 Qinghong Lin1,2,3,4,5,6†
Qinghong Lin1,2,3,4,5,6† Xuejun Wang1,2,3,4,5†
Xuejun Wang1,2,3,4,5† Xiaoliao Peng1,2,3,4,5†
Xiaoliao Peng1,2,3,4,5† Xiaosong Han1,2,3,4,5
Xiaosong Han1,2,3,4,5 Xiaoyu Zhang1,2,3,4,5
Xiaoyu Zhang1,2,3,4,5 Ling Sun1,2,3,4,5Yan Wang6
Ling Sun1,2,3,4,5Yan Wang6 Shengtao Liu1,2,3,4,5*
Shengtao Liu1,2,3,4,5* Xingtao Zhou1,2,3,4,5*
Xingtao Zhou1,2,3,4,5*- 1Department of Ophthalmology, Eye and ENT Hospital of Fudan University, Shanghai, China
- 2Eye Institute and Department of Ophthalmology, Eye and ENT Hospital, Fudan University, Shanghai, China
- 3NHC Key Laboratory of Myopia (Fudan University), Key Laboratory of Myopia, Chinese Academy of Medical Sciences, Shanghai, China
- 4Shanghai Research Center of Ophthalmology and Optometry, Shanghai, China
- 5Shanghai Engineering Research Center of Laser and Autostereoscopic 3D for Vision Care (20DZ2255000), Shanghai, China
- 6Refractive Surgery Department, Bright Eye Hospital, Fuzhou, China
Background: This study reports a three-generation Chinese family with polymorphous corneal dystrophy subtype 3 (PPCD3) and keratoconus (KC) aggravation induced by corneal refractive surgery, specifically small incision lenticule extraction (SMILE), in the context of genetic variations.
Methods: The history of illnesses and blood samples of all family members were collected. One hundred healthy individuals served as normal controls. We conducted whole exome sequencing on genomic DNA and sanger sequencing to verify the variants between all controls and family members.
Results: Three family members were previously diagnosed with subclinical keratoconus (III1 and III2 preoperatively, and II2). Both the proband (III1) and her younger brother (III2) underwent SMILE to correct refractive errors. One year later, visual acuity of III1 decreased significantly with KC aggravation and corneal opacification. The KC of III2 progressed significantly 6 months after surgery. Both were subsequently diagnosed with PPCD3. We detected both Zinc finger E-box-binding homeobox 1 (ZEB1) gene and zinc finger protein 469 (ZNF469) gene pathogenic variant in the proband and another two patients in this family, including a heterozygous missense variation c.13C>G (p.P5A, rs753301298) in the ZEB1 gene, and a heterozygous non-frameshift variant c. 3093_3104del (p.D1035_K1038del) in the ZNF469 gene. The variants including c.13C>G in ZEB1 and c.3093_3104del in ZNF469 were speculated to be pathogenic or a variant of uncertain significance by online prediction software.
Conclusion: This study demonstrated the importance of a thorough ocular examination, especially the cornea, and a gene screening before SMILE.
1 Introduction
Corneal refractive surgery includes various modalities, including laser-assisted in situ keratomileusis (LASIK), photorefractive keratectomy (PRK), and small-incision lenticule extraction (SMILE). Recent studies have suggested that post-refractive keratoconus (KC) occurs from the lowest to highest rate in eyes that have undergone SMILE, PRK and LASIK, respectively. However, given the increasing number of people who have undergone SMILE, post-SMILE keratoconus is a growing concern. Since keratoconus is a spectrum of disease, pre-existing keratoconus is more important in postoperative ectasia than previously thought (Moshirfar et al., 2021; Zhang et al., 2022).
Keratoconus is a progressive disease characterized by corneal thinning, lack of inflammation, irregular curvature, and scar formation, which can lead to severe visual loss in later stages and is often accompanied with other systemic and/or eye diseases (Bykhovskaya et al., 2016; Loukovitis et al., 2018). Variations of ZEB1 and ZNF469, which are identified in corneal dystrophy, have been suggested to be related to keratoconus (Karolak et al., 2020; Zhang et al., 2021). In addition, environmental factors also play important roles (Crawford et al., 2020).
Posterior polymorphous corneal dystrophy (PPCD) and keratoconus have been co-occurrent in many patients (Burdon and Vincent, 2013; Fernández-Gutiérrez et al., 2022). In this study, a three generation Chinese family with keratoconus and PPCD subtype 3 (PPCD3) was studied. A total of three members in the family were observed to have keratoconus and PPCD3, among which two members (including the proband) had undergone SMILE to correct refractive errors. Keratoconus is a progressive corneal disorder that may be associated with genetic factors, and new pathogenetic variants of ZEB1 and ZNF469 were identified in this study. Additionally, PPCD3 can further complicate the clinical course of KC, especially after surgical interventions. The increasing understanding of the genetic underpinnings of these conditions highlights the importance of early diagnosis and gene screening, particularly for patients who may be at risk of postoperative complications like keratectasia or the progression of KC.
2 Materials and methods
2.1 Participants and examinations
There were 105 participants in this study, including five living family members from a three-generation Chinese family with PPCD3 and KC, and 100 unrelated healthy Chinese individuals, who were not diagnosed with PPCD, KC or other inherited corneal disorders. In the family, two members, including the proband (III1) and her younger brother (III2), underwent SMILE to correct refractive errors. All family members denied allergies, habitual eye rubbing, and trauma.
All participants provided written informed consent, and underwent detailed ophthalmic (i.e., best-corrected visual acuity (BCVA), biomicroscopy, and fundus examination) and physical examinations. In addition, the Scheimpflug camera system (Pentacam; Oculus Optikgeräte GmbH, Wetzlar, Germany) and optical coherence tomography (OCT), (Heidelberg Spectralis Heidelberg Engineering Gmbh, Germany) were used for corneal examination. The corneal endothelium cell density (ECD) was measured by non-contact specular microscopy (SP-2000P, Topcon Corporation, Japan). This study was approved by the Institutional Review Board of Fudan University (Shanghai, China) (approval no. 2022128) and was performed in compliance with the Declaration of Helsinki.
2.2 Whole exome sequencing
Exome sequencing (ES) was performed for 3 participants (III:1, III:2 and II:2) using the method previously described (Lin et al., 2022). Genomic DNA was extracted from leukocytes, and the exonic sequences were enriched, after which data processing and analyses were conducted. The 1000 Genomes Project was used to examine reported variants and those presented in patients with corneal dystrophy or KC at frequencies ≤1%. Only the variants shared in affected family members, namely III:1, III:2 and II:2 were considered as candidate variants.
2.3 Variant validation and analysis
Variant validation and analyses were performed. All variations were analyzed using online software, including Polyphen2 (genetics.bwh.harvard.edu), SIFT (sift.jcvi.org), fathmm-MKL (http://fathmm.biocompute.org.uk), CADD v1.4 (cadd.gs.washington.edu), Variant Taster (varianttaster.org) and ACMG guidelines (American College of Medical Genetics and Genomics). Subsequently, candidate variants were confirmed using polymerase chain reaction (PCR) and Sanger sequencing. The PCR primers were designed using Primer3. The validation and analyses were conducted according to the NCBI VARIANT (https://www.ncbi.nlm.nih.gov/clinvar/), NCBI HomoloGene (https://www.ncbi.nlm.nih.gov/guide/howto/find-homolog-gene/), and 1000 Genomes Project (https://www.internationalgenome.org/) databases. Three-dimensional (3D) protein structures of the variants were generated using the online server I-TASSER (https://zhanggroup.org/I-TASSER/).
2.4 Analysis of the protein-protein interaction network
Search Tool for the Retrieval Interacting Genes 11.5 (STRING) (https://cn.string-db.org) was used for online analysis of the protein-protein interaction network (PPI), which was then imported into Cytoscape (v3.9.0). Degree ≥5 was set to select significant proteins among the networks.
3 Results
3.1 Clinical manifestations
The pedigree is shown in Figure 1, and the corresponding clinical data are summarized in Table 1. In this family, three living members developed KC and PPCD3, among which III:2 and III:1 were diagnosed with subclinical KC preoperatively and underwent SMILE surgery in October 2021. One year later, the visual acuity of the proband (III:1) declined significantly due to keratoconus aggravation. Her uncorrected distance visual acuities (UDVAs) were 10/50 in both eyes, corrected distance visual acuities (CDVAs) were 30/50 (right eye) with −1.00DS/-1.50 DC × 175° correction and 30/50 (left eye) with −1.50 DS/-1.50 DC × 15° correction (Supplementary Figure S1). For subject III:2, keratoconus aggravated significantly 6 months after SMILE; the UDVAs were 20/50 in both eyes, and the CDVAs were 40/50 (right eye) with −1.00DS/-1.00 DC × 170° correction and 30/50 (left eye) with −2.00 DS/-2.00 DC × 175° correction. A postoperative evaluation found that the maximum anterior surface curvature (MASC) and posterior elevation of the cornea (PEC) at the thinnest point of the cornea significantly increased, indicating that the keratoconus had worsened (Supplementary Figure S2). Moreover, these values showed a continuous upward trend in the subsequent follow-up.

Figure 1. The genogram of a three-generation Chinese family affected by posterior polymorphous corneal dystrophy (PPCD3) and keratoconus (KC). The squares represent male members, while circles indicate female members. The solid symbols show that the individual has both PPCD3 and KC. The open symbols represent the unaffected family members. Subject III.1 was the proband (arrow).

Table 1. The data of family members.
Greyish opacification of the corneal endothelium and stroma in the subnasal region and endothelial rail tracks were observed in the left eye by biomicroscopy and OCT (Figures 2a,b). OCT showed the corneal endothelial and stromal opacification of the left eye. Moreover, ECD of the proband reduced significantly to only 1914 cells/mm2 in the right eye and 1414 cells/mm2 left eye. Polymorphous, giant endothelial cells with some nucleated cells and hypo-reflective vesicular lesions, in the form of a crater with hyperreflective deposits around the lesions, were shown in both eyes. which is consistent with phenotypes of PPCD3. Subject II:2, the mother of the proband, was also diagnosed with subclinical keratoconus through a pentacam examination. The rough image displayed abnormal increase of the posterior corneal surface. (Supplementary Figure S3).
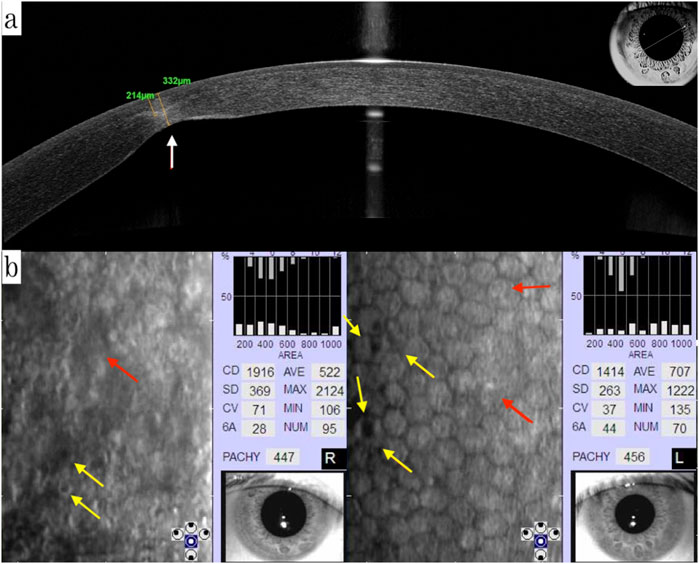
Figure 2. (a) OCT showing the corneal endothelial and stromal opacification of the left eye (white arrow); (b) ECD of the proband was reduced significantly to only 1914 cells/mm2 in the right eye and 1414 cells/mm2 in the left eye. Polymorphous, giant endothelial cells with some nucleated cells (red arrow), and hypo-reflective vesicular lesions, in the form of a crater with hyperreflective deposits around the lesions, were shown in both eyes (yellow arrow). OCT: optical coherence tomography, ECD: endothelium cell density.
3.2 Identification and analysis of the new variants
Two new variants were identified in this three-generation family: a heterozygous non-frameshift variant c. 3093_3104del (p.D1035_K1038del) in the ZNF469 gene, and a heterozygous missense variation c.13C>G (p.P5A, rs753301298) in the ZEB1 gene (Figures 3a,b). Variant c.3093_3104del (delCCCCAGGAAGGA) in ZNF469 is in exon 3, which leads to a four amino acid deletion from 1035 to 1038 in the zinc finger protein. p.P5A in ZEB1 is in exon 1, the amino-terminus (NZF) of the zinc finger E-box-binding homeobox 1 protein. These two variations were detected in III:2, III:1 and II:2. The other two healthy members in this family did not carry these variations. Family member I:2 had passed away 2 years prior to this study but had experienced corneal opacity resulting in blindness prior to her death. All variants were absent in the 100 random controls.
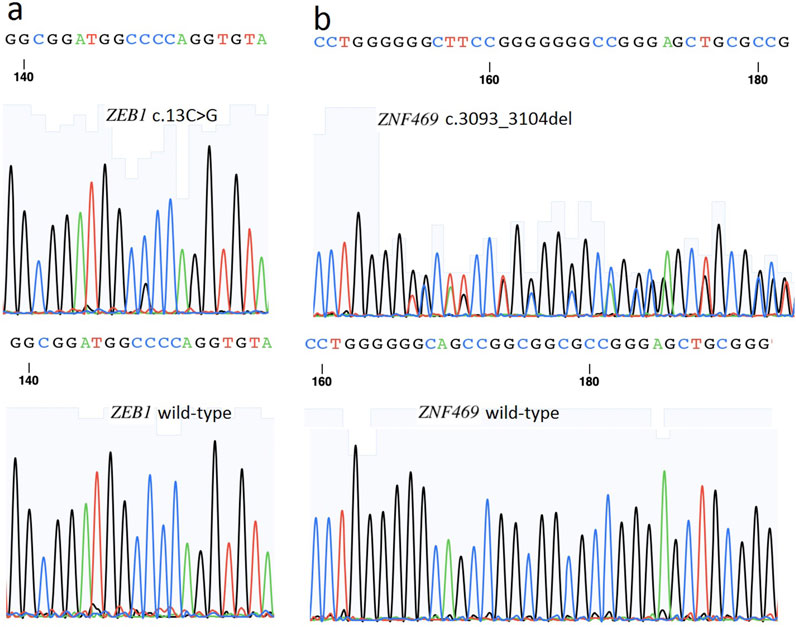
Figure 3. (a) A heterozygous non-frameshift variant c. 3093_3104del (p.D1035_K1038del) in ZNF469 gene is shown (arrow). (b) A heterozygous missense variant c. 13C>G (p.P5A, rs753301298) in ZEB1 gene is shown (arrow).
The variant c. 3093_3104del (p.D1035_K1038del) in ZNF469 was predicted to be a “Polymorphism” by Varianttaster Prediction, however, this variation resulted in the deletion of four amino acids from 1035 to 1038. According to the ACMG guidelines, the variant was predicted to be variant of uncertain significance (VUS), for it had not been reported in Human Gene Variant Database (HGMD) (PM2) and the change in protein length was the result of an in-frame deletion (PM4). The 3D modeling of the wild-type protein and the variation clearly displayed the conformational changes induced by the variant (Figure 4a). A single nucleotide polymorphism (SNPs) c.13C>G (p.P5A, rs753301298) in ZEB1 gene was suggested to be pathogenic by online prediction programs, including Polyphen2, SIFT, VariantTaster and Fathmm-MKL. The CADD (Combined Annotation Dependent Depletion) score is 25.1, indicating “Probably Deleterious” (range from 25.0 to 29.9) (Table 2). Moreover, conformational changes related to the variant were exhibited by the 3D modeling of ZEB1 wild-type and its variant (Figure 4b). The allele frequency of the SNP rs753301298 in the normal population was 0.00001 (database: gnomAD_exome_EAS), which is very rare.
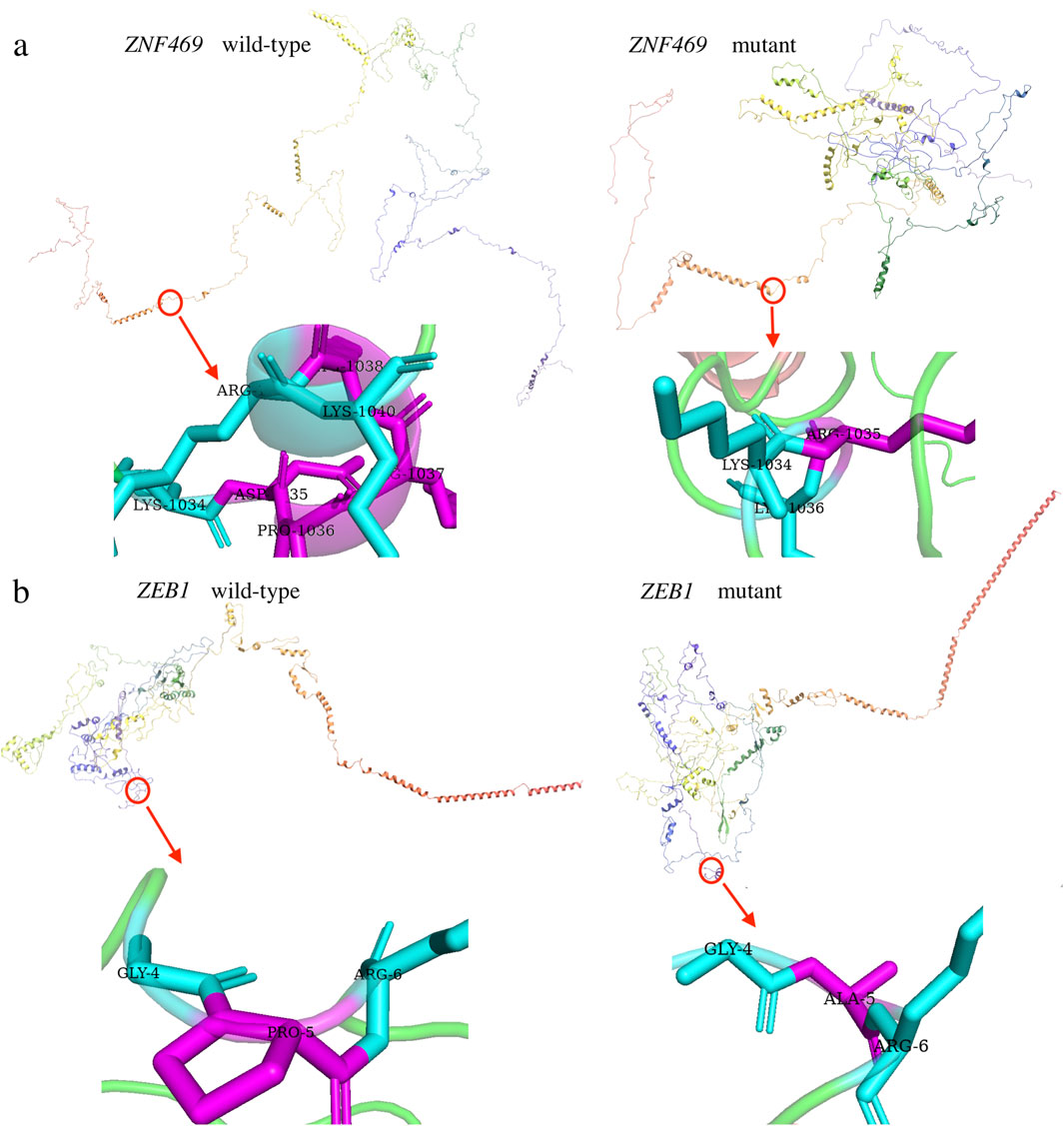
Figure 4. Three dimensional (3D) models of the proteins highlighting the variant sites. The inset pictures show the variants. (a) 3D modeling of wild-type ZNF469 and p.D1035_K1038del variant. (b) 3D modeling of wild-type ZEB1 and p.P5A variant.
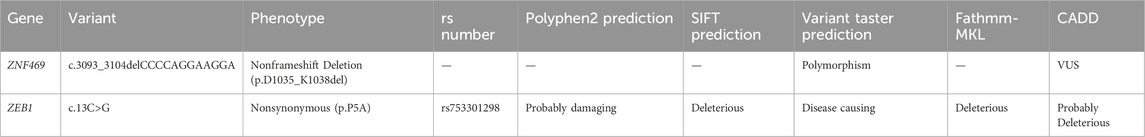
Table 2. Identification and analysis of the new variants.
3.3 Analysis of the protein-protein interaction network
Sixteen and twenty significantly enriched genes were uploaded to STRING to generate the PPI network for ZNF469 and ZEB1, respectively, and the results were subsequently imported to Cytoscape to construct sub-networks. In the network, ZNF469 and ZEB1 are placed and highlighted in the middle (ZNF469 proteins with the high degree of 13, close to COL5A1 and COL8A2, and ZEB1 proteins with the degree of 15.0, which is next to HDAC1, EP300 and CDH1) (Figures 5a,b).

Figure 5. (a) The protein interaction network constructed by STRING and the network of PPI network performed using Cytoscape for ZNF469; (b) The protein interaction network constructed by STRING and the network of PPI network performed using Cytoscape for ZEB1.
4 Discussion
4.1 Pathogenic variants in ZEB1 and ZNF469 are significantly associated with both PPCD3 and KC
Posterior polymorphous corneal dystrophy (PPCD) is a rare autosomal dominant disorder with genetic heterogeneity, primarily characterized by corneal endothelial abnormalities. These defects manifest as opacities and distinct lesions, including Descemet’s membrane bands and endothelial cell vesicles in the posterior corneal layers (Weiss et al., 2015; Lin et al., 2016). Currently, three genetically distinct subtypes have been identified: The gene(s) responsible for the PPCD1 subtype is OVOL2. PPCD3 is due to mutations in ZEB1. PPCD4 is linked to the gene GRHL2, and accounts for about 30% of individuals with PPCD and often is associated with corneal steepening (Davidson et al., 2020).
Notably, the clinical heterogeneity observed in PPCD3 patients, ranging from remaining asymptomatic throughout life to requiring corneal transplantation during adolescence, may be associated with the diversity of mutations or deletions in the coding region of the ZEB1 gene (Jang et al., 2014; Chung et al., 2017; Fernández-Gutiérrez et al., 2022). Studies have demonstrated that ZEB1 (OMIM #189909), functioning as a transcriptional repressor, plays a pivotal role in epithelial-endothelial cell lineage transition and the development of neural crest-derived structures (particularly corneal endothelium) during embryogenesis (Evans et al., 2015). ZEB1 contains three core domains: a N-terminal zinc finger (NZF), central homeodomain (HD), and C-terminal zinc finger cluster (CZF), along with functional domains (SBD, CBD, CID) that mediate protein interactions. Through SMARCA4/BRG1 recruitment to its N-terminus, ZEB1 induces epithelial-mesenchymal transition (EMT) by repressing epithelial markers, such as E-cadherin (Schmalhofer et al., 2009). Mutations in NZF and other sites may affect the normal function of ZEB1. For example, a ZEB1 mutation failed to suppress CDH1 in epithelium, and reduced expression of ZEB1 may lead to insufficient binding to the E2 box, and thus repression of COL4A3 in the corneal endothelium of patients with PPCD3 (Chung et al., 2016). In this study, we identified a heterozygous missense variant c.13C>G (p.P5A, rs753301298) in ZEB1’s exon 1/NZF domain, causing a Pro5Ala substitution (Figure 6). Computational analyses (Polyphen2, SIFT, MutationTaster, Fathmm-MKL, and CADD) (Table 2) and structural modeling suggested pathogenicity, supported by high evolutionary conservation (GERP++ = 4.12), low gnomAD frequency (PM2) and predicted structural perturbations. Moreover, according to the ACMG guidelines (Richards et al., 2015), the variant is likely to be a VUS, though its potential functional impact (PP3) correlates with our patients’ endothelial dystrophy. Notably, although ZEB1 missense variants typically associate with KC/FECD (vs. truncating mutations in PPCD3), our findings and recent reports (Mazzotta et al., 2014; Bykhovskaya et al., 2016), suggest missense variants may also contribute to PPCD3 pathogenesis, particularly in cases of overlap of KC and PPCD3.
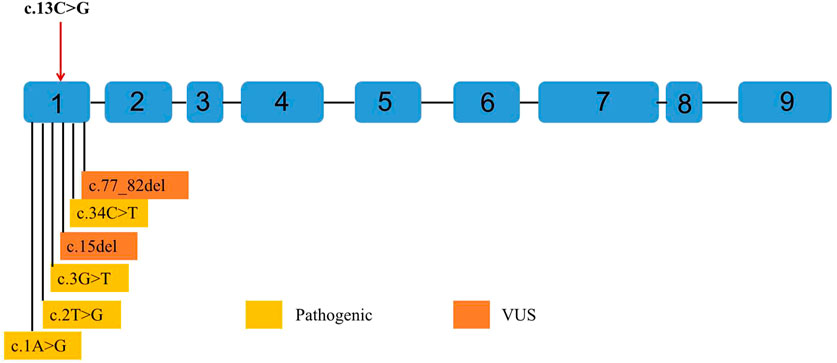
Figure 6. Point variants classified as pathogenic and variants of uncertain significance (VUS) by the American College of Medical Genetics and Genomics (ACMG) observed in ZEB1 exons up to June 2023. Variants classified as pathogenic are shown in yellow. Variants classified as VUS are shown in orange. Any point variant classified as “likely pathogenic” has been reported in the Leiden Open Variation Database (LOVD). The reference transcript employed is NM_030751.6.
In addition to ZEB1, variations in the ZNF469 gene have been implicated in keratoconus pathogenesis. The ZNF469 gene (NM_001127464) encodes a poorly conserved C2H2 zinc finger protein with five exons. While previously unreported in PPCD3, ZNF469 critically regulates central corneal thickness (CCT), and its variants predispose to keratoconus and brittle cornea syndrome type 1 (Abu et al., 2008; Loukovitis et al., 2018). Here, we identified a Mendelian-inherited heterozygous non-frameshift mutation (c.3093_3104del, p.D1035_K1038del) in ZNF469 exon 3, co-occurring with the ZEB1 rs753301298 variant in affected family members (Figure 3). This in-frame deletion causes the loss of four zinc finger domain residues (1035-1038) and protein conformational changes (Figure 4).
In humans, the corneal stroma accounts for 90% of the corneal thickness, and resident keratinocytes deposit a collagen-rich extracellular matrix. ZNF469 mutations downregulate stromal ECM genes, including EGF-like repeat and discoidin I-like domain-containing protein 3 (EDIL3), collagen alpha-1 (IV) chain (COL4A1), collagen alpha-1 (XI) chain (COL11A1), transforming growth factor beta-2 (TGFb2) and hyaluronan and proteoglycan link protein 1 (HAPLN1) (Lechner et al., 2013). Our PPI network analysis also revealed interactions between ZNF469 and CCT candidate genes, including COL5A1, COL1A1, and other genes that regulate eyeball development, such as VSX1 (causing KC and corneal dystrophy) and CHST14 (Ehlers-Danlos Syndrome candidate gene, Figure 5). Therefore, variants in ZNF469 could induce abnormal corneal development through disturbing these pathways. The protein sequence of ZNF469 shows 30% homology with the helical parts of COL1A1, COL1A2 and COL4A1, all of which are abundantly expressed in the cornea (Abu et al., 2008). There is evidence to suggest that patients with KC may have a dysregulation of collagen homeostasis, because 70% of the components in the cornea are collagen, especially type I collagen (Critchfield et al., 1988; Kenney et al., 1997).
4.2 The pathogenic synergy between ZEB1 and ZNF469 in both PPCD3 and KC
Numerous cases have demonstrated a correlation between PPCD3 and KC within the same patient’s cornea (Zhang et al., 2021). The two conditions share similar pathological features, including corneal stromal thinning, extracellular matrix (ECM) remodeling, and inflammatory cell infiltration (Jeang et al., 2021; Santodomingo-Rubido et al., 2022), suggesting potential involvement of common underlying mechanisms. Previous studies have focused primarily on separate genetic analyses of PPCD3 or KC. For instance, Liyan Xu et al. identified eight hub genes (LAMB3, LAMA3, LAMA1, ITGA6, ITGA3, COL6A3, COL6A2, and COL6A1) as key candidate genes for KC (Ren et al., 2023), while Shaowei Li et al. proposed that eleven genes (CAT, COL12A1, FLG, HKDC1, HSPG2, PLOD1, ITGA2, TFAP2B, USH2A, WNT10A, and COL6A5) might be associated with KC pathogenesis in Chinese patients (Song et al., 2024). However, there have been few reports focusing on familial cases with PPCD3-KC overlapping symptoms and their associated genetic analyses. The present study identified pathogenic mutations in ZEB1 and ZNF469 in a pedigree exhibiting PPCD3-KC overlapping symptom (Figures 3, 4). Recently, studies have established that ZEB1 (Lin K. et al., 2024) and ZNF469 (Bao et al., 2023) are critically involved in ECM homeostasis. Furthermore, our protein-protein interaction (PPI) analysis revealed a close association between ZEB1 and HDAC1. HDAC1 regulates ECM stability by deacetylating histones (e.g., H3K27ac), thereby suppressing the transcription of collagen genes (e.g., COL1A1, COL3A1) (Li et al., 2017). This mechanism mitigates fibrosis, as seen in corneal scarring, where HDAC1 upregulation reduces type I/III collagen deposition. Notably, COL1A1 and COL3A1 also exhibit significant interaction with ZNF469 (Figure 5). These findings suggest that the co-mutation of ZEB1 and ZNF469 may cooperatively regulate ECM dyshomeostasis in PPCD3-KC overlapping symptoms, potentially through disrupting HDAC1-mediated transcriptional dysregulation of collagen genes (e.g., COL1A1 and COL3A1).
In addition, the involvement of ZEB1 in ocular inflammatory processes remains poorly characterized. Li et al. demonstrated that ZEB1, in conjunction with CREB, binds to promoters of pro-inflammatory cytokines IL-1β and IFN-γ, upregulating their expression in corneal epithelial cells via p38 MAPK signaling (Li et al., 2010). This finding establishes ZEB1 and CREB as critical regulators in immune-mediated ocular surface squamous metaplasia. Complementing this, Park et al. showed that Epstein-Barr virus infection activates both Snail and ZEB1, promoting their nuclear translocation, a process that may drive epithelial-mesenchymal transition through loss of epithelial characteristics and acquisition of mesenchymal traits (Park et al., 2014). However, studies investigating the potential role of ZNF469 in corneal inflammation remain scarce. Consequently, whether these two genes share common mechanisms in mediating inflammatory responses in both PPCD3 and keratoconus warrants further investigation.
4.3 Clinical translation value and significance of our genetic findings
The largest published cohort study to date identified ZEB1 mutations in 25% of PPCD cases (8/32 probands) (Aldave et al., 2007), with reported detection rates varying significantly across studies (9.1%–45.4%) (Aldave et al., 2007; Dudakova et al., 2019). Notably, Zhou et al. demonstrated that only 1.6% of patients with keratoconus (KC) harbor ZNF469 mutations (Lin Q. et al., 2024). These findings collectively underscore the limited phenotypic contribution of single-gene mutations. In our pedigree, 100% of family members (3/3 probands) carrying dual ZEB1 and ZNF469 mutations exhibited PPCD3-KC overlapping phenotypes (Table 1). Although the generalizability of single-family studies remains constrained, this striking association strongly implicates ZEB1-ZNF469 digenic interactions in driving the PPCD3-KC phenotypic spectrum. For clinical management and preoperative evaluation of refractive surgery candidates in this cohort, meticulous consideration must be given to their elevated risk of developing PPCD3 or KC postoperatively, warranting risk-stratified surveillance protocols and tailored pre-/intraoperative prophylactic interventions.
Two novel variants in ZEB1 and ZNF469 were identified in this study as genetic factors associated with KC and PPCD. This finding highlights the importance of screening for ZEB1 and ZNF469 in patients who are considering refractive surgery and have a family history of KC and PPCD, as well as those exhibiting high corneal astigmatism or irregular corneal morphology. Such screening could facilitate preoperative risk assessment for refractive surgeries. For individuals with subtle corneal abnormalities but harboring mutations in ZEB1 or ZNF469, targeted dynamic monitoring is recommended (e.g., gene-specific matrix) (Henderson et al., 2024). We can develop personalized testing protocols based on the results of genetic screening. Additionally, implementing multimodal intervention strategies has been shown to significantly reduce the incidence of KC and PPCD following refractive surgery. These strategies may include combined corneal cross-linking (Santodomingo-Rubido et al., 2022), genetic counseling, and interdisciplinary management.
5 Conclusion
In conclusion, this study demonstrated the importance of a thorough ocular examination, especially the cornea, and a gene screening before cornea refractive surgery. Genetic abnormalities would increase the risk of post refractive complications, such as KC. Two novel variants in ZEB1 and ZNF469 were identified in this study as genetic factors associated with KC and PPCD. With the application of advanced genetic analyses in the clinic, ocular hereditary disorders such as KC or PPCD can be detected and diagnosed very early before clinical onset to avoid the risk of cornea refractive surgery.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SCV003842308, SCV003842309, SCV003842310, SCV003842311 and SCV003842314.
Ethics statement
The studies involving humans were approved by Institutional Review Board of Fudan University (Shanghai, China). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
QL: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Validation, Writing – original draft. XW: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – original draft. XP: Formal Analysis, Investigation, Methodology, Project administration, Software, Validation, Writing – review and editing. XH: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – original draft. XaZ: Formal Analysis, Investigation, Methodology, Software, Writing – review and editing. LS: Formal Analysis, Investigation, Methodology, Software, Writing – review and editing. YW: Formal Analysis, Investigation, Methodology, Software, Writing – review and editing. SL: Conceptualization, Project administration, Supervision, Validation, Writing – review and editing. XnZ: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study received partial funding support from the following projects. The National Natural Science Foundation of China for Young Scholars (No. 82000929); the National Natural Science Foundation of China (Grant No. 82305379), the National Natural Science Foundation of China (No. 81770955); the Shanghai Sailing Program (No. 20YF1405000); the Project of Shanghai Science and Technology (No. 20410710100); the Clinical Research Plan of SHDC (No. SHDC2020CR1043B); the Project of Shanghai Xuhui District Science and Technology (No. 2020-015); the Project of Shanghai Xuhui District Science and Technology (No. XHLHGG202104); the Shanghai Engineering Research Center of Laser and Autostereoscopic 3D for Vision Care (No. 20DZ2255000); and the construction of a 3D digital intelligent prevention and control platform for the whole life cycle of highly myopic patients in the Yangtze River Delta (No. 21002411600).
Acknowledgments
The authors would like to express their gratitude to all of the study participants for their cooperation The authors also thank Medjaden Inc. for the scientific editing of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1603019/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | The corneal topography (Pentacam) reports for family member III.1(proband). 1a: Pentacam preoperative. The means of posterior elevation of the cornea (PEC) were 9 µm (right eye) and 14 µm (left eye). The central corneal thicknesses (CCT) were 519 and 528 µm in the right and left eye, respectively. 1b: Pentacam 6 months postoperative. The means of posterior elevation of the cornea (PEC) were 6 µm (right eye) and 29 µm (left eye). The central corneal thicknesses (CCT) were 433 and 455 µm in the right and left eye, respectively. 1c: Pentacam 1 year postoperative. The means of posterior elevation of the cornea (PEC) were 19 µm (right eye) and 44 µm (left eye). The central corneal thicknesses (CCT) were 423 and 445 µm in the right and left eye, respectively. 1d: Pentacam 1.5 years postoperative. The means of posterior elevation of the cornea (PEC) were 21 µm (right eye) and 45 µm (left eye). The central corneal thicknesses (CCT) were 426 and 445 µm in the right and left eye, respectively.
SUPPLEMENTARY FIGURE S2 | The corneal topography (Pentacam) reports for family member III.2. 2a: Pentacam preoperative. The means of posterior elevation of the cornea (PEC) were 14 µm (right eye) and 13 µm (left eye). The central corneal thicknesses (CCT) were 564 and 567 µm in the right and left eye, respectively. 2b: Pentacam 1 month postoperative. The means of posterior elevation of the cornea (PEC) were 18 µm (right eye) and 15 µm (left eye). The central corneal thicknesses (CCT) were 454 and 449 µm in the right and left eye, respectively. 2c: Pentacam 6 months postoperative. The means of posterior elevation of the cornea (PEC) were 26 µm (right eye) and 30 µm (left eye). The central corneal thicknesses (CCT) were 465 and 461 µm in the right and left eye, respectively.
SUPPLEMENTARY FIGURE S3 | The corneal topography (Pentacam) reports for family member II.2 The means of posterior elevation of the cornea (PEC) were 12 µm (right eye) and 9 µm (left eye). The central corneal thicknesses (CCT) were 532 and 528 µm in the right and left eye, respectively.
Abbreviations
LASIK, laser-assisted in situ keratomileusis; PRK, photorefractive keratectomy; SMILE, small-incision lenticule extraction; KC, keratoconus; PPCD, posterior polymorphous corneal dystrophy; PPCD3, posterior polymorphous corneal dystrophy subtype 3; BCVA, best-corrected visual acuity; OCT, optical coherence tomography; ECD, endothelium cell density; ES, exome sequencing; PCR, polymerase chain reaction; PPI, protein-protein interaction; UDVAs, uncorrected distance visual acuities; PEC, posterior elevation of the cornea; VUS, variant of uncertain significance; HGMD, Human Gene Variant Database; NZF, amino-terminal zinc finger; HD, homeodomain; SBD, SMAD binding domain; CBD, CAF binding domain; CID, CtBP interacting domain; EMT, Epithelial-mesenchymal transition; FECD, Fuchs endothelial corneal dystrophy; CCT, central corneal thinning; BCS, brittle cornea syndrome; EDIL3, EGF-like repeat and discoidin I-like domain-containing protein 3; TGFb2, transforming growth factor beta-2; HAPLN1, hyaluronan and proteoglycan link protein 1; IVCM, in vivo confocal microscopy.
References
Abu, A., Frydman, M., Marek, D., Pras, E., Nir, U., Reznik-Wolf, H., et al. (2008). Deleterious mutations in the Zinc-Finger 469 gene cause brittle cornea syndrome. Am. J. Hum. Genet. 82 (5), 1217–1222. doi:10.1016/j.ajhg.2008.04.001
Aldave, A. J., Yellore, V. S., Yu, F., Bourla, N., Sonmez, B., Salem, A. K., et al. (2007). Posterior polymorphous corneal dystrophy is associated with TCF8 gene mutations and abdominal hernia. Am. J. Med. Genet. A 143a (21), 2549–2556. doi:10.1002/ajmg.a.31978
Bao, J., Yu, X., Ping, X., Shentu, X., and Zou, J. (2023). Znf469 plays a critical role in regulating synthesis of ECM: a zebrafish model of brittle cornea syndrome. Invest Ophthalmol. Vis. Sci. 64 (5), 29. doi:10.1167/iovs.64.5.29
Burdon, K. P., and Vincent, A. L. (2013). Insights into keratoconus from a genetic perspective. Clin. Exp. Optom. 96 (2), 146–154. doi:10.1111/cxo.12024
Bykhovskaya, Y., Margines, B., and Rabinowitz, Y. S. (2016). Genetics in Keratoconus: where are we? Eye Vis. (Lond) 3, 16. doi:10.1186/s40662-016-0047-5
Chung, D. D., Frausto, R. F., Lin, B. R., Hanser, E. M., Cohen, Z., and Aldave, A. J. (2017). Transcriptomic profiling of posterior polymorphous corneal dystrophy. Invest Ophthalmol. Vis. Sci. 58 (7), 3202–3214. doi:10.1167/iovs.17-21423
Chung, D. W., Frausto, R. F., Chiu, S., Lin, B. R., and Aldave, A. J. (2016). Investigating the molecular basis of PPCD3: characterization of ZEB1 regulation of COL4A3 expression. Invest Ophthalmol. Vis. Sci. 57 (10), 4136–4143. doi:10.1167/iovs.16-19533
Crawford, A. Z., Zhang, J., Gokul, A., McGhee, C. N. J., and Ormonde, S. E. (2020). The enigma of environmental factors in keratoconus. Asia Pac J. Ophthalmol. (Phila) 9 (6), 549–556. doi:10.1097/apo.0000000000000334
Critchfield, J. W., Calandra, A. J., Nesburn, A. B., and Kenney, M. C. (1988). Keratoconus: I. Biochemical studies. Exp. Eye Res. 46 (6), 953–963. doi:10.1016/s0014-4835(88)80047-2
Davidson, A. E., Hafford-Tear, N. J., Dudakova, L., Sadan, A. N., Pontikos, N., Hardcastle, A. J., et al. (2020). CUGC for posterior polymorphous corneal dystrophy (PPCD). Eur. J. Hum. Genet. 28 (1), 126–131. doi:10.1038/s41431-019-0448-8
Dudakova, L., Evans, C. J., Pontikos, N., Hafford-Tear, N. J., Malinka, F., Skalicka, P., et al. (2019). The utility of massively parallel sequencing for posterior polymorphous corneal dystrophy type 3 molecular diagnosis. Exp. Eye Res. 182, 160–166. doi:10.1016/j.exer.2019.03.002
Evans, C. J., Liskova, P., Dudakova, L., Hrabcikova, P., Horinek, A., Jirsova, K., et al. (2015). Identification of six novel mutations in ZEB1 and description of the associated phenotypes in patients with posterior polymorphous corneal dystrophy 3. Ann. Hum. Genet. 79 (1), 1–9. doi:10.1111/ahg.12090
Fernández-Gutiérrez, E., Fernández-Pérez, P., Boto-De-Los-Bueis, A., García-Fernández, L., Rodríguez-Solana, P., Solís, M., et al. (2022). Posterior polymorphous corneal dystrophy in a patient with a novel ZEB1 gene mutation. Int. J. Mol. Sci. 24 (1), 209. doi:10.3390/ijms24010209
Henderson, J., O'Callaghan, J., and Campbell, M. (2024). Gene therapy for glaucoma: targeting key mechanisms. Vis. Res. 225, 108502. doi:10.1016/j.visres.2024.108502
Jang, M. S., Roldan, A. N., Frausto, R. F., and Aldave, A. J. (2014). Posterior polymorphous corneal dystrophy 3 is associated with agenesis and hypoplasia of the corpus callosum. Vis. Res. 100, 88–92. doi:10.1016/j.visres.2014.04.007
Jeang, L. J., Margo, C. E., and Espana, E. M. (2021). Diseases of the corneal endothelium. Exp. Eye Res. 205, 108495. doi:10.1016/j.exer.2021.108495
Karolak, J. A., Ginter-Matuszewska, B., Tomela, K., Kabza, M., Nowak-Malczewska, D. M., Rydzanicz, M., et al. (2020). Further evaluation of differential expression of keratoconus candidate genes in human corneas. PeerJ 8, e9793. doi:10.7717/peerj.9793
Kenney, M. C., Nesburn, A. B., Burgeson, R. E., Butkowski, R. J., and Ljubimov, A. V. (1997). Abnormalities of the extracellular matrix in keratoconus corneas. Cornea 16 (3), 345–351. doi:10.1097/00003226-199705000-00016
Lechner, J., Dash, D. P., Muszynska, D., Hosseini, M., Segev, F., George, S., et al. (2013). Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation. Invest Ophthalmol. Vis. Sci. 54 (5), 3215–3223. doi:10.1167/iovs.13-11781
Li, S., Gallup, M., Chen, Y. T., and McNamara, N. A. (2010). Molecular mechanism of proinflammatory cytokine-mediated squamous metaplasia in human corneal epithelial cells. Invest Ophthalmol. Vis. Sci. 51 (5), 2466–2475. doi:10.1167/iovs.09-4677
Li, Z., Wei, S., Li, H., Wu, K., Cai, Z., Li, D., et al. (2017). Genome-wide genetic structure and differentially selected regions among Landrace, Erhualian, and Meishan pigs using specific-locus amplified fragment sequencing. Sci. Rep. 7 (1), 10063. doi:10.1038/s41598-017-09969-6
Lin, K., Zhao, Y., Tang, Y., Chen, Y., Lin, M., and He, L. (2024a). Collagen I-induced VCAN/ERK signaling and PARP1/ZEB1-mediated metastasis facilitate OSBPL2 defect to promote colorectal cancer progression. Cell Death Dis. 15 (1), 85. doi:10.1038/s41419-024-06468-1
Lin, Q., Wang, X., Han, T., Peng, X., and Zhou, X. (2024b). Variants in the ZNF469 gene in families with Brittle cornea syndrome and keratoconus. Heliyon 10 (5), e27052. doi:10.1016/j.heliyon.2024.e27052
Lin, Q., Zheng, L., and Shen, Z. (2022). A novel variant in TGFBI causes keratoconus in a two-generation Chinese family. Ophthalmic Genet. 43 (2), 159–163. doi:10.1080/13816810.2021.2015788
Lin, Z. N., Chen, J., and Cui, H. P. (2016). Characteristics of corneal dystrophies: a review from clinical, histological and genetic perspectives. Int. J. Ophthalmol. 9 (6), 904–913. doi:10.18240/ijo.2016.06.20
Loukovitis, E., Sfakianakis, K., Syrmakesi, P., Tsotridou, E., Orfanidou, M., Bakaloudi, D. R., et al. (2018). Genetic aspects of keratoconus: a literature review exploring potential genetic contributions and possible genetic relationships with comorbidities. Ophthalmol. Ther. 7 (2), 263–292. doi:10.1007/s40123-018-0144-8
Mazzotta, C., Traversi, C., Raiskup, F., Rizzo, C. L., and Renieri, A. (2014). First identification of a triple corneal dystrophy association: keratoconus, epithelial basement membrane corneal dystrophy and fuchs' endothelial corneal dystrophy. Case Rep. Ophthalmol. 5 (3), 281–288. doi:10.1159/000367937
Moshirfar, M., Tukan, A. N., Bundogji, N., Liu, H. Y., McCabe, S. E., Ronquillo, Y. C., et al. (2021). Ectasia after corneal refractive surgery: a systematic review. Ophthalmol. Ther. 10 (4), 753–776. doi:10.1007/s40123-021-00383-w
Park, G. B., Kim, D., Kim, Y. S., Kim, S., Lee, H. K., Yang, J. W., et al. (2014). The Epstein-Barr virus causes epithelial-mesenchymal transition in human corneal epithelial cells via Syk/src and Akt/Erk signaling pathways. Invest Ophthalmol. Vis. Sci. 55 (3), 1770–1779. doi:10.1167/iovs.13-12988
Ren, S., Yang, K., Fan, Q., Wang, Q., Zhu, M., Yin, S., et al. (2023). Bioinformatics analysis of key candidate genes and pathways in Chinese patients with keratoconus. Exp. Eye Res. 231, 109488. doi:10.1016/j.exer.2023.109488
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Santodomingo-Rubido, J., Carracedo, G., Suzaki, A., Villa-Collar, C., Vincent, S. J., and Wolffsohn, J. S. (2022). Keratoconus: an updated review. Cont. Lens Anterior Eye 45 (3), 101559. doi:10.1016/j.clae.2021.101559
Schmalhofer, O., Brabletz, S., and Brabletz, T. (2009). E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 28 (1-2), 151–166. doi:10.1007/s10555-008-9179-y
Song, C., Li, L., Liu, C., Hu, L., Bai, J., Liang, W., et al. (2024). Whole-exome sequencing screening for candidate genes and variants associated with primary sporadic keratoconus in Chinese patients. Exp. Eye Res. 245, 109978. doi:10.1016/j.exer.2024.109978
Weiss, J. S., Møller, H. U., Aldave, A. J., Seitz, B., Bredrup, C., Kivelä, T., et al. (2015). IC3D classification of corneal dystrophies--edition 2. Cornea 34 (2), 117–159. doi:10.1097/ico.0000000000000307
Zhang, D., Tian, L., Zhang, H., Zheng, Y., Fu, C., Zhai, C., et al. (2022). Differences of corneal biomechanics among thin normal cornea, forme-fruste keratoconus, and cornea after SMILE. Front. Bioeng. Biotechnol. 10, 861924. doi:10.3389/fbioe.2022.861924
Keywords: keratoconus (KC), ZNF469, Zeb1, posterior polymorphous corneal dystrophy (PPCD), corneal refractive surgery
Citation: Lin Q, Wang X, Peng X, Han X, Zhang X, Sun L, Wang Y, Liu S and Zhou X (2025) Polymorphous corneal dystrophy subtype 3 and keratoconus aggravation after corneal refractive surgery in a three-generation family carrying both ZEB1 and ZNF469 pathogenic variant. Front. Genet. 16:1603019. doi: 10.3389/fgene.2025.1603019
Received: 31 March 2025; Accepted: 27 May 2025;
Published: 06 June 2025.
Edited by:
Qi Dai, Wenzhou Medical University, ChinaReviewed by:
Xuefeng Shi, Tianjin Eye Hospital, ChinaQian Zhang, AmCare Genomics Lab, China
Xianyang Liu, Chongqing Medical University, China
Copyright © 2025 Lin, Wang, Peng, Han, Zhang, Sun, Wang, Liu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengtao Liu, MjgyOTg1NzY2QHFxLmNvbQ==; Xingtao Zhou, WmhvdXhpbmd0YW9kb2N0QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship