 Jelle Vlaeminck1†
Jelle Vlaeminck1† Sophie Uyttebroeck1†
Sophie Uyttebroeck1† Elke De Schutter1
Elke De Schutter1 Ann Cordenier2Shauni Wellekens3Erwin Ströker4Kelly De Rooms1Christine Helsen1Frederik J. Hes1
Ann Cordenier2Shauni Wellekens3Erwin Ströker4Kelly De Rooms1Christine Helsen1Frederik J. Hes1 Philippe Giron1*
Philippe Giron1*- 1Centre for Medical Genetics, Research Group Genetics, Reproduction and Development (GRAD), Clinical Sciences, Universitair Ziekenhuis Brussel (UZ Brussel) - Vrije Universiteit Brussel (VUB), Brussels, Belgium
- 2Department of Neurology, Center for Neurosciences, Universitair Ziekenhuis Brussel (UZ Brussel) - Vrije Universiteit Brussel (VUB), Jette, Belgium
- 3Department of Respiratory Medicine, Universitair Ziekenhuis Brussel (UZ Brussel) - Vrije Universiteit Brussel (VUB), Jette, Belgium
- 4Heart Rhythm Management Centre, Universitair Ziekenhuis Brussel (UZ Brussel) - Vrije Universiteit Brussel (VUB), Jette, Belgium
Desmin-related myofibrillar myopathy is a hereditary disorder caused by pathogenic variants in the DES gene (MIM*125660), altering desmin, a muscle-specific intermediate filament which is crucial for sarcomere integrity. This condition presents with skeletal myopathy, cardiomyopathy, and conduction abnormalities. Genetic counselling for index patients and their family members is complicated by variable expressivity, incomplete penetrance, and de novo occurrence. Mosaicism in asymptomatic parents can obscure inheritance patterns, particularly when low-grade mosaic variants in blood may be missed. In case of DES, mosaic carriership has not been described before. We describe a case of a 24-year-old female diagnosed with desmin-related myopathy due to a heterozygous pathogenic NM_001927.4 (DES):c.1216C>T, p.Arg406Trp variant. Cascade testing using targeted Sanger sequencing of her asymptomatic parents suggested the mother is a mosaic carrier of the pathogenic variant, which was confirmed though next-generation sequencing. The proband’s siblings did not carry the DES c.1216C>T variant. We report the first documented case of mosaic carriership of a pathogenic DES variant in an asymptomatic individual and subsequent inheritance by the offspring, leading to desmin-related myopathy. This report highlights the importance of cascade testing in hereditary disorders with a focus on mosaicism, even when the index’s biological parents are asymptomatic, and de novo emergence is suspected.
1 Introduction
Desmin-related myofibrillar myopathy is a progressive, hereditary disorder, that primarily affects skeletal, cardiac, and respiratory muscles. The condition is caused by pathogenic aberrations in the desmin gene (DES, MIM *125660), which encodes a muscle-specific type III intermediate filament protein. Desmin plays a crucial role in regulating sarcomere architecture, ensuring proper muscle function and structural integrity (Brodehl et al., 2018). Pathogenic variants in DES, which can be inherited or occur de novo, disrupt desmin filament formation, leading to defects in muscle cells, particularly in intercalated discs, which are vital for muscle contraction and communication. This is not due to reduced desmin production, as expression is often not significantly decreased, but rather a dominant negative effect, where the mutated desmin interferes with the normal function of wild type desmin. This disruption affects essential cellular processes, including protein interactions, mitochondrial function, and the filament network (Clemen et al., 2013).
The disease often manifests with progressive muscle weakness and atrophy, initially affecting the proximal skeletal muscles (e.g., shoulders and hips) and later involving distal muscles (e.g., hands and feet). Involvement of bulbar muscles can lead to dysphagia (difficulty swallowing) and dysarthria (difficulty speaking), while respiratory muscles can be impaired, leading to restrictive lung disease and respiratory insufficiency (Goldfarb et al., 2004). Cardiac involvement is common and typically presents with dilated cardiomyopathy or less frequently arrhythmogenic right ventricular cardiomyopathy, leading to heart failure and arrhythmias. These arrhythmias include ventricular arrhythmias and atrial fibrillation, significantly increasing the risk of sudden cardiac death, often at a young age (Brodehl et al.,2018; Wilde et al., 2022).
Desmin-related myopathy follows an autosomal dominant inheritance pattern, although cases with autosomal recessive inheritance patterns have been described (Onore et al., 2022). Furthermore, the disease exhibits incomplete penetrance and variable expressivity, meaning that not all individuals with the mutation will develop symptoms, and that symptom severity varies. The variability in expression is partially explained by the location of the genetic variants within the DES gene (Bär et al., 2007; Goldfarb et al., 2008; Clemen et al., 2013).
In this report, we describe a case of a young woman diagnosed with desmin-related myopathy after genetic testing. Targeted sequencing of the pathogenic DES c.1216C>T variant in peripheral leukocytes of the asymptomatic parents was inconclusive. Next-generation sequencing (NGS) showed mosaic carriership in the mother of the same DES variant, leading to further family cascade testing.
2 Case description
The proband was a 24-year-old female (Figure 1, III-1) with a history of atrioventricular conduction disorder and fasciculoventricular bypass tracts. Prior to this diagnosis in 2016, the proband had no cardiovascular symptoms and maintained a healthy, sportive lifestyle. At the age of 16, she suffered an unprovoked syncope with visual disturbances, heat flashes, dizziness, and loss of consciousness. Upon cardiac evaluation, electrocardiogram showed ventricular pre-excitation, suggesting hypertrophic cardiomyopathy. Transthoracic ultrasound revealed a borderline proximal septal thickness of 9 mm. A bicycle ergometry test was planned but had to be aborted due to vasovagal syncope with bradycardia. During a provocative ajmaline test, the proband suffered a cardiopulmonary arrest, but was resuscitated successfully. A double-chamber pacemaker (DDD pacemaker, Medtronic, United States) was implanted after which the proband had no further cardiac symptoms and attended regular follow-up. Familial anamnesis showed no familial cardiovascular illnesses apart from the maternal grandmother using a beta-blocker for tachycardia (Figure 1). Given the young age of the proband, genetic testing for hereditary cardiovascular diseases was proposed and the proband was referred to a clinical geneticist.
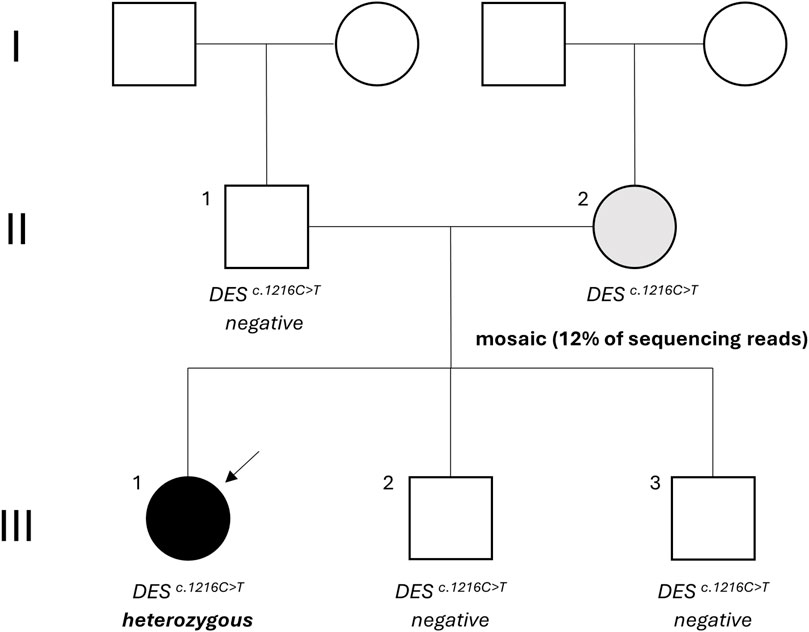
Figure 1. Family tree. The proband is indicated by the arrow and status of the NM_001927.4 (DES):c.1216C>T variant is mentioned. Black: desmin-related myofibrillar myopathy, grey: asymptomatic carrier, white: unaffected. DES: desmin.
3 Diagnostic assessment
3.1 Genetic workup
Following informed consent, leukocyte DNA was extracted from whole peripheral blood and whole exome sequencing (WES) performed on a Novaseq 6000 (Illumina, Inc., United States) platform. An in silico hereditary cardiac disorder gene panel, consisting of 279 genes, was analysed using GRCh37. Three missense variants, one in DES and two in TTN, were identified (Table 1).
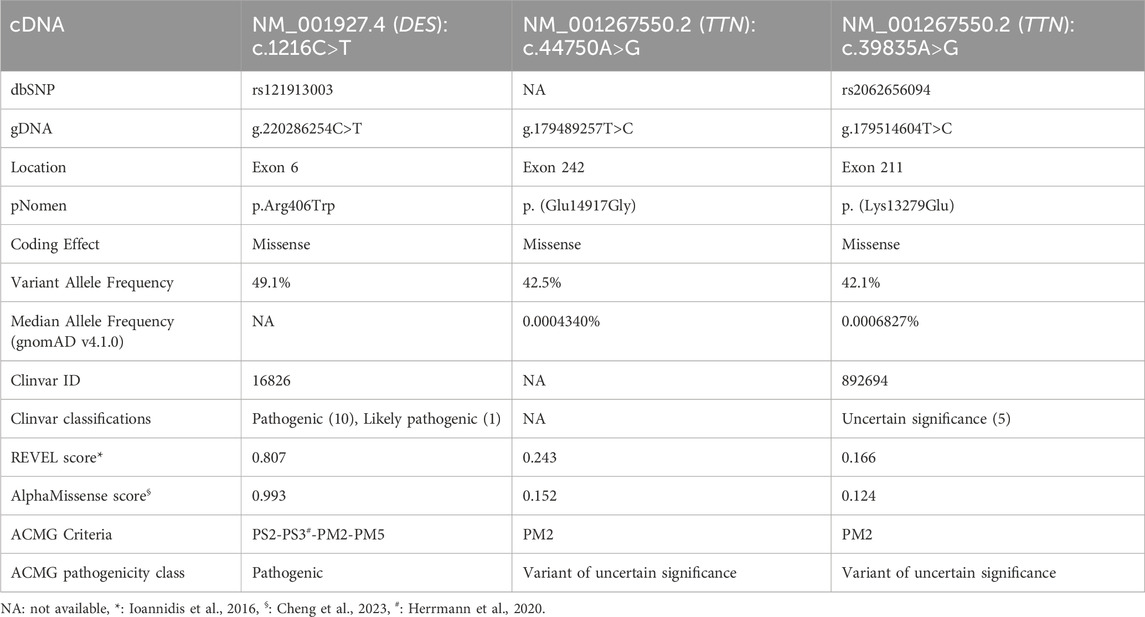
Table 1. Variant features of detected variants in the proband.
The NM_001927.4 (DES):c.1216C>T, p.Arg406Trp variant was described several times with de novo occurrence in patients with severe, early onset cardiomyopathy with or without additional myopathy (Dalakas et al., 2000; Park et al., 2000; Dagvadorj et al., 2004). Additionally, a functional study demonstrated that the Arg406Trp variant resulted in impaired desmin assembly and destabilized filamentous networks in vitro, as shown by immunofluorescence microscopy. Moreover, knock-in mice carrying the Arg406Trp variant developed both myopathy and cardiomyopathy, which were associated with severe intercalated disc derangement. These findings led to the conclusion that DES Arg406Trp is causative for desmin-related myopathy (Herrmann et al., 2020). Given all indications, the variant was classified as “pathogenic” based on the American College of Medical Genetics (ACMG) guidelines (Table 1). The heterozygous state of the c.1216C>T variant was confirmed through Sanger sequencing (Figure 2A), and a diagnosis of desmin-related myopathy was concluded, consistent with the patient’s physical complaints. The two detected TTN variants were both classified as variants of uncertain significance (VUS) (Table 1).
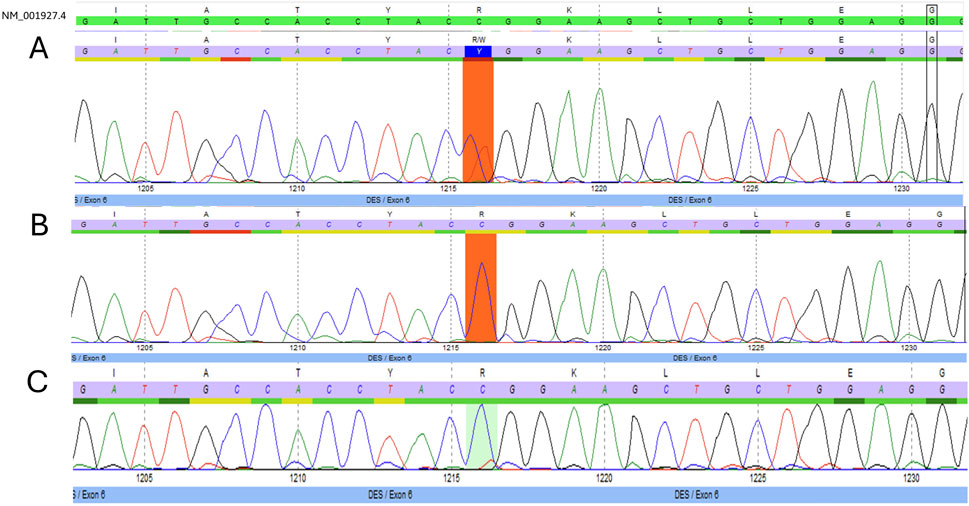
Figure 2. Electropherograms. Targeted Sanger sequencing of exon six of the desmin gene (DES) with position c.1216 marked. (A) Presence of the NM_001927.4 (DES):c.1216C>T variant in heterozygous form in the proband. (B) Absence of the NM_001927.4 (DES):c.1216C>T variant in the proband’s father. (C) Suspected mosaicism of the NM_001927.4 (DES):c.1216C>T variant in the proband’s asymptomatic mother.
Considering the incomplete penetrance and variable expressivity of DES variants, cascade testing was initiated performing targeted Sanger sequencing on the proband’s asymptomatic parents. While the proband’s father (Figure 1, II-1) carried only the wild-type allele (Figure 2B), the mother (Figure 1, II-2) showed a slight but reproducible T signal elevation at c.1216 in the electropherogram (Figure 2C). Suspecting mosaicism, WES on maternal blood DNA (coverage: 332x) confirmed the C>T variant in 12% (41/332) of reads, establishing her mosaic status (Figure 3). To further assess mosaicism in other tissues, we collected two additional buccal-swab samples and performed WES as described above. The c.1216C>T variant was detected at mosaic levels of 4% (4/228 reads) and 15% (30/197 reads) in the two samples, respectively (Supplementary Figure S1).
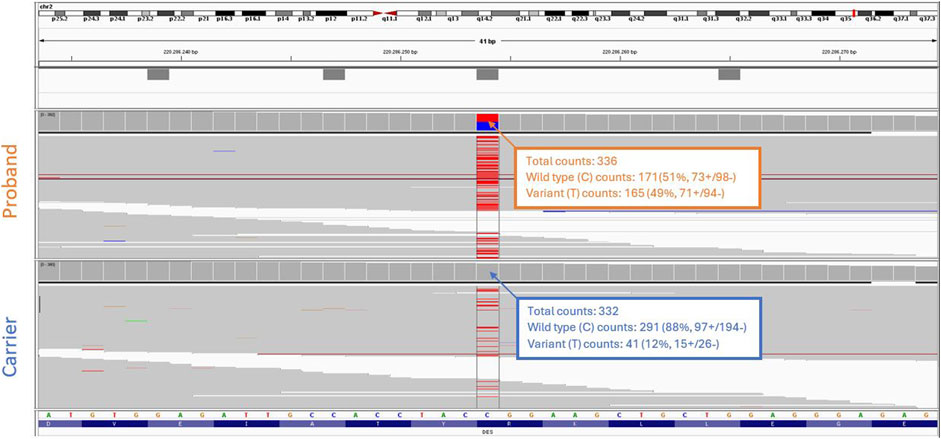
Figure 3. Whole exome sequencing analysis. Integrative genomics viewer (IGV) screenshot shows the NM_001927.4 (DES):c.1216C>T variant present in heterozygous form in the proband (top), and in mosaicism in the blood of the proband’s asymptomatic mother (bottom).
Further cascade testing via targeted Sanger sequencing of the proband’s younger asymptomatic siblings showed that neither had inherited the c.1216C>T variant (Figure 1, III-2 and III-3; Supplementary Figure S2).
3.2 Follow-up
Following diagnosis of desmin-related myopathy, the proband was referred to a neurologist and pneumologist, as neuromuscular and respiratory involvement can be expected in patients with desmin-related myopathy. During the neurological consultation it became apparent that the patient had engaged in regular recreational strength training since the age of 16. However, following cessation of this activity at the age of 21, she began to experience progressive difficulties in daily activities. She reported a slowing of her walking pace, increased fatigue, and a decline in proximal muscle strength of the lower limbs. Additionally, she noted difficulties with articulation and swallowing. Upon neurological examination, the patient demonstrated signs of proximal and distal muscle weakness, most prominently in the lower limbs. She displayed a bilateral steppage gait, and difficulties rising from a seated position. There was noticeable weakness of the facial muscles and the neck flexors. Her speech was dysarthric, with a hyper nasal speech pattern. During a pneumological consultation, the patient reported shortness of breath with minimal exertion, such as climbing two flights of stairs. These symptoms also began following the interruption of her fitness routine at the age of 21. Pulmonary function testing revealed a restrictive pattern with associated respiratory muscle weakness as confirmed by a reduced Maximum Inspiratory Pressure (MIP) and Maximum Expiratory Pressure (MEP). Further evaluation with combined oximetry and capnography demonstrated a nocturnal alveolar hypoventilation for which home non-invasive ventilation was initiated.
The proband was administered to a multidisciplinary neuromuscular reference centre for further treatment. At the time of writing, the proband attends regular cardiologic, neurologic, psychological, pneumological, and physiotherapeutic follow-up for her symptoms, with her current status being stable based on both anamnestic and clinical examination.
Her mother, who is a mosaic carrier, underwent neurophysiological, cardiac, and pulmonary examination. All showed no apparent symptoms or clinical signs indicative of desmin-related myopathy.
4 Discussion
A substantial number of disease-causing variants in DES have been documented, with a current total of 164 variants labelled “(likely) pathogenic” in the Clinvar database and 153 variants labelled “damaging” in the Human Gene Mutation Database (Qiagen, Germany). These include variants leading to premature termination of translation (frameshift and stopgain) as well as splice variants and missense variants. The Arg406Trp variant is a well-known pathogenic variant that has both been described in symptomatic patients and functionally characterized as damaging to the DES protein using in vitro and in vivo models (Dalakas et al., 2000; Park et al., 2000; Dagvadorj et al., 2004; Herrmann et al., 2020).
An important phenomenon complicating genetic diagnostics, not only in desmin-related myopathy but in many genetic conditions, is mosaicism. Mosaicism occurs when a genetic variant is present and/or expressed in only a subset of an organism’s cells (Biesecker and Spinner, 2013), and has been described in several types of myopathy including those related to aberrations in MYH7 (Bader et al., 2022), LMNA (Wang et al., 2023), RYR1 (Estévez-Arias et al., 2024), TPM2 (Tasca et al., 2013), ACTA1 (Miyatake et al., 2014; Lehtokari et al., 2024), and collagen VI-related proteins (Armaroli et al., 2015; Donkervoort et al., 2015). A mosaic carrier may appear asymptomatic or display a milder phenotype, making the diagnosis more challenging. Detecting low-grade mosaic variants through targeted Sanger sequencing of DNA extracted from peripheral blood can be difficult, as these variants may be absent or present at such low levels in the blood cells’ genetic material that they go undetected. Additionally, low-grade mosaic variants can be missed due to the limitation of the performed test (Rohlin et al., 2009).
The diagnosis of mosaic carriers is crucial, as missing such cases has significant implications. When mosaics are overlooked, not only is there a lack of clinical follow-up for the carrier—who may still develop symptoms later in life—but the diagnosis may also be missed for siblings. This can result in incorrect recurrence risk counselling for future offspring and, more critically, the missed opportunity for prenatal testing (Rahbari et al., 2016; Biesecker and Spinner, 2013). If the variant is present in the germ cells, it can be transmitted to offspring, leading to disease in the next-generation. Failing to trace the variant back to an asymptomatic parent may lead to the erroneous conclusion of de novo emergence, preventing necessary actions for the family, such as further genetic testing and family planning (Rahbari et al., 2016). Therefore, accurately diagnosing mosaicism is essential for comprehensive care and genetic counselling.
Additional genetic analyses of buccal-swab DNA also revealed mosaicism for the c.1216C>T variant, indicating that this carrier harbours the variant at varying levels in tissues beyond peripheral blood. However, a limitation of this study is the lack of data on the presence of the c.1216C>T variant in cardiac tissue of the asymptomatic mother. Given the early-onset nature of desmin-related cardiomyopathy, which often manifests in the second or third decade of life (Goldfarb et al., 2004), her lack of clinical signs suggests either a lack of variant expression in cardiac tissue or reduced disease penetrance. A transthoracic ultrasound conducted after her daughter’s initial cardiological event showed no signs of cardiomyopathy. However, incomplete penetrance and variable expressivity could still explain her asymptomatic status. No heart biopsy was performed, as this would neither confirm the diagnosis nor be advisable in an asymptomatic individual (Clemen et al., 2013). Additionally, neurological examination, including electromyography, revealed no signs of myopathy. Given these factors, regular cardiological and neurological follow-up is important for the mother, as she may still develop symptoms at a later age.
Given that the variant was detected in a heterozygous state in the blood of the proband, it indicates that the c.1216C>T variant was present in the germline of the mother, leading to the inheritance. Interestingly, the mosaic variant identified here is a C>T transition in a CG sequence. In somatic APC mosaicism, a higher occurrence of C>T transitions in patient cases with mosaicism compared to non-mosaic cases has been described (Hes et al., 2008).
In addition to the DES c.1216C>T variant, we identified two missense VUS in the TTN gene. While TTN is a known causative gene for myofibrillar myopathy, the missense VUS identified in this case do not meet the criteria for a (likely) pathogenic variant based on current ACMG guidelines. Most known (likely) pathogenic TTN variants constitute a premature termination of translation (frameshift, stopgain) or splicing aberration with only a handful of missense variants. TTN missense variants are common in the general population with over 60.000 variants identified in the 1000 Genomes Project, obscuring their pathogenic potential (Jolfayi et al., 2024). However, we acknowledge that TTN missense variants can contribute to the phenotype in some individuals, either as a primary or secondary factor, as cases have been described (Domínguez et al., 2023). In this case, the role of the TTN missense VUS remains unclear. Strictly speaking they could potentially contribute to the clinical presentation in combination with the pathogenic DES variant, especially given the overlapping features of myofibrillar myopathy and the known variable expressivity of both genes. However, given the monogenic nature of DES related diseases, they could very well be passenger variants. Further functional studies and larger cohort analyses are needed to better understand the clinical relevance of such TTN VUS variants in myofibrillar myopathy (Pfeffer et al., 2014).
Another point highlighted by this case is the importance of cascade testing, even when relatives are asymptomatic, and de novo emergence is expected. In this case, the DES c.1216C>T variant had been mainly described as a de novo event in previous cases (Dalakas et al., 2000; Park et al., 2000; Dagvadorj et al., 2004), however, testing of the asymptomatic parents revealed the mosaic status of the pathogenic variant in the mother. This finding yielded an important indication to also test the younger, at that moment asymptomatic, siblings. One could have assumed neither of the parents carried the pathogenic c.1216C>T variant due to them being asymptomatic at advanced age, while desmin-related myopathy is known to have onset early in life, and therefore not test them, leading to a potential missed inheritance to other siblings and/or future offspring. Nevertheless, the likelihood of recurrence in siblings after a seemingly sporadic mutation varies by gene and relies on empirical data, which is often unavailable. In these cases, a risk estimation of 1%–2% is appropriate, and cascade testing in case of a presumed de novo variant to exclude the possibility of mosaic carriership is strongly recommended (Rahbari et al., 2016).
A prominent issue that needs to be considered hereby is the limitation of Sanger sequencing to detect mosaicism. As it depends on the intensity of the fluorescent signal in the electropherogram, low grade mosaicism could easily be missed (Rohlin et al., 2009). This shortcoming can be overcome by applying NGS techniques, such as WES, as these are able to detect DNA variants at low allele frequencies (Rohlin et al., 2009; Erickson, 2014; Qin et al., 2016). With the continuously lowering cost of NGS, this has become a valid alternative to Sanger sequencing. However, the current tendency to sequence at lower coverage poses challenges to detect mosaic variants. In clinical diagnostics, a coverage of 150x is routinely utilized when performing WES. As WES library preparation usually contains a PCR-based amplification step to enrich the captured exonic sequences, a significant number of duplicate reads are generated during sequencing. Depending on the grade of mosaicism and the depth of sequencing, the relevant variant might therefore be missed. Also, with the ongoing transition from WES to whole genome sequencing (WGS), this will become even more pronounced as WGS coverages are routinely 30–42x. Although WGS omits the need for PCR-amplification during library preparation, sequencing at sufficiently high coverage will still be essential to detect low-grade mosaicism. Furthermore, to confirm mosaicism, it is advisable to perform the analysis on several tissue types such as oral mucosa or skin biopsies. In the case presented here, this was not pursued as the carrier showed no clinical indications of disease. Finally, one other important caveat that must be taken into consideration is that the mosaic variant must be present in the blood when performing routine germline analyses, otherwise only tissue-specific analysis will be able to detect a mosaic carriership.
In conclusion, to our knowledge, this report offers the first documented confirmed case of a pathogenic DES variant harboured in mosaicism in the blood of an asymptomatic individual. Additionally, this report describes the first documented case of desmin-related myopathy caused by inheritance of this mosaic pathogenic DES variant. Furthermore, we stress both the importance of cascade testing of asymptomatic probands, even when de novo occurrence is suspected, and the limitations of targeted Sanger sequencing in detecting low-grade mosaicism.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of this case report including any potentially identifiable images or data.
Author contributions
JV: Conceptualization, Visualization, Investigation, Writing – original draft, Formal Analysis. SU: Formal Analysis, Conceptualization, Writing – original draft, Investigation. ED: Investigation, Writing – review and editing, Conceptualization. AC: Investigation, Writing – review and editing. SW: Investigation, Writing – review and editing. ES: Writing – review and editing, Investigation. KD: Writing – review and editing, Investigation. CH: Writing – review and editing, Investigation. FH: Writing – review and editing, Supervision. PG: Investigation, Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors would like to thank the participants/patients and medical and technical personnel of Universitair Ziekenhuis Brussel (UZ Brussel) involved in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1597851/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Whole exome sequencing analysis of additional tissues of mosaic carrier. Integrative genomics viewer (IGV) screenshot shows the mosaic status of the NM_001927.4(DES):c.1216C>T variant in two mouth mucosa samples of the asymptomatic carrier.
SUPPLEMENTARY FIGURE S2 | Electropherograms of the proband’s siblings. Targeted Sanger sequencing of exon 6 of the desmin gene (DES) with position c.1216 marked. The NM_001927.4(DES):c.1216C>T variant is absent in the proband’s male siblings (A, B).
Abbreviations
ACMG, Americal College of Medical Genetics; MEP, Maximum Expiratory Pressure; MIP, Maximum Inspiratory Pressure; NGS, Next-generation sequencing; VUS, Variant of unknown significance; WES, Whole exome sequencing; WGS, Whole genome sequencing.
References
Armaroli, A., Trabanelli, C., Scotton, C., Venturoli, A., Selvatici, R., Brisca, G., et al. (2015). Paternal germline mosaicism in collagen VI related myopathies. Eur. J. Paediatr. Neurol. 19 (5), 533–536. Epub 2015 Apr 30. PMID: 25978941. doi:10.1016/j.ejpn.2015.04.002
Bader, I., Freilinger, M., Landauer, F., Waldmüller, S., Mueller-Felber, W., Rauscher, C., et al. (2022). A recurrent single-amino acid deletion (p.Glu500del) in the head domain of ß-cardiac myosin in two unrelated boys presenting with polyhydramnios, congenital axial stiffness and skeletal myopathy. Orphanet J. Rare Dis. 17 (1), 279. PMID: 35854315; PMCID: PMC9295345. doi:10.1186/s13023-022-02421-7
Bär, H., Goudeau, B., Wälde, S., Casteras-Simon, M., Mücke, N., Shatunov, A., et al. (2007). Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum. Mutat. 28 (4), 374–386. PMID: 17221859. doi:10.1002/humu.20459
Biesecker, L. G., and Spinner, N. B. (2013). A genomic view of mosaicism and human disease. Nat. Rev. Genet. 14 (5), 307–320. PMID: 23594909. doi:10.1038/nrg3424
Brodehl, A., Gaertner-Rommel, A., and Milting, H. (2018). Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 10 (4), 983–1006. Epub 2018 Jun 20. PMID: 29926427; PMCID: PMC6082305. doi:10.1007/s12551-018-0429-0
Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulytė, A., Applebaum, T., et al. (2023). Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381 (6664), eadg7492. PMID: 37733863. doi:10.1126/science.adg7492
Clemen, C. S., Herrmann, H., Strelkov, S. V., and Schröder, R. (2013). Desminopathies: pathology and mechanisms. Acta Neuropathol. 125 (1), 47–75. Epub 2012 Nov 11. PMID: 23143191; PMCID: PMC3535371. doi:10.1007/s00401-012-1057-6
Dagvadorj, A., Olivé, M., Urtizberea, J. A., Halle, M., Shatunov, A., Bönnemann, C., et al. (2004). A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J. Neurol. 251 (2), 143–149. PMID: 14991347. doi:10.1007/s00415-004-0289-3
Dalakas, M. C., Park, K. Y., Semino-Mora, C., Lee, H. S., Sivakumar, K., and Goldfarb, L. G. (2000). Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 342 (11), 770–780. PMID: 10717012. doi:10.1056/NEJM200003163421104
Domínguez, F., Lalaguna, L., Martínez-Martín, I., Piqueras-Flores, J., Rasmussen, T. B., Zorio, E., et al. (2023). Titin missense variants as a cause of familial dilated cardiomyopathy. Circulation 147 (22), 1711–1713. Epub 2023 May 30. PMID: 37253077. doi:10.1161/CIRCULATIONAHA.122.062833
Donkervoort, S., Hu, Y., Stojkovic, T., Voermans, N. C., Foley, A. R., Leach, M. E., et al. (2015). Mosaicism for dominant collagen 6 mutations as a cause for intrafamilial phenotypic variability. Hum. Mutat. 36 (1), 48–56. PMID: 25204870; PMCID: PMC4601573. doi:10.1002/humu.22691
Erickson, R. P. (2014). Recent advances in the study of somatic mosaicism and diseases other than cancer. Curr. Opin. Genet. Dev. 26, 73–78. Epub 2014 Jul 20. PMID: 25050467. doi:10.1016/j.gde.2014.06.001
Estévez-Arias, B., Matalonga, L., Martorell, L., Codina, A., Ortez, C., Carrera-García, L., et al. (2024). Improving diagnostic precision: phenotype-driven analysis uncovers a maternal mosaicism in an individual with RYR1-congenital myopathy. J. Neuromuscul. Dis. 11 (3), 647–653. PMID: 38489196; PMCID: PMC11091619. doi:10.3233/JND-230216
Goldfarb, L. G., Olivé, M., Vicart, P., and Goebel, H. H. (2008). Intermediate filament diseases: desminopathy. Adv. Exp. Med. Biol. 642, 131–164. PMID: 19181099; PMCID: PMC2776705. doi:10.1007/978-0-387-84847-1_11
Goldfarb, L. G., Vicart, P., Goebel, H. H., and Dalakas, M. C. (2004). Desmin myopathy. Brain 127 (Pt 4), 723–734. Epub 2004 Jan 14. PMID: 14724127. doi:10.1093/brain/awh033
Herrmann, H., Cabet, E., Chevalier, N. R., Moosmann, J., Schultheis, D., Haas, J., et al. (2020). Dual functional states of R406W-desmin assembly complexes cause cardiomyopathy with severe intercalated disc derangement in humans and in knock-in mice. Circulation 142 (22), 2155–2171. Epub 2020 Oct 7. PMID: 33023321. doi:10.1161/CIRCULATIONAHA.120.050218
Hes, F. J., Nielsen, M., Bik, E. C., Konvalinka, D., Wijnen, J. T., Bakker, E., et al. (2008). Somatic APC mosaicism: an underestimated cause of polyposis coli. Gut 57 (1), 71–76. Epub 2007 Jun 29. PMID: 17604324. doi:10.1136/gut.2006.117796
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99 (4), 877–885. Epub 2016 Sep 22. PMID: 27666373; PMCID: PMC5065685. doi:10.1016/j.ajhg.2016.08.016
Jolfayi, A. G., Kohansal, E., Ghasemi, S., Naderi, N., Hesami, M., MozafaryBazargany, M., et al. (2024). Exploring TTN variants as genetic insights into cardiomyopathy pathogenesis and potential emerging clues to molecular mechanisms in cardiomyopathies. Sci. Rep. 14 (1), 5313. PMID: 38438525; PMCID: PMC10912352. doi:10.1038/s41598-024-56154-7
Lehtokari, V. L., Sagath, L., Davis, M., Ho, D., Kiiski, K., Kettunen, K., et al. (2024). A recurrent ACTA1 amino acid change in mosaic form causes milder asymmetric myopathy. Neuromuscul. Disord. 34, 32–40. Epub 2023 Nov 30. PMID: 38142473. doi:10.1016/j.nmd.2023.11.009
Miyatake, S., Koshimizu, E., Hayashi, Y. K., Miya, K., Shiina, M., Nakashima, M., et al. (2014). Deep sequencing detects very-low-grade somatic mosaicism in the unaffected mother of siblings with nemaline myopathy. Neuromuscul. Disord. 24 (7), 642–647. Epub 2014 Apr 24. PMID: 24852243. doi:10.1016/j.nmd.2014.04.002
Onore, M. E., Savarese, M., Picillo, E., Passamano, L., Nigro, V., and Politano, L. (2022). Bi-allelic DES gene variants causing autosomal recessive myofibrillar myopathies affecting both skeletal muscles and cardiac function. Int. J. Mol. Sci. 23 (24), 15906. PMID: 36555543; PMCID: PMC9785402. doi:10.3390/ijms232415906
Park, K. Y., Dalakas, M. C., Semino-Mora, C., Lee, H. S., Litvak, S., Takeda, K., et al. (2000). Sporadic cardiac and skeletal myopathy caused by a de novo desmin mutation. Clin. Genet. 57 (6), 423–429. PMID: 10905661. doi:10.1034/j.1399-0004.2000.570604.x
Pfeffer, G., Barresi, R., Wilson, I. J., Hardy, S. A., Griffin, H., Hudson, J., et al. (2014). Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J. Neurol. Neurosurg. Psychiatry 85 (3), 331–338. Epub 2013 Mar 13. PMID: 23486992; PMCID: PMC6558248. doi:10.1136/jnnp-2012-304728
Qin, L., Wang, J., Tian, X., Yu, H., Truong, C., Mitchell, J. J., et al. (2016). Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J. Mol. Diagn 18 (3), 446–453. Epub 2016 Mar 2. PMID: 26944031. doi:10.1016/j.jmoldx.2016.01.002
Rahbari, R., Wuster, A., Lindsay, S., Hardwick, R., Alexandrov, L., Al Turki, S., et al. (2016). Timing, rates and spectra of human germline mutation. Nat. Genet. 48 (2), 126–133. Epub 2015 Dec 14. PMID 26656846. doi:10.1038/ng.3469
Rohlin, A., Wernersson, J., Engwall, Y., Wiklund, L., Björk, J., and Nordling, M. (2009). Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum. Mutat. 30 (6), 1012–1020. PMID: 19347965. doi:10.1002/humu.20980
Tasca, G., Fattori, F., Ricci, E., Monforte, M., Rizzo, V., Mercuri, E., et al. (2013). Somatic mosaicism in TPM2-related myopathy with nemaline rods and cap structures. Acta Neuropathol. 125 (1), 169–171. Epub 2012 Sep 27. PMID: 23015096. doi:10.1007/s00401-012-1049-6
Wang, G., Hou, Y., Lv, X., Yan, C., and Lin, P. (2023). Somatic and germinal mosaicism in a Han Chinese family with laminopathies. Eur. J. Hum. Genet. 31 (9), 1073–1077. Epub 2022 Dec 16. PMID: 36526864; PMCID: PMC10474091. doi:10.1038/s41431-022-01266-9
Wilde, A. A. M., Semsarian, C., Márquez, M. F., Shamloo, A. S., Ackerman, M. J., Ashley, E. A., et al. (2022). European heart rhythm association (EHRA)/Heart rhythm society (HRS)/Asia pacific heart rhythm society (APHRS)/Latin American heart rhythm society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Europace 24 (8), 1307–1367. doi:10.1093/europace/euac030
Keywords: desmin-related myopathy, mosaicism, DES, c.1216C>T, R406W, Arg406Trp, case report
Citation: Vlaeminck J, Uyttebroeck S, De Schutter E, Cordenier A, Wellekens S, Ströker E, De Rooms K, Helsen C, Hes FJ and Giron P (2025) Case Report: A first case of desmin-related myofibrillar myopathy due to inheritance from a confirmed mosaic asymptomatic carrier. Front. Genet. 16:1597851. doi: 10.3389/fgene.2025.1597851
Received: 21 March 2025; Accepted: 04 June 2025;
Published: 18 June 2025.
Edited by:
Marco Savarese, University of Helsinki, FinlandReviewed by:
Minttu Marttila, University of Helsinki, FinlandMaria Francesca Di Feo, University of Genoa, Italy
Copyright © 2025 Vlaeminck, Uyttebroeck, De Schutter, Cordenier, Wellekens, Ströker, De Rooms, Helsen, Hes and Giron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe Giron, UGhpbGlwcGUuR2lyb25AdXpicnVzc2VsLmJl
†These authors have contributed equally to this work and share first authorship