 Malarmathi Muthusamy1
Malarmathi Muthusamy1 Oludayo Michael Akinsola1,2
Oludayo Michael Akinsola1,2 Pritam Pal3
Pritam Pal3 Chitra Ramasamy1Saravanan Ramasamy1
Chitra Ramasamy1Saravanan Ramasamy1 Abdulraheem Arome Musa4*Aranganoor Kannan Thiruvenkadan5
Abdulraheem Arome Musa4*Aranganoor Kannan Thiruvenkadan5- 1Department of Animal Genetics and Breeding, Veterinary College and Research Institute, TANUVAS, Namakkal, India
- 2Department of Theriogenology and Production, Faculty of Veterinary Medicine, University of Jos, Jos, Nigeria
- 3Animal Genetics and Breeding Division, Indian Council of Agricultural Research (ICAR)-National Dairy Research Institute (NDRI), Karnal, India
- 4Research Institute for Farm Animal Biology (FBN), Dummerstorf, Germany
- 5College of Poultry Production and Management, TANUVAS, Hosur, India
Background: India’s indigenous sheep breeds have evolved under extreme and diverse agro-ecological pressures, yet the genomic basis of their resilience and local adaptation remains poorly understood.
Method: This study combines genomic inbreeding estimates, runs of homozygosity (ROH), population structure analyses, and composite selection scans to investigate three native Indian breeds—Changthangi, Deccani, and Garole—within a panel of nine breeds that also includes populations from Africa (Ethiopian Menz), East and South Asia (Tibetan, Chinese Merino, Bangladesh Garole, Bangladesh East), and Europe (Suffolk).
Results: ROH and heterozygosity estimates revealed strong contrasts: Bangladesh East sheep exhibited high genomic inbreeding (FROH≈14.4%) and low observed heterozygosity (∼30.6%), whereas Deccani sheep showed low inbreeding (FROH≈1.1%) and high observed heterozygosity (∼35.6%), consistent with broader gene flow and larger flock sizes. Changthangi and Garole showed moderate inbreeding and distinct ROH length profiles. Population structure analyses confirmed ecological clustering and gene flow shaped by geography and husbandry practices: high-altitude breeds clustered together, while directional migration edges traced admixture from European Suffolk into Changthangi and from Chinese Merino into Ethiopian Menz. Historical effective population sizes showed sharp declines in most breeds, especially those under recent selection. Selection scans identified 118 significant genomic regions across breeds. In Changthangi, key pathways included purinergic signaling, thyrotropin-releasing hormone, and autophagy—consistent with cold and hypoxia adaptation. Deccani showed enrichment for immune adhesion and epidermal regeneration, reflecting parasite resistance and heat stress. Garole displayed signals for gap-junction communication and skeletal development, aligned with high fertility and compact stature.
Conclusion: These findings reveal ecotype-pecific adaptive nature shaped by polygenic selection, gene flow, and demography, offering actionable insights for sustainable smallholder breeding strategies.
1 Introduction
India sustains one of the world’s largest sheep populations, estimated at approximately 80.7 million head, producing more than 11% of the nation’s meat and nearly all of its wool (FAOSTAT, 2024; Singh, 2024). These flocks provide livelihoods to millions of rural households spread across extreme and heterogeneous agro-ecological zones, from the cold deserts of Ladakh in northern India to the saline marshlands of the Sundarbans in the east (Bhateshwar et al., 2022). Indigenous breeds adapt to these environments under traditional management practices that include nomadic pastoralism, smallholder subsistence, and occasional crossbreeding (Banerjee et al., 2011; Sridhar, 2017; Saravanan et al., 2021). Three ecotypes exemplify the range of selective pressures in India: Changthangi, Deccani, and Garole. Changthangi sheep are renowned for their smooth, fine wool—prized in the production of luxurious fabrics—and for their remarkable high-altitude adaptation in the Ladakh region, where persistent hypoxia and subzero temperatures demand exceptional metabolic and thermoregulatory efficiencies (Ganai et al., 2011; Khan et al., 2022). Deccani sheep inhabit the semi-arid Deccan Plateau, facing recurrent heat stress, fodder scarcity, and parasites, which selects for disease resistance and robust meat-producing traits (APDAI, 2015; Sridhar, 2017). Garole sheep thrive in the Sundarbans delta, where salinity levels fluctuate seasonally and environmental resources are limited, thus shaping selection for high fecundity, tolerance to salt-affected grazing, and low-input productivity (Banerjee et al., 2011; Dhar, 2011).
Although India ranks among the leading global producers of sheep and sheep-derived products, the genomic underpinnings of adaptation in these local populations remain incompletely understood. Landmark global analyses such as Kijas et al. (2012) have highlighted broad patterns of ovine diversity but offered limited resolution of how unique environments and gene flows shaped the genomes of Indian sheep specifically. More recent studies have identified signatures of selection in certain indigenous breeds (Ahmad et al., 2021; Saravanan et al., 2021), yet they often relied on narrower panels or single statistical methods such as integrated haplotype score (iHS) or cross-population extended haplotype homozygosity (XP-EHH), potentially overlooking subtle or polygenic selective pressures (Voight et al., 2006; Ma et al., 2015). In addition, historical evidence suggests that cross-border exchanges, facilitated by trade routes and migratory pastoralism, introduced key alleles for traits like disease tolerance and enhanced wool yield (Muigai and Hanotte, 2013). However, systematic genomic comparisons among Indian, neighboring Asian, and more distantly related foreign breeds—particularly in the context of inbreeding, effective population size shifts, and polygenic selection—have received less comprehensive attention.
To address these gaps, the present study expands both the breed panel and analytical toolkit. We incorporate three indigenous breeds (Changthangi, Deccani, and Garole) alongside six additional populations that represent a spectrum of global agro-ecological and breeding objectives: Bangladesh Garole and Bangladesh East for cross-border reference, Tibetan for parallel high-altitude adaptation, Chinese Merino for intensive wool selection, Ethiopian Menz for African highland production, and Suffolk for commercial meat traits. This broader sampling captures how diverse environments and breeding aims drive genetic variation, while enabling more direct inferences on whether historical gene flow from foreign or regional breeds shaped local adaptation in India (Ganai et al., 2011; Rinchen and Nazia, 2023).
Methodologically, we integrate multiple genomic inbreeding metrics (Purcell et al., 2007; Yang et al., 2011; Purfield et al., 2012; Akinsola et al., 2024), reconstruct historical effective population sizes (Barbato et al., 2015), and quantify population differentiation (
We hypothesize that smaller or more geographically isolated breeds will exhibit pronounced inbreeding and more extensive runs of homozygosity, whereas lines experiencing broader gene flow or larger effective population sizes—such as Deccani—will display lower inbreeding. We further postulate that high-altitude breeds, notably Changthangi and Tibetan, will share genomic footprints linked to cold and hypoxia tolerance, reflected by low pairwise
2 Materials and methods
2.1 Data description
Genotypic data were obtained from the Web-Interfaced Next-generation Database for Genetic Diversity Exploration (WIDDE; Sempéré et al., 2015) a publicly accessible repository that requires no further ethical approvals. The dataset encompasses 240 individuals from nine sheep breeds, eight derived from a global ovine diversity survey (Kijas et al., 2012) and the ninth breed, Suffolk, from Rochus et al. (2018). All animals were genotyped on the Illumina Ovine SNP50 BeadChip (approximately 50,000 single nucleotide polymorphisms, SNPs), ensuring uniform coverage across the genome. The chromosomal locations are based on the OAR v3.1 assembly of the ovine genome.
Three indigenous Indian populations—Changthangi (CHA,
2.2 Genotypic quality control
All genotype files were processed in PLINK v1.9 (Purcell et al., 2007). The initial PED/MAP files were converted to binary BED/BIM/FAM format using the--make-bed command. Sample-level filtering excluded any individual with a call rate below 90% (i.e., --mind 0.10), which removed two Deccani samples and left a total of 238 individuals. At the SNP level, markers with a call rate below 95% (i.e., --geno 0.05) were removed, yielding 39,685 autosomal SNPs from the original 50K array. Sex chromosomes were excluded so that analyses focused solely on autosomal variation.
Because runs of homozygosity (ROH) analysis can benefit from maximal SNP density (Meyermans et al., 2020), no further pruning for minor allele frequency (MAF), Hardy–Weinberg equilibrium (HWE), or linkage disequilibrium (LD) was conducted at this stage. The final autosomal dataset averaged one SNP per ∼61 kb, covering more than 99% of the autosomal genome. Where specialized subsets were required (e.g., for ADMIXTURE, TreeMix,
2.3 Runs of homozygosity and inbreeding coefficients
ROH were identified using PLINK, guided by small-ruminant-oriented recommendations (Meyermans et al., 2020). We set a minimum ROH length of 1,000 kb (i.e., --homozyg-kb 1,000) and allowed gaps of up to 1,000 kb (i.e., --homozyg-gap 1,000) to reduce artificial fragmentation in genomic regions with moderate SNP spacing. Each sliding window of SNPs could not contain any heterozygous calls (--homozyg-window-het 0) and could tolerate one missing call (--homozyg-window-missing 1). A density threshold of one SNP per 150 kb (i.e., --homozyg-density 150) helped maintain consistency in coverage. We applied breed-specific minimum SNP thresholds (i.e., --homozyg-snp) using the L-parameter approach to account for variation in local linkage disequilibrium. The final ROH segments were sorted into categories of 1–5, 5–10, 10–15, 15–20, and >20 Mb in length to distinguish older from more recent inbreeding events (Curik et al., 2014).
Multiple genomic inbreeding coefficients were computed to capture different facets of autozygosity.
2.4 Population structure and demographic analyses
ADMIXTURE v1.3.0 (Alexander et al., 2009) was used to investigate genomic clusters. To limit the confounding impact of LD, we created a subset of unlinked SNPs in PLINK with a sliding window of 50 SNPs, a step of 5 SNPs, and an
Historical gene flow patterns were explored using TreeMix v1.13 (Pickrell and Pritchard, 2012). We removed SNPs with more than 5% missingness, then used the--freq option in PLINK to generate the required allele-count input. We tested models allowing 0 to 4 migration edges
LD-based historical effective population size (
Population differentiation was assessed via the Weir and Cockerham (1984)
2.5 Selection scans and DCMS integration
Within each breed, we integrated multiple selection statistics into a DCMS framework (Ma et al., 2015). Haplotype-based metrics were computed on phased data produced by Beagle v5.4 (Browning et al., 2018; Browning et al., 2021).
The iHS was calculated in rehh v3.2.1 (Gautier et al., 2017) using chromosome-wise standardization, excluding SNPs with MAF <5%. The H12 statistic (Garud et al., 2015) was determined in 25-SNP windows with a 1-SNP step, while ZHp (Hofmeister et al., 2023) was calculated in 200 kb windows overlapping by 50%. The nucleotide diversity (π) and Tajima’s D were each estimated in 300 kb windows via VCFtools (Danecek et al., 2011), and windows containing fewer than 10 SNPs were excluded. iHS, H12, and π were rank-transformed for right-tailed
For DCMS, we adopted 500 kb non-overlapping windows into which these five statistics were merged. The choice of a 500 kb window was based on the need to ensure adequate SNP representation per window for robust estimation of all five statistics. Given the SNP density of the ovine array (∼1 SNP per 61 kb), this window size provides a suitable balance between genomic resolution and statistical reliability. The sample covariance matrix among iHS, H12, ZHp, π, and Tajima’s D ensured that correlated signals did not artificially inflate composite scores. The DCMS analysis was performed in R using MINOTAUR v0.0.9000 (Verity et al., 2017), and each DCMS score was compared to a normal distribution parameterized by the sample mean and standard deviation. Benjamini–Hochberg adjustment was applied to control for multiple testing (Benjamini and Hochberg, 1995), and windows with
2.6 Candidate gene annotation and functional analysis
Significant DCMS windows were extended by ±500 kb and queried against the Ensembl Representational State Transfer (REST) application programming interface (Yates et al., 2020), mapped to OAR v3.1 Gene symbols and annotations were refined using BiomaRt v2.60.1 (Durinck et al., 2009). Putative functional roles were examined with the Database for Annotation, Visualization and Integrated Discovery (DAVID; Huang et al., 2009) at a nominal
3 Results
3.1 Inbreeding, heterozygosity, and ROH
Table 1 summarizes
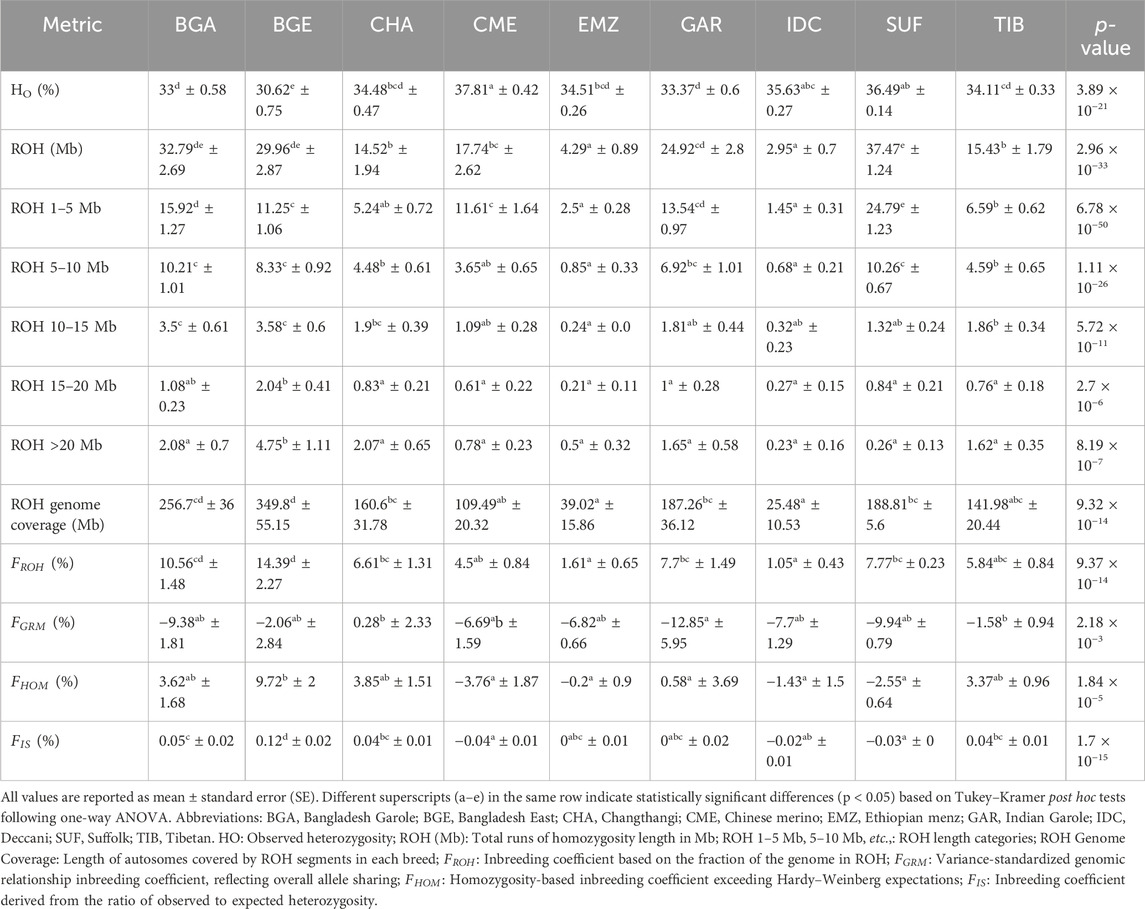
Table 1. Summary of runs of homozygosity (ROH) metrics and genomic inbreeding coefficients (mean ± se) in Indian and foreign sheep populations.
Analyses of total ROH length indicated that SUF, BGA, and BGE had the greatest ROH coverage, whereas IDC and EMZ had lower ROH totals. A large proportion of BGE’s ROH consisted of segments over 20 Mb, contributing to its elevated
Negative or near-zero inbreeding estimates arose in certain metrics. In GAR,
3.2 Population structure and demographics
Cross-validation identified
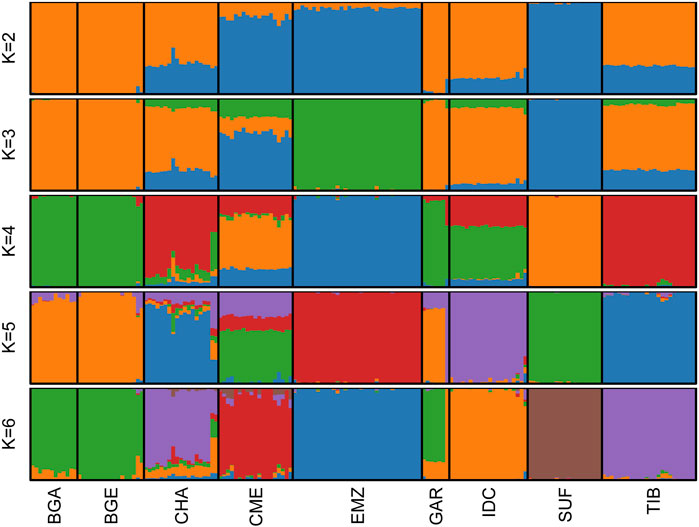
Figure 1. Population structure of Indian and foreign sheep breeds. ADMIXTURE plots showing genetic ancestry proportions at varying ancestral clusters (K = 2–6) for nine sheep populations. Each vertical bar represents a single individual, and the colors indicate ancestry fractions from inferred ancestral sources. Cross-validation analysis identified K = 6 as the optimal number of clusters (see Supplementary Figure S1). Breeds include Changthangi (CHA) from the high-altitude Ladakh region in northern India, Deccani (IDC) from the semi-arid Deccan Plateau in India, Indian Garole (GAR) from the Sundarbans delta in India, Bangladesh Garole (BGA) from southwestern Bangladesh, Bangladesh East (BGE) from eastern Bangladesh, Chinese Merino (CME) from northern China, Ethiopian Menz (EMZ) from the Ethiopian Highlands, Suffolk (SUF) from the United Kingdom, and Tibetan (TIB) from Himalayan regions of Asia.
The fully resolved
TreeMix (Figure 2) supported these findings through a four-edge migration model that minimized residual errors (Supplementary File S1). IDC appeared on the deepest branch, with no incoming gene flow. Two high-weight edges connected the reference breeds: one from SUF to CHA and another from CME to EMZ. In South Asia, BGA, BGE, and GAR formed a close cluster; a medium-weight edge ran from GAR to BGA and a weaker edge extended from the same Garole cluster towards CHA.
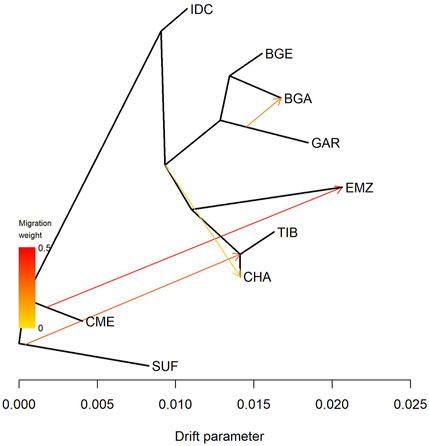
Figure 2. Maximum-likelihood phylogeny of Indian and foreign sheep breeds. This phylogeny was constructed using TreeMix with four migration edges that minimized residual errors (Supplementary File S1). Breeds include Changthangi (CHA) from the high-altitude Ladakh region in northern India, Deccani (IDC) from the semi-arid Deccan Plateau in India, Indian Garole (GAR) from the Sundarbans delta in India, Bangladesh Garole (BGA) from southwestern Bangladesh, Bangladesh East (BGE) from eastern Bangladesh, Chinese Merino (CME) from northern China, Ethiopian Menz (EMZ) from the Ethiopian Highlands, Suffolk (SUF) from the United Kingdom, and Tibetan (TIB) from Himalayan regions of Asia.
Pairwise
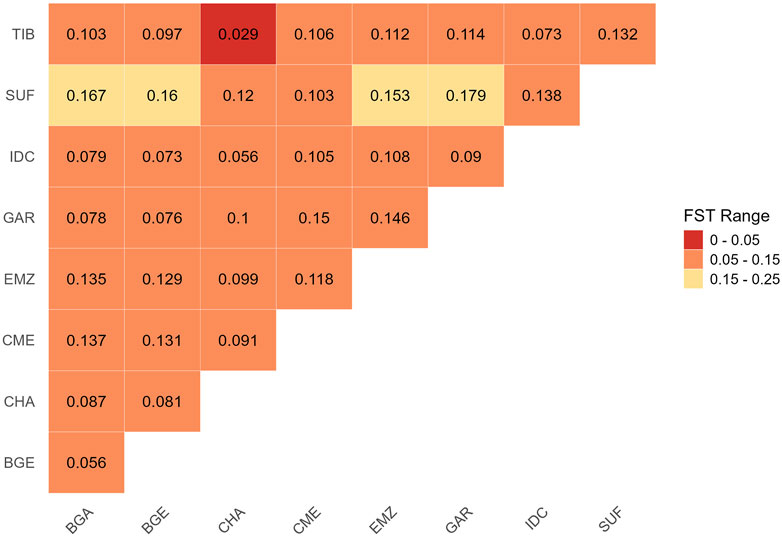
Figure 3. Pairwise genetic differentiation (
Estimates of historical
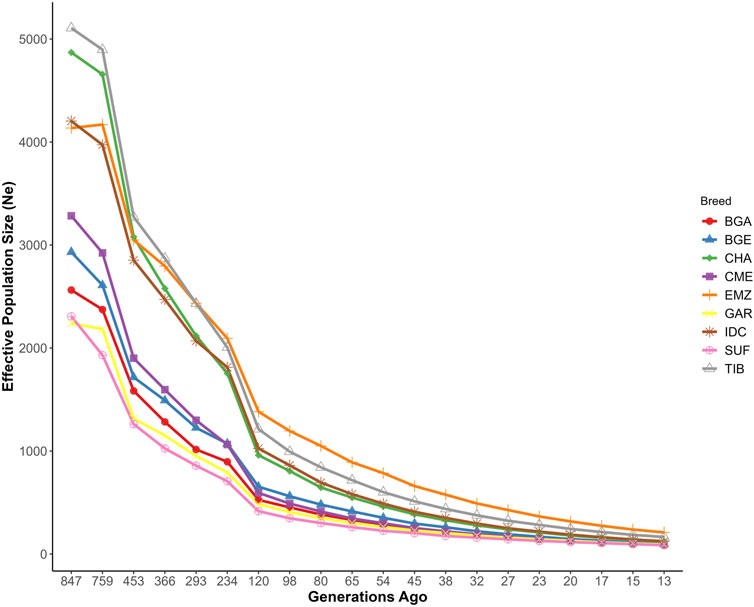
Figure 4. Historical trends in effective population size of Indian and foreign sheep breeds. Breeds include Changthangi (CHA) from the high-altitude Ladakh region in northern India, Deccani (IDC) from the semi-arid Deccan Plateau in India, Indian Garole (GAR) from the Sundarbans delta in India, Bangladesh Garole (BGA) from southwestern Bangladesh, Bangladesh East (BGE) from eastern Bangladesh, Chinese Merino (CME) from northern China, Ethiopian Menz (EMZ) from the Ethiopian Highlands, Suffolk (SUF) from the United Kingdom, and Tibetan (TIB) from Himalayan regions of Asia.
3.3 Selection signatures identified by DCMS
The DCMS analysis integrated five within-population statistics (iHS, H12, ZHp, π, and Tajima’s D), each calculated in 500 kb non-overlapping windows (Figure 5; Supplementary Table S1). In total, 118 windows surpassed the significance threshold (
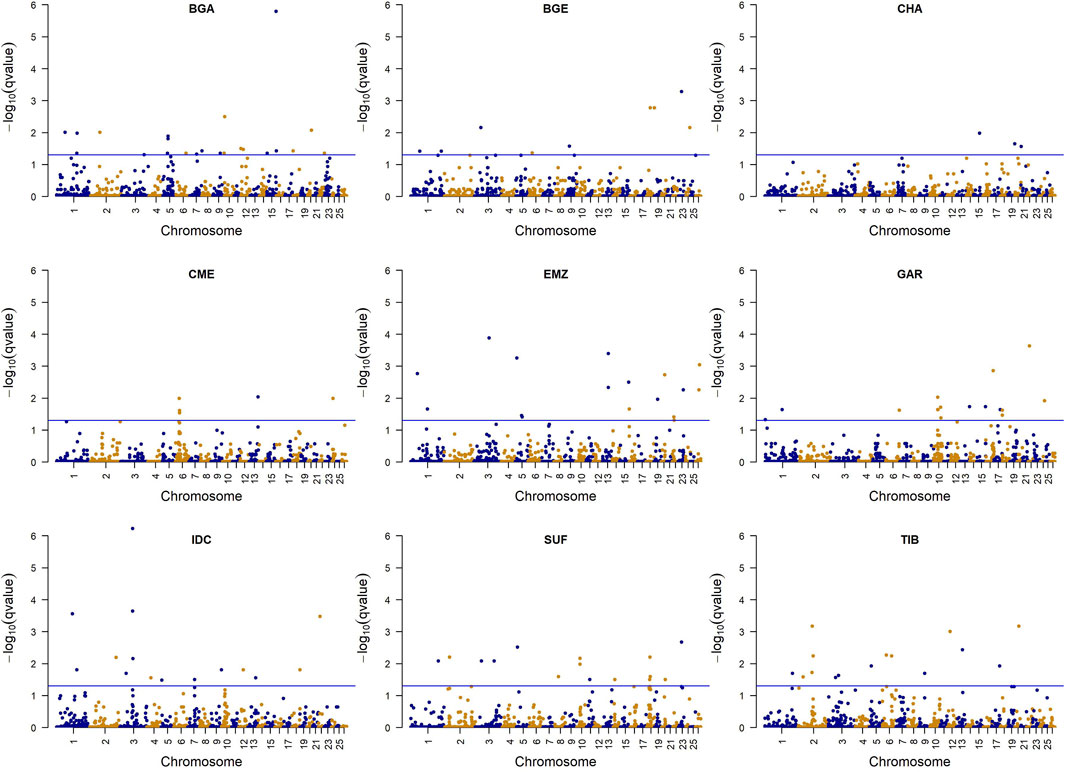
Figure 5. Manhattan plots show selection signals detected using the decorrelated composite of multiple signals for sheep breeds. The horizontal blue lines represent the false discovery rate threshold at q = 0.05. The points represent windows with varying statistical significance. Breeds include Changthangi (CHA) from the high-altitude Ladakh region in northern India, Deccani (IDC) from the semi-arid Deccan Plateau in India, Indian Garole (GAR) from the Sundarbans delta in India, Bangladesh Garole (BGA) from southwestern Bangladesh, Bangladesh East (BGE) from eastern Bangladesh, Chinese Merino (CME) from northern China, Ethiopian Menz (EMZ) from the Ethiopian Highlands, Suffolk (SUF) from the United Kingdom, and Tibetan (TIB) from Himalayan regions of Asia.
GO and KEGG pathway analysis (Supplementary Table S3) uncovered 73 enriched categories at
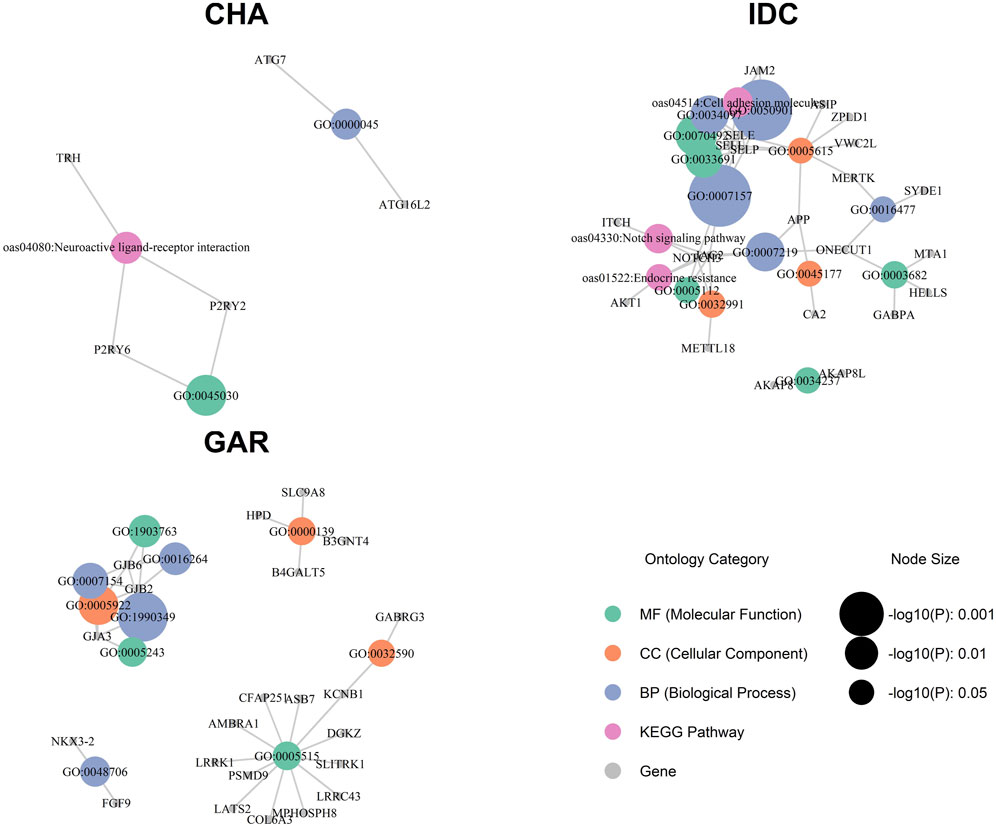
Figure 6. Gene ontology (GO) and KEGG pathway enrichment analysis for Indian sheep breeds: Changthangi (CHA), Deccani (IDC), and Indian Garole (GAR).
The foreign or reference breeds exhibited distinctive pathways (Supplementary Table S3; Supplementary File S2). BGA shared only one category with an Indian sheep, namely, “Neuroactive ligand–receptor interaction” (oas04080;
Although some pathways overlapped partially among the foreign lines, only the “Neuroactive ligand–receptor interaction” category was shared across an Indian breed (CHA) and a foreign breed (BGA). Adhesion-related processes detected in IDC, gap-junction themes in GAR, and desmosomal signals in SUF did not appear in other populations, underscoring different selection histories and adaptive pressures among these breeds. All gene-level annotations and associated p-values are detailed in Supplementary Table S3.
4 Discussion
The integration of inbreeding metrics, population structure analyses, and composite selection scans provides a multifaceted view of how ecological pressures, demographic history, and human-mediated gene flow have shaped the genomes of Indian sheep—CHA, GAR and IDC—and related breeds. The patterns observed do not point to a single axis of differentiation—such as isolation or selection—but rather reflect the interplay of multiple forces acting at different intensities across breeds and landscapes. While some breeds display hallmarks of isolation and constrained diversity, others retain clear genomic evidence of admixture and broader mating networks. In parallel, the detected selection signatures are largely polygenic, with moderate-effect loci clustering within physiological and developmental pathways, reflecting adaptation to specific stressors like hypoxia, heat, parasites, or saline foraging. These insights add depth to earlier surveys of Indian sheep (Ahmad et al., 2021; Saravanan et al., 2021) by resolving finer-scale variation in genomic structure and by revealing the physiological systems most shaped by local environments and breeding regimes.
4.1 Inbreeding and heterozygosity
The combined analysis of genomic inbreeding metrics and heterozygosity revealed contrasting genetic profiles closely tied to breed-specific management practices and ecological contexts (Table 1). The BGE sheep exhibited exceptionally high genomic inbreeding, with approximately 14.39% of their autosomal genome encompassed by extensive ROH segments exceeding 20 Mb, alongside notably low observed heterozygosity (≈30.62%). Such genomic architecture typically arises in populations maintained in small, closed flock systems or under stringent selection practices. Comparable patterns are well-documented in intensively managed or isolated breeds, including Nguni and Blackhead Persian sheep (Dzomba et al., 2021), the miniature Ouessant breed from France (Ma et al., 2025), the improved Awassi line (Getachew et al., 2020), and isolated Mozambican river buffalo (Macciotta et al., 2021). Consistent with these examples, the exceptionally small flock sizes typical of indigenous sheep populations in Bangladesh, often limited to 5–30 individuals per household (Asaduzzaman et al., 2021), substantially elevate the risk of inbreeding accumulation and associated reductions in genetic diversity.
Conversely, IDC sheep demonstrated the lowest genomic inbreeding among the studied populations, with minimal ROH coverage (
CHA and GAR populations displayed intermediate levels of genomic autozygosity and heterozygosity. The moderate genomic inbreeding observed in CHA sheep (
These distinct genomic architecture highlight the strong interplay between flock size, genetic management strategies, and ecological setting in shaping breed-specific patterns of genetic diversity. Specifically, small, isolated flocks (e.g., BGE) risk rapid genomic erosion, while larger, periodically admixed populations (e.g., IDC) effectively maintain genetic health and resilience. Intermediate scenarios, as represented by CHA and GAR sheep, underscore the delicate balance between isolation-induced adaptive specialization and necessary genetic admixture to maintain long-term viability.
4.2 Population structure and demographic patterns
Population-structure analyses based on ADMIXTURE, TreeMix, and pairwise
The TreeMix model provided complementary insights into directional gene flow patterns. IDC occupied the longest, edge-free branch, indicative of minimal recent admixture, consistent with documented selection sweeps associated with heat and drought tolerance (Saravanan et al., 2021). In contrast, the GAR population shared gene flow edges with Bangladeshi populations (BGA and BGE), supporting historical records of frequent trade in breeding rams between Indian and Bangladeshi Sundarbans communities (Banerjee et al., 2011). Additionally, moderate gene flow signals connecting SUF and CME to geographically distant breeds (e.g., CHA and EMZ) underscore historical crossbreeding efforts to introduce improved wool or meat traits, paralleling global sheep breeding trends (Kijas et al., 2012; Rochus et al., 2018; Da Silva et al., 2024).
Historical
From a breeding management perspective, these demographic and structural insights underline two critical considerations. Firstly, breeds with stable admixture levels and historical gene inflow, exemplified by IDC, maintain sufficient genetic diversity and resilience to adaptively buffer against environmental stresses. Secondly, breeds with declining effective population sizes, such as GAR and CHA, require cautious management to balance ongoing selection pressures with controlled gene inflow strategies that preserve critical adaptive genetic variants (Ganai et al., 2011). Thus, strategic genetic improvement program tailored to each breed’s demographic history and ecological context emerges as pivotal for long-term sustainability and adaptive potential.
4.3 Selection signatures and polygenic adaptation
The DCMS framework applied here revealed nuanced selection signatures across the nine breeds, supporting the hypothesis that local adaptation in Indian sheep operates through polygenic architectures rather than single, hard sweeps. This composite approach, integrating haplotype- and frequency-based metrics, was particularly effective in uncovering subtle selection pressures, which might remain undetected by isolated statistics such as iHS alone—an issue highlighted in previous studies (Ahmad et al., 2021; Saravanan et al., 2021). A complete list of all enriched GO and KEGG terms, together with the underlying genes, is provided in Supplementary Table S3.
In CHA, the most compelling DCMS outlier regions included the purinergic receptors P2RY2 and P2RY6, alongside TRH and autophagy-related genes ATG7 and ATG16L2. These loci collectively reflect a multi-scale physiological response to high-altitude stress. Purinergic signalling plays a well-characterized role in endothelial vasodilation and oxygen delivery under hypoxic conditions (Erlinge and Burnstock, 2008; Burnstock and Pelleg, 2015), while TRH is integral to thermogenic control via thyroid hormone activation, a pathway upregulated under acute cold stress in mammals (Cabral et al., 2012). Meanwhile, the autophagy-related genes are known targets of HIF-1α-mediated hypoxia response, modulating cell survival through enhanced mitochondrial recycling (Chen et al., 2012). Although “Autophagosome assembly” did not survive FDR correction, the co-location of ATG7/ATG16L2 with P2RY2/6 and TRH inside the KEGG term “Neuroactive ligand–receptor interaction” indicates that CHA has tuned an integrated sensor-effector loop: purinergic receptors sense shear-stress ATP, TRH drives thyroidal heat, and mitophagy protects mitochondrial output, collectively potentially supporting ewe mobility at extremely low temperatures and lambing at high altitudes. The co-occurrence of these pathways suggests that CHA sheep have not adapted via single major-effect loci, but rather through subtle modulation of diverse pathways coordinating vasodilation, thermogenesis, and cellular protection in a hypoxic environment. Comparable high-altitude sweeps involving ARHGEF17, a mitotic-checkpoint regulator detected by XP-EHH in the same population (Ahmad et al., 2021), lend further support to the notion that multiple cell-survival pathways are co-opted in this breed.
IDC sheep exhibited a distinct immunological and thermotolerance profile. Notably, selectin genes (SELP, SELL, SELE), junctional adhesion molecule JAM2, and Notch pathway components (NOTCH3, JAG2, APP) were prominent within enriched categories linked to leukocyte trafficking and cell–cell communication. These molecules orchestrate the rapid mobilisation of neutrophils, a response critical under chronic parasite exposure—a well-known challenge in the semi-arid Deccan Plateau (McEver and Zhu, 2010; Vestweber, 2015; Sridhar, 2017). The inclusion of APP and the ubiquitin ligase ITCH—both regulators of γ-secretase turnover—implies that IDC has fine-tuned Notch signal duration rather than merely boosting receptor copy number, an adjustment likely advantageous for repeated tick infestation cycles. The associated Notch signalling further supports keratinocyte turnover and skin repair, traits that would be valuable under conditions of intense solar radiation and ectoparasite pressure (Bray, 2016). A supplementary finding, involving AKAP8/AKAP8L, points to scaffolded activation of the PKA cascade, a central axis in mammalian heat-shock response (Pidoux and Taskén, 2010).
In addition, nominal enrichment for “chromatin binding” (HELLS, MTA1, ONECUT1, GABPA) signals an epigenetic layer of immune regulation. HELLS encodes a chromatin helicase that remodels chromatin to facilitate DNA methylation by supporting de novo DNA methyltransferase activity (Zocchi et al., 2020) and is essential for ectopic proliferation in the developing retina, suggesting that selection on HELLS may help optimize chromatin control under intense solar irradiance. The functional breadth of IDC’s response is widened by TBC1D12 and several RAB-family GTPases (RAB17/21/24/28) detected in additional nominal windows; these genes modulate vesicle traffic and cellular energy balance, traits previously associated with climate-mediated adaptation in sheep (Lv et al., 2014) and broadly relevant to intracellular adaptability and resilience in mammals (Homma et al., 2021). Together, these candidate loci and pathways suggest that IDC sheep have adapted to their environment through enhanced epithelial resilience, immune agility, and heat-responsive intracellular signalling.
GAR’s selective landscape is distinguished by its reproductive and morphological adaptations. The leading DCMS signal was driven by connexin genes (GJB2, GJA3, GJB6), which regulate intercellular metabolic cooperation in the ovary and epidermis (Goodenough and Paul, 2009). Connexins are essential for oocyte–granulosa cell communication, and their disruption impairs meiotic progression and ovulation, indicating that selection on these genes may underpin the breed’s high fecundity (Kidder and Mhawi, 2002). The same genes may also reinforce epidermal integrity, relevant in the Sundarbans’ saline and water-logged habitat. A second cluster of outliers included NKX3-2 and FGF9—genes involved in skeletal patterning and chondrocyte proliferation, respectively (Hellemans et al., 2009; Ornitz and Itoh, 2015). These loci may support the compact, lightweight conformation characteristic of GAR sheep, facilitating mobility in swampy terrain. This constellation of traits—high fertility, skin integrity, and efficient locomotion—likely constitutes an integrated adaptive strategy tailored to the Sundarbans’ extreme and fluctuating conditions. Human studies indicate that mutations in GJB2 and GJB6 cause the majority of genetic cases of non-syndromic hearing loss (Chan and Chang, 2014), underscoring the conserved physiological importance of these gap-junction genes across mammals. Parallel signatures of selection on GJB2 and GJB6 have been detected in sheep and goats adapted to arid environments (Kim et al., 2016), possibly suggesting a shared evolutionary strategy among small ruminants in response to harsh ecological conditions. Another distinct genomic region under selection in Garole sheep includes RNF17 and PARP4: RNF17 is associated with germ-cell development, potentially enhancing reproductive resilience, whereas PARP4 plays a critical role in DNA strand-break detection and repair and is notably expressed in sheep adipose tissue (Jean et al., 1999), implicating a role in oxidative stress resilience, possibly related to environmental challenges such as saline inundation. In the comparative reference populations, additional breed-specific adaptations were evident. The Ganges delta breed BGA showed signals in the neuroactive ligand–receptor pathway, particularly involving hypoxia-responsive receptors P2RX3 and APLNR, which may regulate cardiorespiratory responses and vascular perfusion during oxygen deprivation. The GABRR3, GABRG3, and prolactin-releasing peptide signals suggest potential neuroendocrine modulation of water–salt balance, possibly complementing the P2X3–apelin perfusion system under prolonged submersion scenarios. These signals likely reflect physiological responses to the periodic flooding and high humidity that typify deltaic ecosystems. In BGE, stress-related genes BAG5 and HSPB1 were associated with antioxidant and anti-apoptotic responses, a plausible adaptation to oxidative stress induced by seasonal heat and waterlogging (Kalia et al., 2004; Webster et al., 2019). The same genomic region also contains EXT1, which encodes a glycosyltransferase essential for heparan sulfate biosynthesis and is implicated in tissue morphogenesis and developmental regulation (Okada et al., 2010). Its role in extracellular matrix formation and signaling could plausibly influence follicle morphogenesis and wool fiber diameter, potentially providing a link between flood-plain nutrition and fleece quality.
CME, shaped by decades of selection for wool traits, showed FDR-significant enrichment for ECM–receptor interaction pathways involving SDC4, IBSP, and MEPE. These genes contribute to matrix mineralisation and skin-follicle anchoring (Bouleftour et al., 2014; Carneiro et al., 2014), potentially supporting high wool density and structural robustness. SUF presented a highly significant signal for the desmosome complex, with enriched cadherins (DSG1–4, DSC1–3, CDH15) known to support epidermal cohesion under shear stress and contribute to muscle fibre integrity—attributes critical for a fast-growing meat breed managed under intensive systems (Yin and Green, 2004; Garrod and Chidgey, 2008).
EMZ, from the Ethiopian highlands, demonstrated polygenic enrichment for cytoskeletal and antioxidant genes, notably DNAH9 and MSRB3. DNAH9 encodes dynein chains crucial for mucociliary clearance, while MSRB3 protects against hypoxia-induced oxidative stress (Takeuchi et al., 2021; Chandran and Binninger, 2024; Seifu et al., 2024). Additional partners (PPP2R2B, TACC1, FRMD4B) strengthen microtubule stability, further potentially supporting high-altitude endurance. These findings align with earlier reports of altitude-driven adaptation in sheep (Wei et al., 2016). In TIB, while no FDR-significant hits were found, nominal enrichment for axonal structure genes (NEFL, NEFM, SLC8A1, SNCA) implies possible selection on peripheral nerve conductivity under cold-stress conditions, consistent with the breed’s highland origins (Yuan et al., 2017). Together with signatures of selection at ATP12A, a gene implicated in trophectoderm development and possibly linked to placental efficiency in cattle (Wei et al., 2017), these loci suggest coordinated selection on reproductive and metabolic pathways in sheep inhabiting East African highland environments. Altogether, the DCMS scan across these breeds elucidates a common theme: adaptive traits in sheep are not governed by singular, easily identifiable loci but emerge from small-effect variants distributed across multiple physiological pathways. This finding reinforces the utility of composite methods in livestock genomics, particularly for dissecting complex traits such as cold tolerance, parasite resistance, and reproductive efficiency—traits critical for flock sustainability in marginal environments.
4.4 Complementarity with single-metric studies
The added value of our DCMS-based approach becomes clearer when compared directly with earlier genome-wide selection scans that relied on single metrics. Saravanan et al. (2021), for example, used iHS alone to detect candidate sweeps in Indian sheep breeds. That study yielded important insights into loci under recent directional selection but was inherently biased toward detecting strong, ongoing sweeps with long haplotypes. In contrast, our composite method integrated iHS with four additional statistics—H12, ZHp, π, and Tajima’s D—allowing it to capture not only these canonical hard sweeps but also incomplete or diffuse signals consistent with soft sweeps or polygenic adaptation (Voight et al., 2006; Ma et al., 2015).
A direct juxtaposition of the two approaches (Supplementary Table S4) demonstrates both overlap and expansion. Nearly all of the high-confidence iHS signals reported by Saravanan et al. (2021) reappear in our DCMS analysis, reaffirming their biological relevance and underscoring the robustness of our pipeline. Notably, however, DCMS identifies an additional 19 outlier windows not captured by iHS alone. These novel windows are enriched for pathways involved in immune regulation (e.g., leukocyte adhesion, Notch signalling), thermotolerance (e.g., AKAP-mediated PKA activation), and tissue homeostasis (e.g., gap-junction and desmosomal integrity). Their biological plausibility is strengthened by earlier findings in animals exposed to analogous environmental challenges (Garrod and Chidgey, 2008; McEver and Zhu, 2010).
This layered discovery reflects the theoretical strengths of DCMS. While iHS performs well under assumptions of long, unbroken haplotypes rising rapidly in frequency, it is less effective at capturing older selection events or signals arising from subtle shifts in allele frequency. ZHp and π, for instance, are particularly sensitive to reductions in heterozygosity due to long-term selection but may miss more recent signals unless paired with haplotype-based tests. Tajima’s D, meanwhile, is informative for identifying population-level deviations in allele-frequency spectrum caused by balancing or directional selection but lacks spatial resolution on its own. The H12 statistic excels at detecting soft sweeps from standing variation, especially when multiple haplotypes are under selection. By decorrelating these metrics and aggregating them in a single composite, DCMS balances their complementary strengths while mitigating redundant signals (Ma et al., 2015; Verity et al., 2017).
Our findings illustrate that composite tests are not simply additive but synergistic: they can detect functionally relevant genomic regions that remain invisible to any single approach. Particularly in livestock species like sheep, where complex traits such as reproductive performance, parasite resilience, or thermal adaptation arise from distributed genetic architectures, reliance on single metrics risks underestimating the scope and heterogeneity of adaptive evolution.
4.5 Implications for breeding, conservation, and rural livelihoods
The selection signatures revealed in this study are not just of academic interest; they point to ecotype-specific constellations of genes—adaptive nature—that underpin real-world fitness and productivity under marginal conditions. These multi-gene configurations offer a genomic blueprint for tailoring breeding strategies that preserve key local adaptations while selectively enhancing production traits.
For CHA, the ensemble of purinergic receptors (P2RY2, P2RY6), TRH, and autophagy-related genes (ATG7, ATG16L2) represents a cold-climate physiological toolkit: vasodilation ensures tissue oxygenation, TRH promotes thermogenesis, and autophagy protects cellular function during hypoxic stress. These adaptations may be critical for winter survival in the Ladakh highlands. In IDC sheep, signals linked to neutrophil tethering (SELP, SELE, SELL, JAM2), Notch-mediated epidermal homeostasis (NOTCH3), and AKAP-anchored heat-shock signaling suggest a strong selection for immune responsiveness, skin repair, and acute stress response—traits that are indispensable in semi-arid environments with high parasite loads and radiant heat. In GAR, the prominence of gap-junction genes (GJB2, GJB6, GJA3) alongside mild enrichment in skeletal morphogenesis genes (FGF9, NKX3-2) supports a profile oriented toward reproductive efficiency, physical resilience in flooded terrain, and a compact frame suited to low-input production.
Preserving these local adaptations requires careful management. Unlike hard sweeps, which often involve high-frequency haplotypes at a few loci, polygenic traits are vulnerable to erosion through indiscriminate crossbreeding. Introgression of commercial traits—e.g., Merino fleece density or Suffolk carcass yield—into these populations is not inherently detrimental, but must be accompanied by strategies to monitor and retain key indigenous haplotypes. Selective breeding programs that use low-density SNP panels centered on the 118 DCMS outlier windows could achieve this balance at low cost.
The inbreeding patterns observed offer additional management cues. Breeds like BGE, with elevated
Breeds with historically large but recently contracted effective sizes, such as CHA and IDC, may retain enough diversity to support future selection programs, provided that key adaptive blocks are not lost. Conversely, lines with small long-term
From a rural livelihoods perspective, these genomic insights translate into concrete benefits. Adaptive traits—cold resilience, parasite defense, fertility—directly affect flock survival and productivity, especially under low-input conditions where veterinary care and supplemental feeding are scarce. Genomic conservation of these traits can reduce reliance on external inputs and buffer smallholders against climatic or epidemiological shocks. Aligning these goals with India’s livestock-mission objectives may offer a pathway toward pro-poor, climate-smart genetic improvement.
5 Conclusion
This study provides a comprehensive comparative genomic analysis of indigenous Indian sheep and related breeds across South Asia, Africa, and Europe, integrating metrics of inbreeding, population structure, and composite selection signals. By uniting haplotype- and frequency-based statistics through DCMS, we captured both recent and diffuse polygenic sweeps, identifying candidate genes linked to thermoregulation, immune responsiveness, hypoxia tolerance, fecundity, and oxidative stress resilience. Our results highlight that Indian breeds are not genetically insular; rather, they are shaped by local ecological pressures as well as episodic gene flow—historical and ongoing. The study confirms that adaptive traits are governed by multiple genomic pathways and that preserving these traits will require targeted management strategies to balance productivity gains with genetic conservation. Future work incorporating high-resolution phenotypes and functional validation will be essential to translate these genomic signals into breeding indices that support climate-smart, smallholder-oriented livestock improvement.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: http://widde.toulouse.inra.fr/widde/widde/main.do?module=sheep.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with local legislation and institutional requirements because publicly available data were used.
Author contributions
MM: Formal Analysis, Writing – review and editing, Writing – original draft, Investigation, Methodology. OA: Writing – review and editing, Formal Analysis, Methodology, Writing – original draft, Investigation. PP: Writing – original draft. CR: Writing – original draft, Writing – review and editing, Investigation. SR: Writing – review and editing. AM: Software, Writing – review and editing, Formal Analysis, Methodology, Conceptualization. AT: Supervision, Writing – review and editing, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The publication of this article was funded by the Open Access Fund of the FBN.
Acknowledgments
We are grateful to the Dean, Veterinary College and Research Institute, Namakkal, Tamil Nadu, India for all the necessary help and support for this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1621960/full#supplementary-material
References
Ahmad, S. F., Mehrotra, A., Charles, S., and Ganai, N. A. (2021). Analysis of selection signatures reveals important insights into the adaptability of high-altitude Indian sheep breed Changthangi. Gene 799, 145809. doi:10.1016/j.gene.2021.145809
Akinsola, O. M., Musa, A. A., Muansangi, L., Singh, S. P., Mukherjee, S., and Mukherjee, A. (2024). Genomic insights into adaptation and inbreeding among Sub-Saharan African cattle from pastoral and agropastoral systems. Front. Genet. 15, 1430291. doi:10.3389/fgene.2024.1430291
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi:10.1101/gr.094052.109
APDAI (2015). Reviving Deccani sheep breed for climate resilience by wassan NGO - Issuu. Available online at: https://issuu.com/wassanngo/docs/reviving_deccani_sheep_breed_for_cl (Accessed February 27, 2025).
Asaduzzaman, M., Alam, M. G., Jha, P. K., and Bari, F. (2021). On-farm management, breeding practice and constraints between two sheep breeds in Bangladesh. J. Anim. Prod., 62 (1),15–24. Available online at: https://dergipark.org.tr/en/pub/hayuretim/issue/62749/767083 (Accessed April 14, 2025).
Banerjee, S., Galloway, S. M., and Davis, G. H. (2011). Distribution of prolific Garole sheep in West Bengal, India. Anim. Genet. Resour. 48, 29–35. doi:10.1017/s207863361100004x
Barbato, M., Orozco-terWengel, P., Tapio, M., and Bruford, M. W. (2015). SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 6, 109. doi:10.3389/fgene.2015.00109
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Bhateshwar, V., Rai, D. C., Datt, M., and Aparnna, V. P. (2022). Current status of sheep farming in India. J. Livest. Sci. 13, 135. doi:10.33259/jlivestsci.2022.135-151
Bouleftour, W., Boudiffa, M., Wade-Gueye, N. M., Bouët, G., Cardelli, M., Laroche, N., et al. (2014). Skeletal development of mice lacking Bone Sialoprotein (BSP) - impairment of long bone growth and progressive establishment of high trabecular bone mass. PLoS One 9, e95144. doi:10.1371/journal.pone.0095144
Bray, S. (2016). Notch signalling in context. Nat. Rev. Mol. Cell Biol. 17, 722–735. doi:10.1038/nrm.2016.94
Browning, B. L., Zhou, Y., and Browning, S. R. (2018). A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348. doi:10.1016/j.ajhg.2018.07.015
Browning, B. L., Tian, X., Zhou, Y., and Browning, S. R. (2021). Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 108, 1880–1890. doi:10.1016/j.ajhg.2021.08.005
Burnstock, G., and Pelleg, A. (2015). Cardiac purinergic signalling in health and disease. Purinergic Signal 11, 1–46. doi:10.1007/s11302-014-9436-1
Cabral, A., Valdivia, S., Reynaldo, M., Cyr, N. E., Nillni, E. A., and Perello, M. (2012). Short-term cold exposure activates TRH neurons exclusively in the hypothalamic paraventricular nucleus and raphe pallidus. Neurosci. Lett. 518, 86–91. doi:10.1016/j.neulet.2012.04.059
Carneiro, B. R., Pernambuco Filho, P. C. A., De Sousa Mesquita, A. P., Santos Da Silva, D., Pinhal, M. A. S., Nader, H. B., et al. (2014). Acquisition of anoikis resistance up-regulates syndecan-4 expression in endothelial cells. PLoS One 9, e116001. doi:10.1371/journal.pone.0116001
Chan, D. K., and Chang, K. W. (2014). GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 124, E34–E53. doi:10.1002/lary.24332
Chandran, S., and Binninger, D. (2024). Role of oxidative stress, methionine oxidation and methionine sulfoxide reductases (MSR) in alzheimer’s disease. Antioxidants 13, 21. doi:10.3390/antiox13010021
Chen, B., Longtine, M. S., and Nelson, D. M. (2012). Hypoxia induces autophagy in primary human trophoblasts. Endocrinology 153, 4946–4954. doi:10.1210/en.2012-1472
Corbin, L. J., Liu, A. Y. H., Bishop, S. C., and Woolliams, J. A. (2012). Estimation of historical effective population size using linkage disequilibria with marker data. J. Anim. Breed. Genet. 129, 257–270. doi:10.1111/j.1439-0388.2012.01003.x
Csardi, G., and Tamas, N. (2006). The igraph software package for complex network research. Inter. J. Complex Syst. 1695. 1–9. Available online at: http://igraph.org/ (Accessed April14, 2025).
Csárdi, G., Nepusz, T., Traag, V., Horvát, S., Zanini, F., Noom, D., et al. (2025). Igraph: network analysis and visualization in R. R package. version 2.1.1. Available online at: https://CRAN.R-project.org/package=igraph.
Curik, I., Ferenčaković, M., and Sölkner, J. (2014). Inbreeding and runs of homozygosity: a possible solution to an old problem. Livest. Sci. 166, 26–34. doi:10.1016/j.livsci.2014.05.034
Da Silva, A., Ahbara, A., Baazaoui, I., Jemaa, S. B., Cao, Y., Ciani, E., et al. (2024). History and genetic diversity of African sheep: contrasting phenotypic and genomic diversity. Anim. Genet. 56, e13488. doi:10.1111/age.13488
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi:10.1093/bioinformatics/btr330
Dhar, S. (2011). Impact of climate change on the salinity situation of the Piyali River, sundarbans, India. J. Water Resour. Prot. 03, 495–503. doi:10.4236/jwarp.2011.37059
Durinck, S., Spellman, P., Birney, E., and Huber, W. (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191. doi:10.1038/nprot.2009.97
Dzomba, E. F., Chimonyo, M., Pierneef, R., and Muchadeyi, F. C. (2021). Runs of homozygosity analysis of South African sheep breeds from various production systems investigated using OvineSNP50k data. BMC Genomics 22, 7. doi:10.1186/s12864-020-07314-2
Edea, Z., Dessie, T., Dadi, H., Do, K. T., and Kim, K. S. (2017). Genetic diversity and population structure of Ethiopian sheep populations revealed by high-density SNP markers. Front. Genet. 8, 218. doi:10.3389/fgene.2017.00218
Erlinge, D., and Burnstock, G. (2008). P2 receptors in cardiovascular regulation and disease. Purinergic Signal 4, 1–20. doi:10.1007/s11302-007-9078-7
FAOSTAT (2024). Food and agriculture database. Food Agric. Organ. United Nations. Available online at: https://www.fao.org/faostat/en/#data (Accessed April 14, 2025).
Felipe, M. (2023). Agricolae: statistical procedures for agricultural research. R packag. version 1.3-7. Available online at: https://cran.r-project.org/package=agricolae.
Ganai, T. A., Misra, S. S., and Sheikh, F. D. (2011). Description of Changthangi sheep of Ladakh. Indian J. Small Ruminants 17, 32–40. Available online at: https://www.cabidigitallibrary.org/doi/pdf/10.5555/20113188872 (Accessed April 14, 2025).
Garrod, D., and Chidgey, M. (2008). Desmosome structure, composition and function. Biochim. Biophys. Acta - Biomembr. 1778, 572–587. doi:10.1016/j.bbamem.2007.07.014
Garud, N. R., Messer, P. W., Buzbas, E. O., and Petrov, D. A. (2015). Recent selective sweeps in north American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 11, e1005004–e1005032. doi:10.1371/journal.pgen.1005004
Gautier, M., Klassmann, A., and Vitalis, R. (2017). Rehh 2.0: a reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol. Ecol. Resour. 17, 78–90. doi:10.1111/1755-0998.12634
Getachew, T., Haile, A., Mészáros, G., Rischkowsky, B., Huson, H. J., Gizaw, S., et al. (2020). Genetic diversity, population structure and runs of homozygosity in Ethiopian short fat-tailed and Awassi sheep breeds using genome-wide 50k SNP markers. Livest. Sci. 232, 103899. doi:10.1016/j.livsci.2019.103899
Goodenough, D. A., and Paul, D. L. (2009). Gap junctions. Cold Spring Harb. Perspect. Biol. 1, a002576. doi:10.1101/cshperspect.a002576
Hayes, B. J., Visscher, P. M., McPartlan, H. C., and Goddard, M. E. (2003). Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 13, 635–643. doi:10.1101/gr.387103
Hellemans, J., Simon, M., Dheedene, A., Alanay, Y., Mihci, E., Rifai, L., et al. (2009). Homozygous inactivating mutations in the NKX3-2 gene result in Spondylo-Megaepiphyseal-Metaphyseal dysplasia. Am. J. Hum. Genet. 85, 916–922. doi:10.1016/j.ajhg.2009.11.005
Hofmeister, R. J., Ribeiro, D. M., Rubinacci, S., and Delaneau, O. (2023). Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank. Nat. Genet. 55, 1243–1249. doi:10.1038/s41588-023-01415-w
Homma, Y., Hiragi, S., and Fukuda, M. (2021). Rab family of small GTPases: an updated view on their regulation and functions. FEBS J. 288, 36–55. doi:10.1111/febs.15453
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi:10.1038/nprot.2008.211
Jean, L., Risler, J. L., Nagase, T., Coulouarn, C., Nomura, N., and Salier, J. P. (1999). The nuclear protein PH5P of the inter-α-inhibitor superfamily: a missing link between poly(ADP-ribose)polymerase and the inter-α-inhibitor family and a novel actor of DNA repair? FEBS Lett. 446, 6–8. doi:10.1016/S0014-5793(99)00173-8
Kalia, S. K., Lee, S., Smith, P. D., Liu, L., Crocker, S. J., Thorarinsdottir, T. E., et al. (2004). BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron 44, 931–945. doi:10.1016/j.neuron.2004.11.026
Khan, N. N., Ganai, N., Ahmad, T., and Shah, R. (2022). Changthangi sheep: the pride of Ladakh. 1, 49–51. doi:10.5281/zenodo.6535189
Kidder, G. M., and Mhawi, A. A. (2002). Gap junctions and ovarian folliculogenesis. Reproduction 123, 613–620. doi:10.1530/rep.0.1230613
Kijas, J. W., Lenstra, J. A., Hayes, B., Boitard, S., Neto, L. R., Cristobal, M. S., et al. (2012). Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 10, e1001258. doi:10.1371/journal.pbio.1001258
Kim, E. S., Elbeltagy, A. R., Aboul-Naga, A. M., Rischkowsky, B., Sayre, B., Mwacharo, J. M., et al. (2016). Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity (Edinb) 116, 255–264. doi:10.1038/hdy.2015.94
Kizilaslan, M., Arzik, Y., Behrem, S., White, S. N., and Cinar, M. U. (2024). Comparative genomic characterization of indigenous fat-tailed Akkaraman sheep with local and transboundary sheep breeds. Food Energy Secur 13, e508. doi:10.1002/fes3.508
Liu, S., He, S., Chen, L., Li, W., Di, J., and Liu, M. (2017). Estimates of linkage disequilibrium and effective population sizes in Chinese Merino (Xinjiang type) sheep by genome-wide SNPs. Genes Genomics 39, 733–745. doi:10.1007/s13258-017-0539-2
Lv, F. H., Agha, S., Kantanen, J., Colli, L., Stucki, S., Kijas, J. W., et al. (2014). Adaptations to climate-mediated selective pressures in sheep. Mol. Biol. Evol. 31, 3324–3343. doi:10.1093/molbev/msu264
Lv, F. H., Cao, Y. H., Liu, G. J., Luo, L. Y., Lu, R., Liu, M. J., et al. (2022). Whole-genome resequencing of worldwide wild and domestic sheep elucidates genetic diversity, introgression, and agronomically important loci. Mol. Biol. Evol. 39, msab353. doi:10.1093/molbev/msab353
Ma, Y., Ding, X., Qanbari, S., Weigend, S., Zhang, Q., and Simianer, H. (2015). Properties of different selection signature statistics and a new strategy for combining them. Heredity (Edinb) 115, 426–436. doi:10.1038/hdy.2015.42
Ma, R., Liu, J., Ma, X., and Yang, J. (2025). Genome-wide runs of homozygosity reveal inbreeding levels and trait-associated candidate genes in diverse sheep breeds. Genes (Basel) 16, 316. doi:10.3390/genes16030316
Macciotta, N. P. P., Colli, L., Cesarani, A., Ajmone-Marsan, P., Low, W. Y., Tearle, R., et al. (2021). The distribution of runs of homozygosity in the genome of river and swamp buffaloes reveals a history of adaptation, migration and crossbred events. Genet. Sel. Evol. 53, 20. doi:10.1186/s12711-021-00616-3
McEver, R. P., and Zhu, C. (2010). Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 26, 363–396. doi:10.1146/annurev.cellbio.042308.113238
McQuillan, R., Leutenegger, A.-L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 658. doi:10.1016/j.ajhg.2008.10.009
Meyermans, R., Gorssen, W., Buys, N., and Janssens, S. (2020). How to study runs of homozygosity using plink? A guide for analyzing medium density snp data in livestock and pet species. BMC Genomics 21, 94. doi:10.1186/s12864-020-6463-x
Muigai, A. W. T., and Hanotte, O. (2013). The origin of African sheep: archaeological and genetic perspectives. Afr. Archaeol. Rev. 30, 39–50. doi:10.1007/s10437-013-9129-0
Nimbkar, C., Ghalsasi, P. M., Walkden-Brown, S. W., and Van Der Werf, J. H. J. (2023). FecB carrier Deccani cross bred ewes in Maharashtra, India have moderately higher litter sizes than non-carrier ewes. Proc. Assoc. Advmt. Anim. Breed. Genet. 25, 138–141. Available online at: https://hdl.handle.net/1959.11/56098 (Accessed April 14, 2025).
Ohta, T., and Kimura, M. (1971). On the constancy of the evolutionary rate of cistrons. J. Mol. Evol. 1, 18–25. doi:10.1007/BF01659391
Okada, M., Nadanaka, S., Shoji, N., Tamura, J. I., and Kitagawa, H. (2010). Biosynthesis of heparan sulfate in EXT1-deficient cells. Biochem. J. 428, 463–471. doi:10.1042/BJ20100101
Ornitz, D. M., and Itoh, N. (2015). The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266. doi:10.1002/wdev.176
Pickrell, J. K., and Pritchard, J. K. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967. doi:10.1371/journal.pgen.1002967
Pidoux, G., and Taskén, K. (2010). Specificity and spatial dynamics of protein kinase a signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 44, 271–284. doi:10.1677/JME-10-0010
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi:10.1086/519795
Purfield, D. C., Berry, D. P., McParland, S., and Bradley, D. G. (2012). Runs of homozygosity and population history in cattle. BMC Genet. 13, 70. doi:10.1186/1471-2156-13-70
R Core Team (2024). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available online at: http://www.r-project.org
Rinchen, D., and Nazia, J. (2023). Significance of wool trade in the political and economical development of Ladakh. Int. J. Creat. Res. Thoughts, 11 (5), M425–433. Available online at: https://ijcrt.org/papers/IJCRT23A5467.pdf (Accessed April 14, 2025).
Rochus, C. M., Tortereau, F., Plisson-Petit, F., Restoux, G., Moreno-Romieux, C., Tosser-Klopp, G., et al. (2018). Revealing the selection history of adaptive loci using genome-wide scans for selection: an example from domestic sheep. BMC Genomics 19, 71. doi:10.1186/s12864-018-4447-x
Saravanan, K. A., Panigrahi, M., Kumar, H., Bhushan, B., Dutt, T., and Mishra, B. P. (2021). Genome-wide analysis of genetic diversity and selection signatures in three Indian sheep breeds. Livest. Sci. 243, 104367. doi:10.1016/j.livsci.2020.104367
Seifu, W. D., Bekele-Alemu, A., and Zeng, C. (2024). Genomic and physiological mechanisms of high-altitude adaptation in Ethiopian highlanders: a comparative perspective. Front. Genet. 15, 1510932. doi:10.3389/fgene.2024.1510932
Sempéré, G., Moazami-Goudarzi, K., Eggen, A., Laloë, D., Gautier, M., and Flori, L. (2015). WIDDE: a web-interfaced next generation database for genetic diversity exploration, with a first application in cattle. BMC Genomics 16, 940. doi:10.1186/s12864-015-2181-1
Shi, L., Wang, L., Liu, J., Deng, T., Yan, H., Zhang, L., et al. (2020). Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a large white pig population. J. Anim. Sci. Biotechnol. 11, 46. doi:10.1186/s40104-020-00447-0
Singh, A. (2024). Livestock production statistics of India - 2024. Vet. Extensions. doi:10.13140/RG.2.2.32034.86721
Sridhar, X. (2017). Study on temporal changes of Deccani sheep rearing in mahbubnagar district of Telangana state. India: P.V. Narsimha Rao Telangana Veterinary University. Available online at: http://krishikosh.egranth.ac.in/handle/1/5810021223.
Sved, J. A., and Feldman, M. W. (1973). Correlation and probability methods for one and two loci. Theor. Popul. Biol. 4, 129–132. doi:10.1016/0040-5809(73)90008-7
Takeuchi, K., Xu, Y., Ogawa, S., Ikejiri, M., Nakatani, K., Gotoh, S., et al. (2021). A pediatric case of productive cough caused by novel variants in DNAH9. Hum. Genome Var. 8, 3. doi:10.1038/s41439-020-00134-6
Verity, R., Collins, C., Card, D. C., Schaal, S. M., Wang, L., and Lotterhos, K. E. (2017). Minotaur: a platform for the analysis and visualization of multivariate results from genome scans with R Shiny. Mol. Ecol. Resour. 17, 33–43. doi:10.1111/1755-0998.12579
Vestweber, D. (2015). How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 15, 692–704. doi:10.1038/nri3908
Voight, B. F., Kudaravalli, S., Wen, X., and Pritchard, J. K. (2006). A map of recent positive selection in the human genome. PLoS Biol. 4, e72–e458. doi:10.1371/journal.pbio.0040072
Webster, J. M., Darling, A. L., Uversky, V. N., and Blair, L. J. (2019). Small heat shock proteins, big impact on protein aggregation in neurodegenerative disease. Front. Pharmacol. 10, 1047. doi:10.3389/fphar.2019.01047
Wei, C., Wang, H., Liu, G., Zhao, F., Kijas, J. W., Ma, Y., et al. (2016). Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 6, 26770. doi:10.1038/srep26770
Wei, Q., Zhong, L., Zhang, S., Mu, H., Xiang, J., Yue, L., et al. (2017). Bovine lineage specification revealed by single-cell gene expression analysis from zygote to blastocyst. Biol. Reprod. 97, 5–17. doi:10.1093/biolre/iox071
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population-structure. Evolution (N. Y) 38, 1358–1370. doi:10.1111/j.1558-5646.1984.tb05657.x
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi:10.1016/j.ajhg.2010.11.011
Yates, A. D., Achuthan, P., Akanni, W., Allen, J., Allen, J., Alvarez-Jarreta, J., et al. (2020). Ensembl 2020. Nucleic Acids Res. 48, D682–D688. doi:10.1093/nar/gkz966
Yin, T., and Green, K. J. (2004). Regulation of desmosome assembly and adhesion. Semin. Cell Dev. Biol. 15, 665–677. doi:10.1016/j.semcdb.2004.09.005
Yuan, A., Rao, M. V., and Nixon, R. A. (2017). Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 9, a018309. doi:10.1101/cshperspect.a018309
Keywords: indigenous sheep, genomic inbreeding, runs of homozygosity (ROH), selection signatures, polygenic adaptation, population structure, effective population size, livestock conservation genetics
Citation: Muthusamy M, Akinsola OM, Pal P, Ramasamy C, Ramasamy S, Musa AA and Thiruvenkadan AK (2025) Comparative genomic insights into adaptation, selection signatures, and population dynamics in indigenous Indian sheep and foreign breeds. Front. Genet. 16:1621960. doi: 10.3389/fgene.2025.1621960
Received: 02 May 2025; Accepted: 29 July 2025;
Published: 21 August 2025.
Edited by:
Albano Beja-Pereira, University of Porto, PortugalReviewed by:
Gregorio Miguel Ferreira De Camargo, Federal University of Bahia (UFBA), BrazilMohammed Ali Al Abri, Sultan Qaboos University, Oman
Copyright © 2025 Muthusamy, Akinsola, Pal, Ramasamy, Ramasamy, Musa and Thiruvenkadan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdulraheem Arome Musa, bXVzYUBmYm4tZHVtbWVyc3RvcmYuZGU=