clear main band and no tailing. DNA concentration (≥50 ng/μL, total ≥1.5 μg) and purity (OD260/280 = 1.8–2.0) were measured using a Nanodrop 2000. The DNA library was prepared by randomly fragmenting the DNA into 100–500 bp fragments with Covaris. Following end-repair and A-tailing, Illumina sequencing adapters were ligated to both ends of the library DNA using T4 DNA ligase. The library was then purified and size-selected using the Agencourt SPRLselect kit. Hybridization was performed in the solution phase with SureSelectXT Human All Exon V6 probes. Exon-targeted sequences were captured using streptavidin-coated magnetic beads, followed by PCR amplification. After the library passed quality control, it was further quantified via Qubit, and the insert size was verified by Agilent 2,100. The effective concentration of the library was precisely determined through qPCR (>5 ng/μL). Finally, the library was sequenced on the Illumina Hiseq platform in 2 × 150 bp paired-end mode to generate FastQ data.
Mapping to human genome sequence, variants identification, and annotation
The quality of raw sequencing data was assessed using FastQC (v0.11.9), followed by low-quality read filtering (Phred quality score <20) and adapter removal using Trimmomatic (v0.39). The processed reads were aligned to the human reference genome GRCh37/hg19 using BWA-MEM (v0.7.17), and PCR duplicate reads were marked using Picard (v2.27.5). To further improve alignment accuracy, base quality score recalibration (BQSR) was performed using GATK (v4.2.6.1), and single-sample gVCF files, including single nucleotide variants (SNVs) and insertions/deletions (InDels), were generated using GATK HaplotypeCaller, following the GATK best practices.
Priority classification for SNV/InDel
For all SNV/InDel variants, first apply the following filters: frequency in 1000Genomes, ExAC03 Asian population, and gnomAD Asian population must be below 0.01, GeneskyExonDB_Freq frequency must be below 0.05, and the variants should be non-synonymous or located in exonic regions. Then, classify the filtered variants into priority categories: First1 requires the variant to be either already present in HGMD or meet the following conditions: conserved (predicted as harmful by SIFT, BayesDel addAF), frequency in 1000Genomes below 0.001, frequency in ESP6500 below 0.01, SNV calling quality not equal to L, homogeneity equal to 1, and mutation frequency in Genesky Database below 0.005. First2 requires the variant to meet the following conditions: frequency in 1000Genomes below 0.001, frequency in ESP6500 below 0.01, SNV calling quality not equal to L, homogeneity equal to 1, and mutation frequency in Genesky Database below 0.005. Second is assigned to variants that have SNV calling quality not equal to L, frequency in 1000Genomes below 0.01, and homogeneity less than 3. Variants that do not meet any of the above conditions are classified as Third.
Identification of pathogenic genes and variants
This study selects candidate pathogenic loci and genes based on the following criteria: First, priority is given to the First1 loci. Next, high-quality variants with an SNP Calling quality of “H” are selected. For functional prediction, loci predicted as damaging (D) by SIFT, PolyPhen V2, and MutationTaster are considered, with the additional requirement that the VEST Score is ≥0.5 to ensure the potential impact of the mutation on gene function. Finally, based on annotation data from databases such as OMIM, HPO, HGMD, and MGI, as well as functional annotations from GO and KEGG, further screening is carried out to identify candidate genes associated with diseases. We can efficiently identify potential pathogenic variants and related genes through this comprehensive filtering approach.
Results
Study patients
The basic characteristics of the participants are shown in Table 1. This study comprised 18 children with PFO, with balanced sex distribution (9 males/9 females) and ages spanning 2 days to 17 years. Echocardiographic measurements revealed PFO measuring 2.0–3.5 mm, with 94.4% (17/18) demonstrating persistent atrial-level left-to-right flow. Valvular dysfunction affected 72.2% (13/18) of cases, predominantly characterized by trivial/mild tricuspid regurgitation (61.1%, 11/18), with isolated mitral and aortic valve involvement observed in one case each.
Comprehensive analysis of WES data
WES was performed on 18 PFO samples using the Illumina HiSeq platform, generating a total of 731,487,944 sequencing reads. The sequencing read lengths ranged from 15 to 151 bp. All samples exhibited high-quality clean reads with optimal Q20 and Q30 scores. The average proportion of bases with Phred quality score of ≥30 (Q30) in the raw data exceeded 97%, confirming the superior sequencing quality suitable for downstream bioinformatics analyses. Detailed statistical outcomes of the exome sequencing data are provided in Supplementary Table S1.
Alignment quality assessment of preprocessed reads from 18 samples was performed using the Picard toolkit (https://broadinstitute.github.io/picard/), with comprehensive metrics presented in Supplementary Table S2. The cohort demonstrated an average of 95,851,924.82 reads participating in genomic alignment, of which 60,767,983 reads (99.5% efficiency) were successfully mapped to the reference genome. Exome coverage analysis revealed that 98.3% of targeted regions achieved ≥10× depth, and 84.8% attained ≥30× depth on average. Following established guidelines, single nucleotide variants detected at positions with ≥10× coverage were considered reliably called.
Using the GATK HaplotypeCaller pipeline, we identified 350,398 genetic variants comprising 291,532 single nucleotide variants (SNVs, 83.2%) and 58,866 insertion-deletion variants (InDels, 16.8%). Genomic region-based classification revealed distinct distribution patterns of SNVs/InDels across coding regions, regulatory elements, and intergenic regions, as illustrated in Figure 1. Functional annotation analysis further categorized these variants into missense, synonymous, and other functional types, with their respective proportions detailed in Figure 2. Systematic genotypic classification (homozygous/heterozygous) of both SNV and InDel loci was performed, with comprehensive quantitative data archived in Supplementary Table S3.
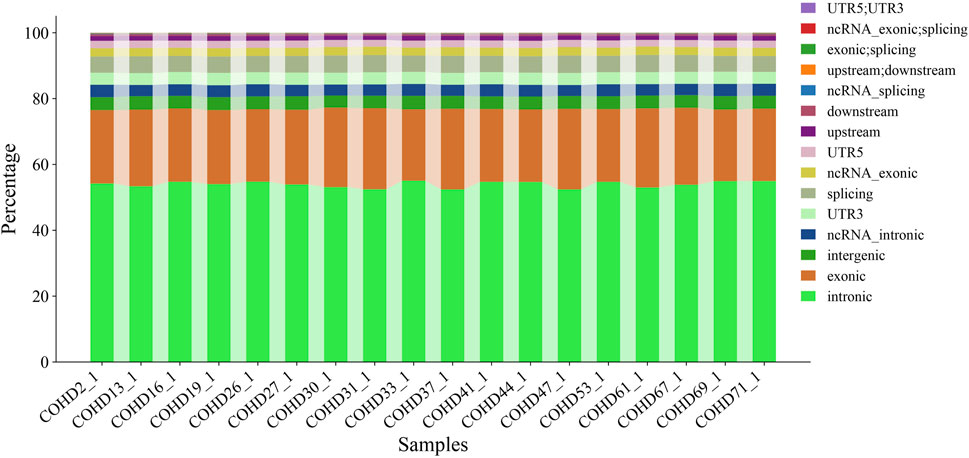
Figure 1. Variant distribution in genomic regions.
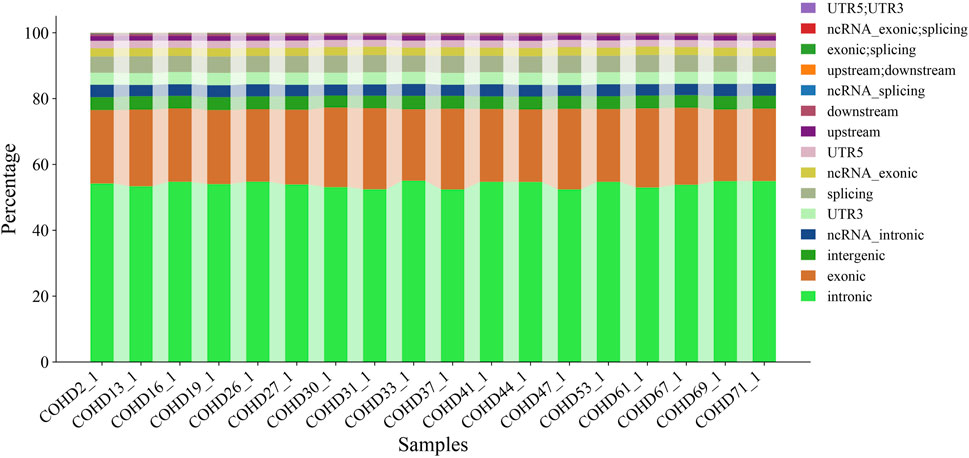
Figure 2. Distribution of functional types for all variants.
A multi-stage filtering pipeline was systematically applied to analyze detected SNVs/InDels. Initial prioritization identified 4,121 variants classified as “First 1″tier in the exome data of PFO patients. Subsequent filtering employed stringent population frequency thresholds: 1,000 Genomes <0.001, 1,000 Genome-EAS <0.01, ExAC03 < 0.01, gnomAD exome <0.01, and ESP6500 < 0.01, with concurrent requirement of high-quality SNP calling (grade H). This refined selection retained 28 rare variants. Pathogenicity prediction was performed using composite algorithms, including SIFT, PolyPhen-2, MutationTaster, and VEST3 (≥0.5 threshold) to identify deleterious mutations. Four exonic nonsynonymous SNVs were ultimately validated: GABRP (c.421C>T: p. R141C), GJB4 (c.292C>T: p. R98C), RTTN (c.5410G>A: E1804K), and USH2A (c.5608C>T: p. R1870W). As illustrated in Figure 3, PolyPhen-2 predictions yielded pathogenic probabilities of 1.000 (GABRP), 0.996 (GJB4), 0.943 (RTTN), and 0.880 (USH2A), respectively. Besides, GJB4 and USH2A were identified as pathogenic mutations through the HGMD database. The results of these four genes are presented in Table 2.
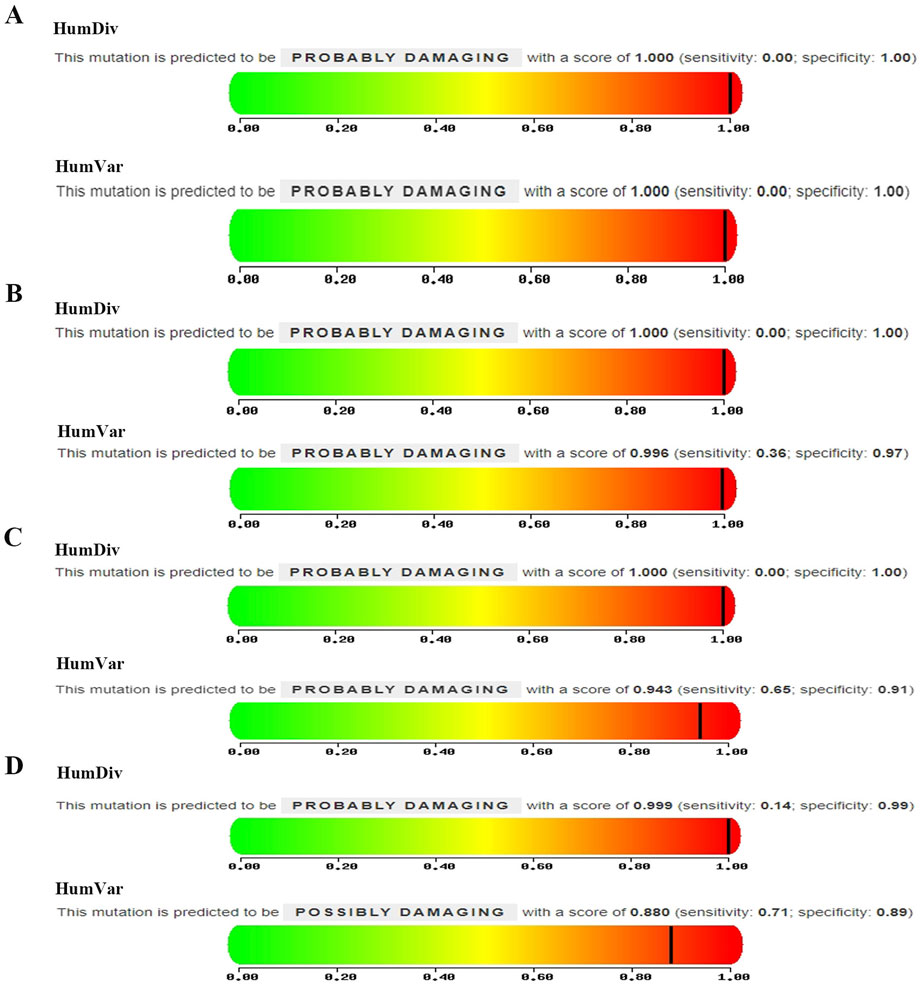
Figure 3. Mutation predictive of (A) GABRP (c.421C>T: p. R141C), (B) GJB4 (c.292C>T: p. R98C), (C) RTTN (c.5410G>A: E1804K), and (D) USH2A (c.5608C>T: p. R1870W).
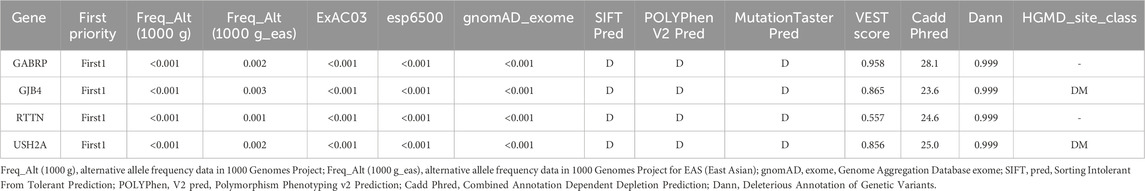
Table 2. Prediction of functional impairment impact on proteins resulting from four potential pathogenic genetic variants.
Table 3 shows detailed information of four mutation sites. These mutations are located in exonic regions and belong to non-synonymous SNVs, including rs201584759 in GABRP, rs200602523 in GJB4, rs199568901 in RTTN, and rs144768593 in USH2A. Additionally, functional annotation analysis indicated that GABRP was significantly associated with congenital heart disease and developmental disorder, while GJB4 was involved in cardiomyopathy. RTTN was related to pulmonary stenosis, whereas USH2A was associated with amyotrophic lateral sclerosis (Table 4).
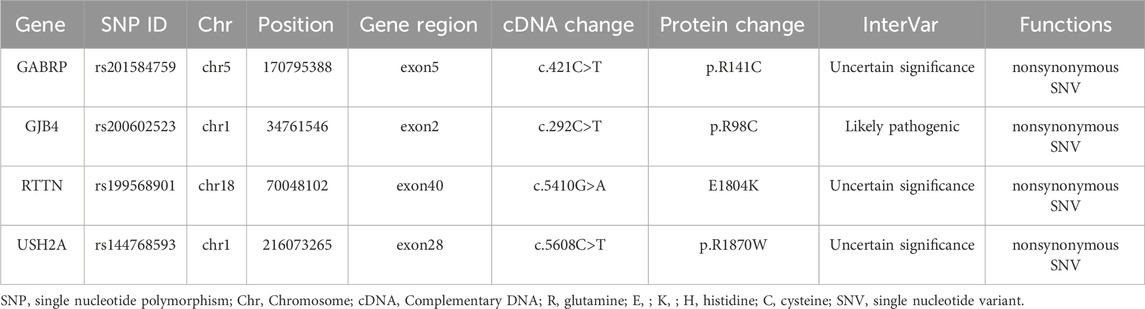
Table 3. Genetic data of predicted genes.
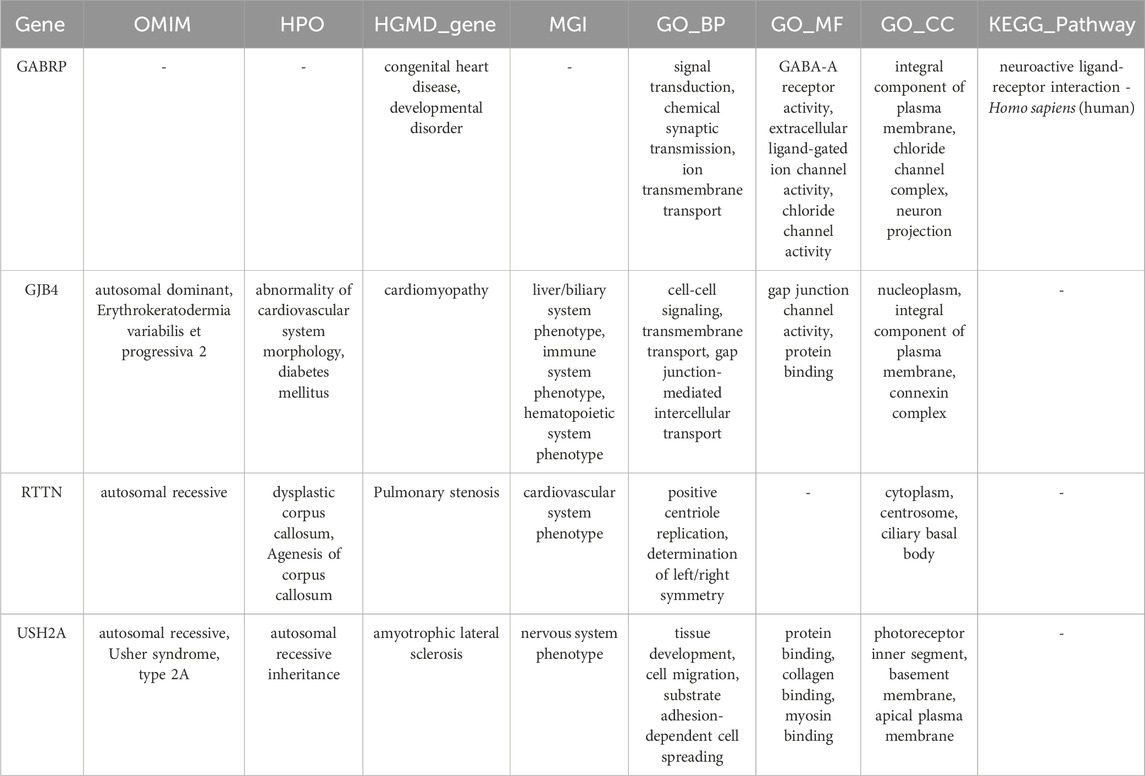
Table 4. Function analysis for the candidate genes.
Discussion
PFO, a prevalent congenital heart defect, remains incompletely understood in its pathogenic mechanisms. Previous studies have shown that genetic factors likely play a significant role in PFO development (Moradi et al., 2023). Utilizing whole exome sequencing, we identified four novel pathogenic variants associated with PFO in the Tibetan population: rs201584759 (c.421C>T: p. R141C) in GABRP, rs200602523 (c.292C>T: p. R98C) in GJB4, rs199568901 (c.5410G>A: E1804K) in RTTN, and rs144768593 (c.5608C>T: p. R1870W) in USH2A. Functional investigations further elucidated the potential roles of GABRP, GJB4, and RTTN in CHD. These findings not only advance the understanding of genetic contributors to PFO in Tibetan populations but also illuminate molecular regulatory networks underlying cardiac developmental abnormalities in high-altitude hypoxic environments.
GABRP, a member of the neurotransmitter receptor family, has functions that extend beyond the traditional GABAergic signaling framework. Research reveals that GABRP orchestrates tumor microenvironment remodeling in pancreatic cancer by modulating KCNN4-dependent calcium ion flux, a mechanism independent of GABA transmission (Jiang et al., 2019). Similarly, GABRP sustains the self-renewal capacity of triple-negative breast cancer stem cells via EGFR pathway activation (Li et al., 2021), while also governing airway epithelial progenitor differentiation by regulating goblet cell formation, thereby maintaining tissue homeostasis (Wang et al., 2021). These findings underscore its pleiotropic regulatory roles in cellular signaling and developmental processes. Given its pivotal position in critical pathways, genetic alterations in GABRP may trigger diverse pathological consequences. For instance, GABRP polymorphisms correlate with susceptibility to systemic lupus erythematosus (Kim et al., 2015), and specific mutations potentially exacerbate immune checkpoint inhibitor-induced hepatotoxicity (Fontana et al., 2024). Notably, our study identified GABRP rs201584759 (c.421C>T: p. R141C) as a pathogenic variant in Tibetan patients with PFO, with functional analyses implicating its involvement in CHD. This evidence supports the hypothesis that GABRP mutations may critically contribute to PFO pathogenesis, though further experimental validation remains essential. Clinically, understanding the role of GABRP in calcium signaling pathways could provide insights into potential dysregulations in cardiac development relevant to PFO and point to pathways that might influence susceptibility to PFO-related complications like arrhythmias in the future.
The GJB4 gene encodes a connexin protein that regulates intercellular electrophysiological signaling by forming gap junctions, which are essential for maintaining cellular coordination and communication, particularly in tissues such as the skin and heart (Lucaciu et al., 2023; Dai et al., 2020). Research indicates that mutations in GJB4 are closely associated with various diseases, notably cutaneous and cardiovascular disorders. For instance, GJB4 mutations are linked to erythrokeratodermia variabilis et progressiva (EKVP), a skin condition characterized by impaired trafficking of connexins, leading to defective gap junction assembly (Zhang et al., 2022). Furthermore, GJB4 plays a critical role in cardiac function, where mutations or deficiencies may disrupt electrical signaling, contributing to arrhythmias and congenital heart defects (Okamoto et al., 2020). Our study has identified the GJB4 rs200602523 variant (c.292C>T: p. R98C) as a pathogenic mutation associated with PFO and cardiomyopathy, highlighting its systemic impact on cardiovascular health. The association of this specific variant with cardiomyopathy in our study underscores a direct clinical link between GJB4 mutations and significant cardiac phenotypes beyond PFO, suggesting that carriers of this variant, particularly in the Tibetan population, may warrant closer cardiac monitoring for potential functional impairment.
RTTN plays a critical role in cellular division, microtubule organization, and neurological development. It encodes a protein involved in essential biological processes such as spindle formation and stabilization, as well as cell cycle regulation (Kheradmand Kia et al., 2012). Mutations in RTTN disrupt these mechanisms, leading to diverse developmental disorders, particularly affecting the nervous and cardiovascular systems. For instance, RTTN variants are associated with primary microcephaly and primordial dwarfism, often manifesting as intellectual disability and developmental delays (Shamseldin et al., 2015). Additionally, RTTN is vital for cerebral cortex formation, with mutations causing structural abnormalities in cortical layering (Guguin et al., 2024). Beyond neurodevelopmental impacts, RTTN mutations may also impair cardiac and brain morphogenesis (Cavallin et al., 2018). Notably, studies link RTTN mutations to infantile dilated cardiomyopathy (IDC), where defective myocardial cell proliferation and differentiation result in cardiac dysfunction (Chun et al., 2023). Importantly, our investigation identified the RTTN rs199568901 variant (c.5410G>A: E1804K) as a pathogenic mutation in PFO. Those findings suggest that RTTN mutations may play a critical role in PFO pathogenesis by disrupting developmental pathways in cardiac septation. The established link between RTTN mutations and severe cardiomyopathies like IDC highlights the potential for RTTN variants, such as the one identified here, to confer risk for broader cardiac developmental issues or functional deficits, emphasizing the importance of comprehensive cardiac evaluation in individuals carrying such mutations.
USH2A encodes a protein critical for maintaining cellular structure and signaling, particularly in the auditory, visual, and cardiovascular systems. By stabilizing intercellular junctions, this protein supports retinal and inner ear development, with mutations linked to Usher syndrome (hearing and vision loss) and keratoconus (Ahmed et al., 2021; Yeo et al., 2025). Recent studies also associate USH2A mutations with congenital heart defects. For instance, rare variants (c.2299delG) were identified in fetuses with ventricular septal defects, suggesting disrupted cardiac cell adhesion or migration during development (Cao et al., 2022). Notably, our research identified pathogenic USH2A mutation (rs144768593, c.5608C>T: p. R1870W) patients with PFO, underscoring its broader role in cardiovascular anomalies. These findings emphasize the pleiotropic effects of USH2A mutations, though their specific mechanisms in cardiac disorders require further validation. The association of USH2A mutations with both structural heart defects (like VSD and now PFO) and sensory disorders (Usher syndrome, keratoconus) suggests that individuals diagnosed with PFO, especially if accompanied by sensory issues, could potentially benefit from genetic screening for USH2A variants, informing broader health management.
This investigation has several limitations. Primarily, the restricted sample size might diminish the broader applicability of the findings. Importantly, our cohort consisted exclusively of pediatric patients, and we recognize that the natural history of PFO closure dynamics differs significantly cross age groups. Besides, the present study is limited to 18 Tibetan pediatric PFO patients identified incidentally via cardiac murmur or routine physical examination; none exhibited clinical stroke or migraine. Consequently, subgroup or sensitivity analyses based on stroke or migraine phenotypes could not be performed. Validation of the association between the identified mutations and late-onset stroke or migraine will require larger, longitudinally followed adult Tibetan cohorts in future work. Additionally, while functional analyses provided initial insights into the association between identified genes and PFO, precise mechanistic pathways require validation through targeted experimental approaches. Notably, this research marks the inaugural discovery of four pathogenic mutations linked to PFO within the Tibetan population, offering novel perspectives on the genetic architecture underlying this cardiac defect. However, given the exploratory nature of this study and its focus on pediatric cases, it is critical to validate these findings in larger, independent cohorts spanning different age groups (especially adults) to confirm the mutations’ prevalence and their specific association with persistent PFO in the Tibetan population.
Conclusion
This study offers critical genetic evidence for pathogenic mutations associated with PFO in Tibetan populations. The identification of four disease-causing variants (GABRP rs201584759, GJB4 rs200602523, RTTN rs199568901, and USH2A rs144768593) may establish novel targets for early detection and personalized therapeutic strategies. Subsequent investigations should focus on elucidating the precise biological effects of these genetic alterations on cardiac morphogenesis and their underlying molecular pathways.
Data availability statement
The original contributions presented in the study are publicly available in the Figshare repository. This data can be found here: https://doi.org/10.6084/m9.figshare.29858897.v1.
Ethics statement
This study was approved by the Ethics Committee of the Hospital of Chengdu Office of People’s Government of Xizang Autonomous Region (Hospital.C.X.). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
HL: Conceptualization, Visualization, Writing – original draft. YH: Resources, Writing – original draft. YW: Data curation, Formal Analysis, Writing – review and editing. LL: Data curation, Formal Analysis, Writing – review and editing. WD: Data curation, Formal Analysis, Writing – review and editing. DW: Data curation, Formal Analysis, Writing – review and editing. ZH: Methodology, Writing – review and editing. LZ: Funding acquisition, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Key Research and Development Program of the Department of Science and Technology of the Tibet Autonomous Region (No. XZ202402ZY0003), the Science and Technology Major Project of Tibetan Autonomous Region of China (No. XZ202201ZD0001G01), and the Key Research and Development Program of the Department of Science and Technology of the Tibet Autonomous Region (No. XZ202403ZY0006).
Acknowledgments
We would like to express our gratitude to the Second People’s Hospital of the Tibet Autonomous Region for providing the blood samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1592306/full#supplementary-material
References
Ahmed, A. N., Tahir, R., Khan, N., Ahmad, M., Dawood, M., Basit, A., et al. (2021). USH2A gene variants cause keratoconus and Usher syndrome phenotypes in Pakistani families. BMC Ophthalmol. 21 (1), 191. doi:10.1186/s12886-021-01957-9
Apostolos, A., Tsiachris, D., Drakopoulou, M., Trantalis, G., Oikonomou, G., Ktenopoulos, N., et al. (2024). Atrial fibrillation after patent foramen ovale closure: incidence, pathophysiology, and management. J. Am. Heart Assoc. 13 (9), e034249. doi:10.1161/JAHA.124.034249
Baik, M., Shim, C. Y., Gwak, S. Y., Kim, Y. D., Nam, H. S., Lee, H. S., et al. (2022). Patent foramen ovale and risk of recurrence in stroke of determined etiology. Ann. neurology 92 (4), 596–606. doi:10.1002/ana.26449
Bolkier, Y., Barel, O., Marek-Yagel, D., Atias-Varon, D., Kagan, M., Vardi, A., et al. (2022). Whole exome sequencing reveals a monogenic cause in 56% of individuals with laterality disorders and associated congenital heart defects. J. Med. Genet. 59 (7), 691–696. doi:10.1136/jmedgenet-2021-107775
Cao, Y., Chau, M. H. K., Zheng, Y., Zhao, Y., Kwan, A. H. W., Hui, S. Y. A., et al. (2022). Exploring the diagnostic utility of genome sequencing for fetal congenital heart defects. Prenat. Diagn. 42 (7), 862–872. doi:10.1002/pd.6151
Cavallin, M., Bery, A., Maillard, C., Salomon, L. J., Bole, C., Reilly, M. L., et al. (2018). Recurrent RTTN mutation leading to severe microcephaly, polymicrogyria and growth restriction. Eur. J. Med. Genet. 61 (12), 755–758. doi:10.1016/j.ejmg.2018.08.001
Cho, K. K., Khanna, S., Lo, P., Cheng, D., and Roy, D. (2021). Persistent pathology of the patent foramen ovale: a review of the literature. Med. J. Aust. 215 (2), 89–93. doi:10.5694/mja2.51141
Chun, Y. W., Miyamoto, M., Williams, C. H., Neitzel, L. R., Silver-Isenstadt, M., Cadar, A. G., et al. (2023). Impaired reorganization of centrosome structure underlies human infantile dilated cardiomyopathy. Circulation 147 (17), 1291–1303. doi:10.1161/CIRCULATIONAHA.122.060985
Dai, S., Wang, H., and Lin, Z. (2020). Novel and recurrent mutations in GJB3 and GJB4 cause erythrokeratodermia variabilis et progressiva. Indian J. dermatology, Venereol. leprology 86 (1), 87–90. doi:10.4103/ijdvl.IJDVL_926_18
Fontana, R. J., Li, Y. J., Chen, V., Kleiner, D., Stolz, A., Odin, J., et al. (2024). Genetic variants associated with immune-mediated liver injury from checkpoint inhibitors. Hepatol. Commun. 8 (9), e0518. doi:10.1097/HC9.0000000000000518
Gao, Y., Wu, D., Chen, B., Chen, Y., Zhang, Q., and Zhao, P. (2022). Rare variants in novel candidate genes associated with nonsyndromic patent ductus arteriosus identified with whole exome sequencing. Front. Genet. 13, 921925. doi:10.3389/fgene.2022.921925
González-Andrade, F. (2020). High altitude as a cause of congenital heart defects: a medical hypothesis rediscovered in Ecuador. High Alt. Med. and Biol. 21 (2), 126–134. doi:10.1089/ham.2019.0110
Gordon, D. M., Cunningham, D., Zender, G., Lawrence, P. J., Penaloza, J. S., Lin, H., et al. (2022). Exome sequencing in multiplex families with left-sided cardiac defects has high yield for disease gene discovery. PLoS Genet. 18 (6), e1010236. doi:10.1371/journal.pgen.1010236
Guguin, J., Chen, T. Y., Cuinat, S., Besson, A., Bertiaux, E., Boutaud, L., et al. (2024). A taybi-linder syndrome-related RTTN variant impedes neural rosette formation in human cortical organoids. PLoS Genet. 20 (12), e1011517. doi:10.1371/journal.pgen.1011517
Jiang, S. H., Zhu, L. L., Zhang, M., Li, R. K., Yang, Q., Yan, J. Y., et al. (2019). GABRP regulates chemokine signalling, macrophage recruitment and tumour progression in pancreatic cancer through tuning KCNN4-mediated Ca(2+) signalling in a GABA-Independent manner. Gut 68 (11), 1994–2006. doi:10.1136/gutjnl-2018-317479
Kheradmand Kia, S., Verbeek, E., Engelen, E., Schot, R., Poot, R. A., de Coo, I. F., et al. (2012). RTTN mutations link primary cilia function to organization of the human cerebral cortex. Am. J. Hum. Genet. 91 (3), 533–540. doi:10.1016/j.ajhg.2012.07.008
Kim, H. S., Jin, E. H., Mo, J. S., Shim, H., Lee, S. S., and Chae, S. C. (2015). The association of the GABRP polymorphisms with systemic lupus erythematosus. J. Immunol. Res. 2015, 602154. doi:10.1155/2015/602154
Li, X., Wang, H., Yang, X., Wang, X., Zhao, L., Zou, L., et al. (2021). GABRP sustains the stemness of triple-negative breast cancer cells through EGFR signaling. Cancer Lett. 514, 90–102. doi:10.1016/j.canlet.2021.04.028
Li, X., Xie, L., Dai, J., Zhou, X., Chen, T., and Mao, W. (2024). A whole exome sequencing study of patent foramen ovale: investigating genetic variants and their association with cardiovascular disorders. Front. Genet. 15, 1405307. doi:10.3389/fgene.2024.1405307
Lucaciu, S. A., Figliuzzi, R., Neumann, R., Nazarali, S., Del Sordo, L., Leighton, S. E., et al. (2023). GJB4 variants linked to skin disease exhibit a trafficking deficiency en route to gap junction formation that can be restored by co-expression of select connexins. Front. cell Dev. Biol. 11, 1073805. doi:10.3389/fcell.2023.1073805
Mac Grory, B., Ohman, E. M., Feng, W., Xian, Y., Yaghi, S., Kamel, H., et al. (2022). Advances in the management of cardioembolic stroke associated with patent foramen ovale. BMJ Clin. Res. ed 376, e063161. doi:10.1136/bmj-2020-063161
Mohan, R. C., and Litwin, S. E. (2023). The intriguing links among patent foramen ovale, patent foramen ovale closure, and the risk for heart failure. J. Am. Soc. Echocardiogr. official Publ. Am. Soc. Echocardiogr. 36 (8), 821–823. doi:10.1016/j.echo.2023.04.012
Möller, F. N., Fan, J. L., Futral, J. E., Hodgman, C. F., Kayser, B., and Lovering, A. T. (2024). Cardiopulmonary haemodynamics in tibetans and han Chinese during rest and exercise. J. physiology 602 (16), 3893–3907. doi:10.1113/jp286303
Moradi, M. M., Kirk, E. P., Patrick, R., Alankarage, D., Humphreys, D. T., Del Monte-Nieto, G., et al. (2023). Quantitative trait and transcriptome analysis of genetic complexity underpinning cardiac interatrial septation in mice using an advanced intercross line. eLife 12, e83606. doi:10.7554/eLife.83606
Morton, S. U., Quiat, D., Seidman, J. G., and Seidman, C. E. (2022). Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 19 (1), 26–42. doi:10.1038/s41569-021-00587-4
Nguyen, A., Nguyen, E., and Kumar, P. (2024). Patent foramen ovale and hypoxemia. Cardiol. Clin. 42 (4), 509–519. doi:10.1016/j.ccl.2024.01.008
Okamoto, R., Goto, I., Nishimura, Y., Kobayashi, I., Hashizume, R., Yoshida, Y., et al. (2020). Gap junction protein beta 4 plays an important role in cardiac function in humans, rodents, and zebrafish. PloS one 15 (10), e0240129. doi:10.1371/journal.pone.0240129
Paolucci, M., Vincenzi, C., Romoli, M., Amico, G., Ceccherini, I., Lattanzi, S., et al. (2021). The genetic landscape of patent foramen ovale: a systematic review. Genes 12 (12), 1953. doi:10.3390/genes12121953
Shamseldin, H., Alazami, A. M., Manning, M., Hashem, A., Caluseiu, O., Tabarki, B., et al. (2015). RTTN mutations cause primary microcephaly and primordial dwarfism in humans. Am. J. Hum. Genet. 97 (6), 862–868. doi:10.1016/j.ajhg.2015.10.012
Sposato, L. A., Albin, C. S. W., Elkind, M. S. V., Kamel, H., and Saver, J. L. (2024). Patent foramen ovale management for secondary stroke prevention: state-Of-The-art appraisal of current evidence. Stroke 55 (1), 236–247. doi:10.1161/STROKEAHA.123.040546
Wang, A., Zhang, Q., Wang, Y., Li, X., Li, K., Li, Y., et al. (2021). Inhibition of gabrp reduces the differentiation of airway epithelial progenitor cells into goblet cells. Exp. Ther. Med. 22 (1), 720. doi:10.3892/etm.2021.10152
Yan, C., and Li, H. (2021). Preliminary investigation of in situ thrombus within patent foramen ovale in patients with and without stroke. Jama 325 (20), 2116–2118. doi:10.1001/jama.2021.4359
Yeh, R. W., and DeFaria Yeh, D. (2021). Patent foramen ovale closure-harnessing clinical trial evidence to inform individualized treatment decisions. Jama 326 (22), 2265–2267. doi:10.1001/jama.2021.21317
Yeo, E. Y. H., Kominami, T., Tan, T. E., Babu, L., Ong, K. G. S., Tan, W., et al. (2025). Phenotypic distinctions between EYS- and USH2A-Associated retinitis pigmentosa in an Asian population. Transl. Vis. Sci. and Technol. 14 (2), 16. doi:10.1167/tvst.14.2.16
Keywords: pathogenetic mutations, whole exome sequencing, patent foramen ovale, Tibetan, genetics
Citation: Li H, He Y, Wu Y, Liu L, Du W, Wang D, He Z and Zhao L (2025) Four novel genetic mutations are associated with patent foramen ovale in Tibetan population using whole exome sequencing. Front. Genet. 16:1592306. doi: 10.3389/fgene.2025.1592306
Received: 12 March 2025; Accepted: 29 July 2025;
Published: 18 August 2025.
Edited by:
Theodoros Karapanayiotides, Aristotle University of Thessaloniki, GreeceReviewed by:
Ioanna Koutroulou, Faculty of Sciences, Aristotle University of Thessaloniki, GreeceVasileios Siokas, University of Thessaly, Greece
Copyright © 2025 Li, He, Wu, Liu, Du, Wang, He and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liming Zhao, ZXJtaW5lMTA0OEAxNjMuY29t
†These authors have contributed equally to this work