 Salvatore Savasta1
Salvatore Savasta1 Fabiola Serra2
Fabiola Serra2 Lucrezia Galimberti2
Lucrezia Galimberti2 Francesco Fabrizio Comisi2*Marcello Cossu2Alessandro Vannelli3Maddalena Masala4Sara Tanca4Stefania Murru4
Francesco Fabrizio Comisi2*Marcello Cossu2Alessandro Vannelli3Maddalena Masala4Sara Tanca4Stefania Murru4- 1Pediatric and Rare Diseases Clinic, Microcitemico Hospital “A. Cao”, Department of Medical Sciences and Public Health, University of Cagliari, Cagliari, Italy
- 2Pediatric and Rare Diseases Clinic, Microcitemico Hospital “A. Cao”, University of Cagliari, Cagliari, Italy
- 3Binaghi Hospital, ASL8, Cagliari, Italy
- 4Genetic and Genomic Laboratory, Pediatric Children Hospital “A. Cao”, ASL 8, Cagliari, Italy
Hereditary neuropathy with liability to pressure palsies (HNPP) is a genetic disorder characterized by recurrent focal neuropathies typically occurring at sites of nerve entrapment or compression. It is classically described as a painless condition; however, pain is frequently reported. Due to its rarity and variable clinical presentation, HNPP is often underdiagnosed or initially misdiagnosed. We report the case of a 14-year-old girl who presented with sudden-onset arm weakness and pain following physical activity. The clinical presentation initially raised suspicion for a hereditary demyelinating neuropathy. Although there was no known family history, the patient’s age and the persistence of symptoms supported the hypothesis of a genetic etiology. Neurophysiological studies were consistent with HNPP, which was subsequently confirmed by genetic testing. The primary aim of this report is to emphasize the importance of recognizing the early manifestations of HNPP—including pain, a symptom often underestimated or overlooked—in order to enable prompt diagnosis, reduce unnecessary diagnostic delays, and ensure timely initiation of appropriate genetic counseling. This case supports the notion that pain may represent an early feature of HNPP and should not lead clinicians away from considering this diagnosis.
Introduction
Hereditary neuropathy with liability to pressure palsies (HNPP) is an autosomal dominant disorder characterized by recurrent peripheral mononeuropathies at sites of nerve entrapment, typically triggered by minor trauma or compression (Mouton et al., 1999). Although HNPP is classically described as a painless neuropathy (Mouton et al., 1999), recent studies have shown that pain is a frequent symptom. In particular, up to 75% of patients report experiencing pain during the disease course, and in approximately 12% of cases, pain represents the initial manifestation (Yilmaz et al., 2015).
First described in 1947 by Dutch neurologist De Jong, the condition was originally referred to as “potato grubbing disease,” in reference to the occupation of the first reported family, whose repetitive work led to compressive neuropathies (Beydoun and Cho, 2013; Attarian et al., 2020). The estimated prevalence of HNPP ranges from 7 to 16 per 100,000 individuals, though it seems to vary considerably across populations and studies (Beydoun and Cho, 2013; Attarian et al., 2020; van Paassen et al., 2014). In fact, more specific data report prevalence rates from approximately 0.84 per 100,000 in Ireland, 2 per 100,000 in Northern England, and 16 per 100,000 in Southwestern Finland. In contrast, a Korean newborn screening study revealed a markedly higher prevalence of 1 in 1,698 (Park et al., 2018): further studies on larger populations would be needed to know the true global prevalence. The clinical presentation typically involves recurrent sensory and motor mononeuropathies, most often beginning in the second or third decade of life (Attarian et al., 2020). Diagnosis is supported by nerve conduction studies, which reveal characteristic delays in distal motor latencies.
HNPP is caused by a deletion on chromosome 17p11.2–p12 involving the PMP22 gene, which encodes peripheral myelin protein 22 (Verhagen et al., 1993). Reduced expression of PMP22 leads to impaired myelin function and clinical manifestations of the disease (Beydoun and Cho, 2013). In addition to the common deletion, other rare mutations in the PMP22 gene, such as point mutations and small deletions, have also been implicated in the pathogenesis of HNPP (Beydoun and Cho, 2013). The disorder is also referred to as “tomaculous neuropathy”, due to the presence of focal “sausage-like” myelin swellings known as tomacula (Saurabh and Ahmad, 2023).
Nerve biopsy, once commonly used for diagnosis, has largely been replaced by genetic testing (Beydoun and Cho, 2013).
The primary aim of this report is to highlight the importance of performing neurophysiological and genetic testing when clinical suspicion of HNPP arises, in order to achieve a definitive diagnosis. Particular attention should be given to the presence of pain, which—despite being frequently underrecognized—may represent an early and relevant symptom of the disease.
Case report
A 14-year-old girl was initially admitted to another hospital due to the sudden onset of weakness and pain in her left arm upon awakening. She had played basketball in the preceding days. She was born at term following an uneventful pregnancy and spontaneous delivery. At 4 months of age, she had been hospitalized for bronchiolitis. No other significant medical history was reported, and her psychomotor development had been normal. There was no family history of neurological disorders, and her parents were not consanguineous.
Neurological examination revealed proximal weakness of the left upper limb, with compensatory recruitment of the parabrachial muscles during extension, flexion, and abduction. Localized tenderness was noted on palpation near the distal head of the biceps tendon, while distal muscle strength remained preserved. Initial blood tests (including complete blood count, creatine phosphokinase, electrolytes, and C-reactive protein) were within normal limits. Cervical spine and left shoulder X-rays were unremarkable. Additional investigations including orthopedic evaluation, brain CT scan, and ophthalmologic examination also yielded normal results. Brain and cervical-thoracic spine MRI revealed a mild dilation of the ependymal canal in the thoracic region between T3–T4 and T8–T9.
An electromyography (EMG) study showed increased distal motor latency of the median and peroneal nerves bilaterally, reduced motor conduction velocities in the median, ulnar, peroneal, and tibial nerves bilaterally, and decreased sensory conduction velocities in the examined nerves. These findings were suggestive of a hereditary demyelinating polyneuropathy of probable genetic origin.
Five months later, the patient was referred to our center for further diagnostic evaluation, as her symptoms had improved but not completely resolved. On admission, her neurological examination was normal; although she reported lingering weakness in her left arm, both muscle strength and sensory function were preserved. Repeat blood tests were within normal limits. Shoulder MRI and ultrasound showed no abnormalities. Neurophysiological assessment revealed normal somatosensory evoked potentials. However, EMG and nerve conduction studies demonstrated a chronic, widespread, mixed sensorimotor neurogenic impairment with demyelinating features. The left ulnar nerve at the elbow showed reduced sensory conduction velocity (38.4 m/s) and motor conduction velocity ranging from 36.4 to 27.4 m/s. Similarly, the right common peroneal nerve showed reduced motor conduction velocities at the fibular head (39.7 m/s) and across the knee (29.9 m/s). These findings were consistent with hereditary neuropathy with liability to pressure palsies (HNPP). Genetic testing subsequently confirmed a heterozygous 1.4 Mb deletion on chromosome 17p12, encompassing the PMP22 gene. Upon discharge, genetic counseling was provided, along with behavioral recommendations aimed at reducing the risk of symptom recurrence (Figure 1).
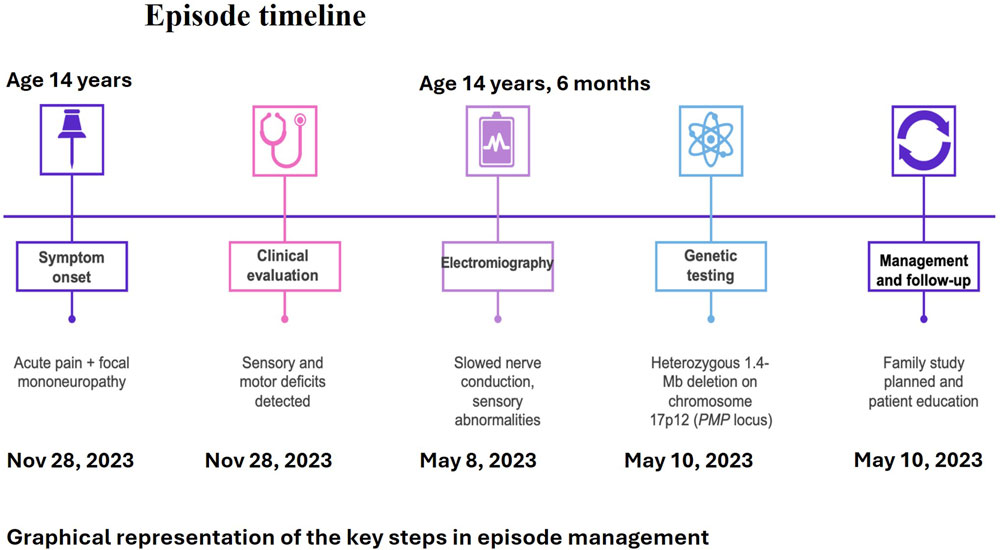
Figure 1. Episode timeline.
Methods
A written informed consent for blood sample collection and genetic analysis was obtained from the proband in compliance with the Declaration of Helsinki. Genomic DNA was extracted from peripheral blood samples using the QIAamp DNA Blood Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. SNP-Array was performed using the Infinium CytoSNP 850K v1.4 microarray, spanning the entire genome, with a resolution of 100 kb, according to the instruction provided by the manufacturer. The Illumina Infinium CytoSNP 850K v1.4 Bead Chip platform and scanning using NextSeq550 was used for SNP-Array analysis. The level of resolution, dependent on the number of SNPs and their distance in the investigated DNA region is between 5 and 1 Kb in areas including relevant genes as indicated by ICCG (International Collaboration for Clinical Genomics) and CCMC (Cancer Cytogenomics Microarray Consortium). The analysis of CNVs (Copy Number Variants) and regions with LOH (loss of heterozygos-ity) is performed using Bluefuse Multi Software v4.3. Map positions refer to the Human Genome Reference Consortium (hg19). Genetic testing identified a heterozygous interstitial microdeletion encompassing 1.4 Mb in the short arm of chromosome 17 with breakpoints in band 17p12 (arr [GRCh37] 17p12 (14095309_15455610)). This region contains the PMP22 gene (Figure 2). Microdeletion was confirmed by Comparative Genomic Hybridization (CGH) Array method using Agilent GenetiSure Cyto CGH + SNP 4 × 180k.
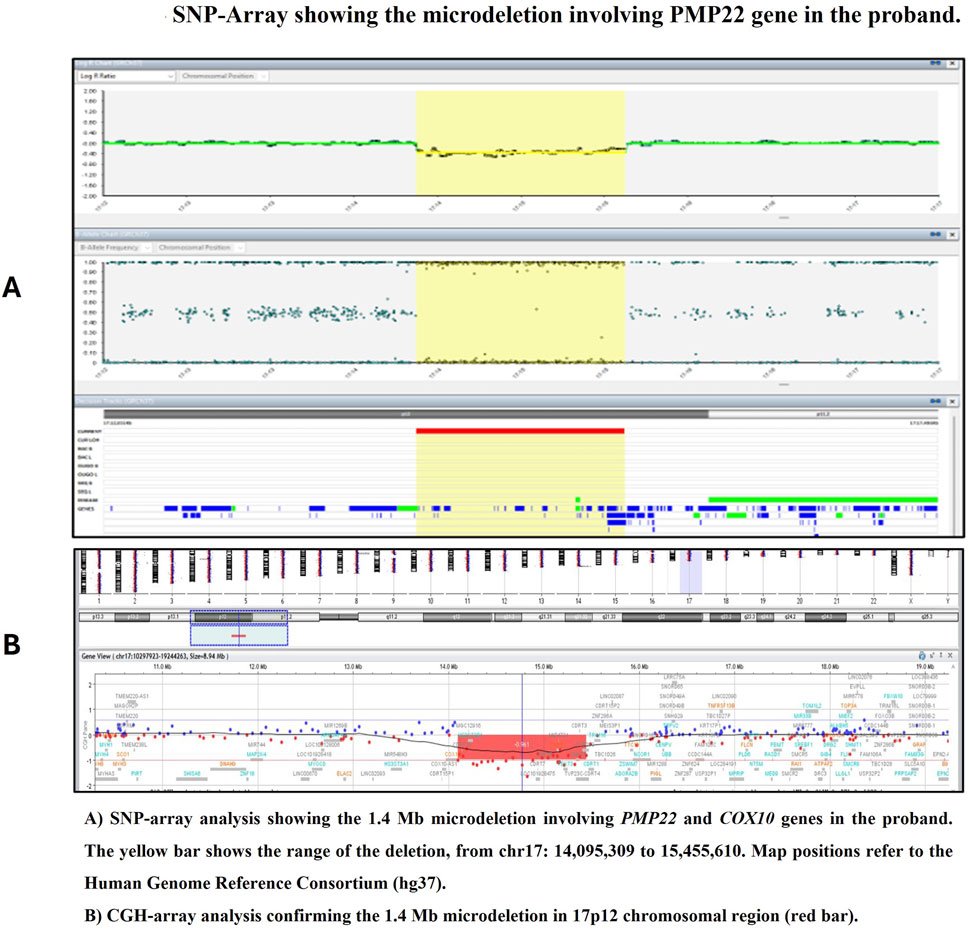
Figure 2. SNP-Array showing the microdeletion involving PMP22 gene in the proband. (A) SNP-array analysis showing the 1.4 Mb microdeletion involving PMP22 and COX10 genes in the proband. The yellow bar shows the range of the deletion, from chr17: 14,095,309 to 15,455,610. Map positions refer to the Human Genome Reference Consortium (hg37). (B) CGH-array analysis confirming the 1.4 Mb microdeletion in 17p12 chromosomal region (red bar).
Discussion
HNPP is an autosomal dominant disorder that is often underdiagnosed (Dubourg et al., 2000). Some studies have highlighted a higher prevalence in males (male-to-female ratio of 1.3:1), probably due to greater exposure to trauma in this population (Tinant et al., 2002). Approximately one-third of individuals carrying the deletion remain asymptomatic, while most symptomatic cases follow a favorable course (Tinant et al., 2002). A family history of compressive neuropathies may be absent in HNPP patients, either due to sporadic mutations or the presence of asymptomatic carriers (Beydoun et al., 2008). The classic presentation of HNPP is characterized by recurrent, transient focal mononeuropathies (Pareyson et al., 1996).
Symptoms are typically triggered by prolonged compression, chronic mild pressure, or trauma (Felice et al., 1999). The age of onset varies widely, ranging from infancy to adulthood, with most cases occurring between 10 and 30 years of age (Mouton et al., 1999; Beydoun and Cho, 2013). In 85% of cases, the initial clinical manifestation is acute and painless; however, as the disease progresses, up to 75% of patients develop persistent pain (Attarian et al., 2020; Beales et al., 2017). According to the literature, our patient falls within the typical age range and experienced a stress-related event preceding symptom onset. However, the presence of pain at onset is uncommon in patients diagnosed before 18 years of age, as highlighted in a recent series of seven pediatric cases (Isik and Odabaşı, 2024). In patients presenting with pain at disease onset, the clinical phenotype may differ from the classical painless presentation, and for this reason diagnosis is often delayed. Symptoms may present with neuropathic or musculoskeletal features, leading to consideration of alternative diagnoses, such as fibromyalgia, when features such as allodynia and widespread discomfort overlap (Yilmaz et al., 2015; Cotelli et al., 2023). Although standard nerve conduction studies remain the principal method for diagnosis, no specific electrophysiological pattern has yet been identified for this presentation. In some cases, initial studies may show only subtle sensory abnormalities or even normal findings, potentially delaying diagnosis (Yilmaz et al., 2015; Cotelli et al., 2023). The nerves most frequently affected by compressive trauma include the ulnar nerve (at the elbow), the peroneal nerve (at the fibular head), and the radial nerve (at the spiral groove), followed by the brachial plexus and median nerve (Potulska-Chromik et al., 2014). Cranial nerve involvement is rare (van Paassen et al., 2014; Dubourg et al., 2000). In approximately 85% of patients, a heterozygous 1.5-Mb deletion on chromosome 17p12 (PMP22 locus) is identified (Beydoun and Cho, 2013). The same locus is duplicated in Charcot–Marie–Tooth disease type 1A, which is also the main differential diagnosis (Yilmaz et al., 2015). The PMP22 protein is involved in myelin production by Schwann cells and plays an essential structural role in stabilizing the myelin sheath, thereby protecting nerves from repetitive trauma (Beydoun and Cho, 2013; Attarian et al., 2020; Li et al., 2013a). Loss of function can result from point or frameshift mutations in about 15% of cases, while de novo mutations account for approximately 20% (Beydoun and Cho, 2013; Saurabh and Ahmad, 2023; Pareyson et al., 1996; Li et al., 2013a). In our case, genetic testing confirmed the diagnosis with a heterozygous 1.4-Mb deletion on chromosome 17p12. A negative family history, as seen in our patient, is common, since approximately half of deletion carriers remain asymptomatic (van Paassen et al., 2014; Infante et al., 2001). Further investigations, including a segregation study and electromyography of the patient’s parents, are planned to determine whether the deletion is inherited or de novo.
While mechanical compression and trauma are recognized as the main triggers for symptom onset in HNPP, several studies have suggested additional modifying factors that may influence disease penetrance. Metabolic stressors, such as diabetes mellitus, hypothyroidism, or vitamin deficiencies, have been proposed as potential risk factors that may unmask subclinical neuropathy in genetically predisposed individuals (Sung et al., 2012; Luigetti et al., 2014). Furthermore, environmental and occupational exposures (for example, repetitive movements, poor ergonomics, or cold temperatures) may contribute to increased vulnerability (Kim et al., 2010). In our patient we detected insufficient Vitamin D levels (11.3 ng/mL) as a side ward, but further studies would be needed to understand whether this could influence symptom onset.
Conversely, potential protective factors associated with the asymptomatic phenotype have been hypothesized but are less well-defined. One proposed factor is the presence of compensatory mechanisms in peripheral myelination, including upregulation of other myelin-related genes or structural variants in modifier loci, though evidence remains preliminary (Li et al., 2013b). Additionally, lifestyle factors that reduce nerve compression risk, such as regular physical activity without overuse, may contribute to the absence of clinical symptoms in some carriers (Kimura, 2013). A better understanding of these modifiers could help stratify risk and guide preventive strategies.
Neurophysiological studies typically show prolonged distal motor latencies, especially in the median and peroneal nerves (van Paassen et al., 2014; Li et al., 2002). Focal motor slowing at entrapment sites is common, particularly in the ulnar nerve at the elbow, though it may also affect the peroneal nerve at the fibular head (van Paassen et al., 2014; Li et al., 2002). Distal sensory conduction velocities are often diffusely reduced, findings that can be present in both symptomatic and asymptomatic nerves (Mouton et al., 1999; Beydoun and Cho, 2013; van Paassen et al., 2014; Li et al., 2002). Acute neuropathies usually resolve within days to weeks (Dubourg et al., 2000). Full recovery occurs in approximately 50% of episodes; if chronic symptoms persist, they are usually mild (van Paassen et al., 2014). However, relapses can be frequent, and paresis may last several months, as observed in our patient (Beydoun and Cho, 2013; Attarian et al., 2020; Dubourg et al., 2000). When symptoms persist or recur, patients may develop distal atrophy, sometimes symmetrical, resembling a CMT phenotype (Attarian et al., 2020; van Paassen et al., 2014; Chance, 2006). Life expectancy remains unaffected (van Paassen et al., 2014). To date, no pharmacological treatment has proven effective, and management remains symptomatic (Attarian et al., 2020; Beales et al., 2017; Potulska-Chromik et al., 2014). Patient education is crucial to prevent recurrences by avoiding positions that promote nerve compression, such as leg-crossing, repetitive wrist movements, prolonged leaning on the elbows, or rapid weight loss (Attarian et al., 2020; van Paassen et al., 2014; Potulska-Chromik et al., 2014). Upon discharge, we provided the patient and her family with the information they needed to understand the origin and course of the disease, reassuring them that it was benign and that she could lead a completely normal life. We also provided her with advice to prevent further episodes, and we prescribed a neurotrophic agent.
Conclusion
Hereditary neuropathy with liability to pressure palsies (HNPP) is a well-known condition that remains significantly underdiagnosed. Neurophysiological studies can reveal sensorimotor demyelinating lesions in affected nerves, while histological examination may demonstrate characteristic myelin thickening (tomacula), resulting from hypermyelination due to PMP22 protein deficiency. Genetic testing now provides a reliable, non-invasive method for confirming the diagnosis, largely replacing the need for nerve biopsy. As this is a single case report, our primary objective was to describe and contextualize a rare clinical presentation within the existing literature, rather than to establish generalizable findings. The case presented highlights not only the frequent delays in HNPP diagnosis, but also the undervalued role of pain as an early symptom. In our patient, several months elapsed before the correct diagnosis was established. Since diagnosis can be challenging, pain should be recognized as a possible presenting symptom, as it can lead to an early genetic diagnosis and help avoid unnecessary investigations.
Given its generally favorable prognosis, early identification of HNPP enables patients and their families to manage the condition effectively and maintain a normal quality of life.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: (https://www.ncbi.nlm.nih.gov/clinvar/submitters/510099).
Ethics statement
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
SS: Data curation, Conceptualization, Investigation, Writing – review and editing, Visualization, Formal Analysis, Writing – original draft, Supervision, Methodology, Validation. FS: Writing – original draft, Investigation, Writing – review and editing. LG: Data curation, Writing – original draft, Writing – review and editing, Investigation. FC: Supervision, Writing – review and editing, Formal Analysis, Conceptualization, Writing – original draft, Visualization. MC: Writing – review and editing, Writing – original draft, Investigation, Validation. AV: Data curation, Writing – original draft, Investigation, Validation, Writing – review and editing. MM: Validation, Writing – review and editing, Investigation, Writing – original draft. ST: Validation, Writing – review and editing, Investigation, Writing – original draft. SM: Investigation, Writing – original draft, Validation, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Attarian, S., Fatehi, F., Rajabally, Y. A., and Pareyson, D. (2020). Hereditary neuropathy with liability to pressure palsies. J. Neurol. 267 (8), 2198–2206. doi:10.1007/s00415-019-09319-8
Beales, D., Fary, R., Little, C., Nambiar, S., Sveinall, H., Yee, Y. L., et al. (2017). Characterisation of pain in people with hereditary neuropathy with liability to pressure palsy. J. Neurol. 264 (12), 2464–2471. doi:10.1007/s00415-017-8648-z
Beydoun, S. R., and Cho, J. (2013). Hereditary neuropathy with liability to pressure palsy: two cases of difficult diagnosis. J. Clin. Neuromuscul. Dis. 15 (1), 28–33. doi:10.1097/CND.0b013e31829e22fe
Beydoun, S. R., Sykes, S. N., Ganguly, G., and Lee, T. S. (2008). Hereditary neuropathy with liability to pressure palsies: description of seven patients without known family history. Acta Neurol. Scand. 117 (4), 266–272. doi:10.1111/j.1600-0404.2007.00935.x
Chance, P. F. (2006). Inherited focal, episodic neuropathies: hereditary neuropathy with liability to pressure palsies and hereditary neuralgic amyotrophy. Neuromolecular Med. 8 (1-2), 159–174. doi:10.1385/NMM:8:1:159
Cotelli, M., Balsamo, E., and Citterio, A. (2023). Painful onset in hereditary neuropathy with liability to pressure palsies: a pediatric and adolescent case series. Neuromuscul. Disord. 33 (2), 128–134. doi:10.1016/j.nmd.2022.11.003
Dubourg, O., Mouton, P., Brice, A., LeGuern, E., and Bouche, P. (2000). Guidelines for diagnosis of hereditary neuropathy with liability to pressure palsies. Neuromuscul. Disord. 10 (3), 206–208. doi:10.1016/s0960-8966(99)00103-0
Felice, K. J., Leicher, C. R., and DiMario, F. J. (1999). Hereditary neuropathy with liability to pressure palsies in children. Pediatr. Neurol. 21 (5), 818–821. doi:10.1016/s0887-8994(99)00086-7
Infante, J., García, A., Combarros, O., Mateo, J. I., Berciano, J., Sedano, M. J., et al. (2001). Diagnostic strategy for familial and sporadic cases of neuropathy associated with 17p11.2 deletion. Muscle Nerve 24 (9), 1149–1155. doi:10.1002/mus.1126
Isik, K., and Odabaşı, Z. (2024). An interesting cause of wrist drop: the Crow position in yoga and hereditary neuropathy with liability to pressure palsies. Turk J. Phys. Med. Rehabil. 70 (2), 282–284. doi:10.5606/tftrd.2024.12006
Kim, S. H., Park, K. S., and Yoon, J. S. (2010). Occupational predisposition in HNPP: a case report and review. J. Occup. Health 52 (4), 256–259. doi:10.1539/joh.L9133
Kimura, J. (2013). Electrodiagnosis in diseases of nerve and muscle: principles and practice. 4th ed. Oxford University Press.
Li, J., Krajewski, K., Shy, M. E., and Lewis, R. A. (2002). Hereditary neuropathy with liability to pressure palsy: the electrophysiology fits the name. Neurology 58 (12), 1769–1773. doi:10.1212/wnl.58.12.1769
Li, J., Parker, B., and Martyn, C. (2013b). Genetic modifiers of PMP22 expression and their impact on CMT1A and HNPP phenotype. Ann. Neurol. 74 (6), 930–939. doi:10.1002/ana.23997
Li, J., Parker, B., Martyn, C., Natarajan, C., and Guo, J. (2013a). The PMP22 gene and its related diseases. Mol. Neurobiol. 47 (2), 673–698. doi:10.1007/s12035-012-8370-x
Luigetti, M., Bisogni, G., and Romano, A. (2014). Hereditary neuropathy with liability to pressure palsies: a clinical and electrophysiological study of 15 patients. Neurol. Sci. 35 (3), 379–384. doi:10.1007/s10072-013-1548-y
Mouton, P., Tardieu, S., Gouider, R., Birouk, N., Maisonobe, T., Dubourg, O., et al. (1999). Spectrum of clinical and electrophysiologic features in HNPP patients with the 17p11.2 deletion. Neurology 52 (7), 1440–1446. doi:10.1212/wnl.52.7.1440
Pareyson, D., Scaioli, V., Taroni, F., Botti, S., Lorenzetti, D., Solari, A., et al. (1996). Phenotypic heterogeneity in hereditary neuropathy with liability to pressure palsies associated with chromosome 17p11.2-12 deletion. Neurology 46 (4), 1133–1137. doi:10.1212/wnl.46.4.1133
Park, J. E., Noh, S. J., Oh, M., Cho, D. Y., Kim, S. Y., and Ki, C. S. (2018). Frequency of hereditary neuropathy with liability to pressure palsies (HNPP) due to 17p11.2 deletion in a Korean newborn population. Orphanet J. Rare Dis. 13 (1), 40. doi:10.1186/s13023-018-0779-5
Potulska-Chromik, A., Sinkiewicz-Darol, E., Ryniewicz, B., Lipowska, M., Kabzińska, D., Kochański, A., et al. (2014). Clinical, electrophysiological, and molecular findings in early onset hereditary neuropathy with liability to pressure palsy. Muscle Nerve 50 (6), 914–918. doi:10.1002/mus.24250
Saurabh, K., and Ahmad, R. (2023). An unusual case of hereditary neuropathy with liability to pressure palsy: a diagnostic challenge. Cureus 15 (1), e33306. doi:10.7759/cureus.33306
Sung, M. S., Kim, Y. J., and Park, H. J. (2012). Hereditary neuropathy with liability to pressure palsy misdiagnosed as diabetic neuropathy. Muscle Nerve 45 (2), 290–293. doi:10.1002/mus.22291
Tinant, F., Zeevaert, B., Benkirane, H., Laurent, L., and Wang, F. (2002). La neuropathie avec hypersensibilité héréditaire à la pression ou neuropathie tomaculaire [Hereditary neuropathy with pressure hypersensitivity or tomaculous neuropathy]. Rev. Med. Liege 57 (10), 651–654.
van Paassen, B. W., van der Kooi, A. J., van Spaendonck-Zwarts, K. Y., Verhamme, C., Baas, F., and de Visser, M. (2014). PMP22 related neuropathies: charcot-marie-tooth disease type 1A and hereditary neuropathy with liability to pressure palsies. Orphanet J. Rare Dis. 9, 38. doi:10.1186/1750-1172-9-38
Verhagen, W. I., Gabreëls-Festen, A. A., van Wensen, P. J., Joosten, E. M., Vingerhoets, H. M., Gabreëls, F. J., et al. (1993). Hereditary neuropathy with liability to pressure palsies: a clinical, electroneurophysiological and morphological study. J. Neurol. Sci. 116 (2), 176–184. doi:10.1016/0022-510x(93)90323-q
Keywords: HNPP, genetics, diagnosis, child, demyelinating, palsy, neuropathy, hereditary
Citation: Savasta S, Serra F, Galimberti L, Comisi FF, Cossu M, Vannelli A, Masala M, Tanca S and Murru S (2025) Case Report: Hereditary neuropathy with liability to pressure palsy (HNPP): the role of genetic investigation in diagnostic assessment. Front. Genet. 16:1613022. doi: 10.3389/fgene.2025.1613022
Received: 16 April 2025; Accepted: 24 June 2025;
Published: 14 August 2025.
Edited by:
Giovanni Stevanin, Institut des Neurosciences Cognitives et Intégratives d'Aquitaine (INCIA), CNRS, FranceReviewed by:
Ahmed Bouhouche, Mohammed V University, MoroccoTakashi Matsukawa, The University of Tokyo, Japan
Copyright © 2025 Savasta, Serra, Galimberti, Comisi, Cossu, Vannelli, Masala, Tanca and Murru. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Fabrizio Comisi, ZnJhbmNlc2NvLmYuY29taXNpQGdtYWlsLmNvbQ==