 Chunrong Gui1,2†Zifeng Cheng3†Yongsheng Chen4†Yunting Ma3Hongfei Chen3Wei Wei1,2
Chunrong Gui1,2†Zifeng Cheng3†Yongsheng Chen4†Yunting Ma3Hongfei Chen3Wei Wei1,2 Xianda Wei1,2Juliang Liu1,2
Xianda Wei1,2Juliang Liu1,2 Xu Zhou3Qianqian Du5Yinghui Lai4*
Xu Zhou3Qianqian Du5Yinghui Lai4* Baoheng Gui1,2,3*
Baoheng Gui1,2,3*- 1Center for Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
- 2The Guangxi Health Commission Key Laboratory of Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
- 3The Second School of Medicine, Guangxi Medical University, Nanning, China
- 4Department of Hematology, The Second Affiliated Hospital of Guangxi Medical University, Nanning, China
- 5Berry Genomics Corporation, Beijing, China
Background: Hemoglobinopathies are a group of autosomal recessive disorders characterized by a high degree of clinical and genetic heterogeneity. Comprehensive genetic screening for hemoglobin variants is crucial for prevention and treatment of these conditions. Single-molecule real-time (SMRT) sequencing enables efficient and reliable analysis of common and complex or rare hemoglobin variants.
Methods: We launched a population-based genetic screening program for hemoglobinopathies in Guangxi, China, using SMRT. The in silico structural predictions based on Alphafold2 were performed for the rare variants identified. Additionally, a comprehensive literature review was conducted to elucidate the origin and genotype-phenotype correlation of these variants.
Results: A total of 11,019 participants throughout Guangxi were recruited via the screening program. In two unrelated families, the variants, Hb O-Arab and Hb D-Punjab at the same genetic locus, were identified with an extremely low frequency of 0.0045% [1/(11,019*2), respectively] in the population. Structural prediction showed Hb O-Arab exerted a relatively significant impact on the hemoglobin structure, whereas the influence of Hb D-Punjab was minimal. This was consistent with findings from the literature review and the two recruited families, which confirmed that individuals with Hb O-Arab presented relatively obvious manifestations compared to those with Hb D-Punjab.
Conclusion: Two rare variants, Hb O-Arab and Hb D-Punjab, were identified in Guangxi, China using SMRT. The first report of Hb O-Arab enriches the spectrum of hemoglobin variants in the Chinese population. Analyzing the frequency, origin and genotype-phenotype correlation of these variants could pave the way for clinical management and genetic counseling for hemoglobinopathies.
Introduction
Hemoglobinopathies are a group of inherited blood disorders that are classified as autosomal recessive genetic hemolytic anemias. These conditions stem from a variety of hemoglobin (Hb) variants, which often result from mutations or deletions within the genes encoding the α- or β-globin chains of hemoglobin. The genetic alterations can lead to either a reduced production of these globin chains or structural changes in the hemoglobin molecule itself. When there is a reduced production of these globin chains, the resulting conditions are collectively known as thalassemia syndromes. Structural changes in hemoglobin cause abnormal hemoglobin, such as Hb S, Hb E, Hb O, and Hb D (Harteveld et al., 2022). Globally, there are approximately 350 million carriers of thalassemia, and over 300,000 newborns are affected by hemoglobinopathies, such as sickle cell anemia or thalassemia, etc. each year (Modell et al., 2008; Weatherall, 2010). Southern China has a particularly high prevalence of thalassemia, with Guangxi Province being a hotspot, where the prevalence rate approaches 20% (Lai et al., 2017). The clinical manifestation of hemoglobinopathies varies widely, from asymptomatic carriers to severe, life-threatening conditions (Kohne, 2011). Heterozygous carriers of these variants are typically asymptomatic but may exhibit hematological characteristics. However, homozygous or compound heterozygous states can result in clinically significant phenotypes with varying degrees of severity, such as thalassemia major, thalassemia intermedia, sickle cell syndrome, and Hb E syndrome.
Hemoglobinopathies are characterized by a remarkable level of genetic diversity. To navigate this genetic landscape, specialized databases like HbVAR and ithanet have been created to document and manage numerous genetic variations associated with hemoglobinopathies (Giardine et al., 2021; Kountouris et al., 2014). The HbVAR database, in particular, has amassed an extensive catalog of over 1800 distinct variants involving hemoglobin gene cluster, including HBA1, HBA2, and HBB (Giardine et al., 2021). Among these variants, changes at the 122nd codon of the β-globin gene, specifically c.364G>A [p.(Glu122Lys)] and c.364G>C [p.(Glu122Gln)], result in Hb O-Arab and Hb D-Punjab, respectively (van Gammeren et al., 2020). These distinct amino acid substitutions lead to different clinical manifestations and effects on red blood cell morphology and function. Both variants have a global distribution, with Hb D-Punjab being more prevalent in regions such as Punjab, India, and also found in Italy, Belgium, Austria, and Turkey (Torres et al., 2015). Hb O-Arab is found in people from the Middle East, and the Mediterranean (Elbashir and Elsayed Yousif, 2023).
Given the high genetic heterogeneity and the complexity of variations in hemoglobinopathies, there is an urgent need for an efficient and reliable method for screening and diagnosing of the hemoglobinopathies. Recently, a cutting-edge approach based on single-molecule real-time (SMRT) sequencing targeting the hemoglobin gene cluster has emerged (Xu et al., 2020). Benefiting from its long-read sequencing, the SMRT method comprehensively encompasses the full spectrum of known structural variations, single nucleotide variants (SNVs), and insertions/deletions (InDels) involving the HBA1, HBA2, and HBB gene clusters. Rigorous retrospective and prospective multi-center cohort analyses have demonstrated its efficiency and reliability in identifying and analyzing the common and even complex or rare hemoglobin variations, enabling accurate diagnosis and comprehensive understanding of hemoglobinopathies (Xu et al., 2020; Liang et al., 2021).
In this study, we launched a population-based genetic screening initiative for hemoglobinopathies in Guangxi, China, using the SMRT sequencing. Two rare hemoglobin variants, Hb O-Arab and Hb D-Punjab, were identified, whose impact on the structure of the hemoglobin were predicted. Furthermore, we conducted a literature review to analyse the origins and genotype-phenotype correlations of the two variants.
Materials and methods
Population and subjects
A population-based genetic screening project for hemoglobinopathies was conducted throughout Guangxi Zhuang Autonomous Region, China, spanning from July 2021 to December 2024. Physical examination and clinical assessment were performed for the subjects and their peripheral blood samples were collected for further hematological screening and genetic analysis.
Hematological screening
Routine hematological indicators were measured by automatic hematological analyzer (LH780, Beckman Co., Nanning, China or BC-6000, Mindray Co., Nanning, China), and standard hemoglobin testing was performed by automatic high-pressure liquid-flow capillary electrophoresis (CAPILLARYS2, Sebia Co., Nanning, China), according to the manufacturer’s instructions. Normal reference ranges of the hematological indicators included mean corpuscular volume (MCV)≥82 fL, mean corpuscular Hb (MCH) ≥27 pg, Hb A2 levels between 2.4% and 3.5%, and Hb F ≤ 2%.
Genetic screening by the SMRT sequencing
The SMRT sequencing was conducted as previously described (Xu et al., 2020; Liang et al., 2021). Briefly, genomic DNA was extracted from peripheral blood and subjected to multiple long range PCR to amplify the hemoglobin gene cluster including the HBA1, HBA2, HBB, etc. The amplified products were input for library preparation and subsequent sequencing on a PacBio Sequel II SMRT sequencer (Pacific Biosciences Inc., Menlo Park, United States), following the manufacturer’s instructions. The generated raw subreads were subsequently processed using circular consensus sequencing software (RRID: SCR_021174, Pacific Biosciences Inc., Menlo Park, United States) and the Pbbioconda package (Pacific Biosciences Inc., Menlo Park, United States) to obtain circular consensus sequencing reads, which were mapped to the GRCh38 reference genome and further used for variant calling using FreeBayes1.3.4 (RRID: SCR_010761, https://www.geneious.com/plugins/freebayes). The pathogenicity of the candidate variants was classified according to the ACMG/AMP guidelines (Richards et al., 2015) and information documented in hemoglobin variant databases, such as HbVar (https://globin.bx.psu.edu/hbvar/), ithanet (https://www.ithanet.eu/), and LOVD (https://www.lovd.nl/).
Validation of the candidate variants identified by the SMRT sequencing
Sanger sequencing was employed to verify SNVs and InDels, followed by locus-specific amplification. Multiplex ligation-dependent probe amplification (MRC Holland, Amsterdam, Netherlands) was conducted to confirm structural rearrangements, including large deletions or duplications within specific hemoglobin gene regions. Additionally, gap polymerase chain reaction (Gap-PCR) (Yilifang Bio, Shenzhen, China) was utilized to detect hotspot deletions in the Chinese population, targeting--SEA, -α3.7, -α4.2, and--THAI, according to the manufacturer’s protocol. A sample containing the known heterozygous -α3.7 variant was used as the positive control. Meanwhile, a sample without any known HBA1 or HBA2 variants, as well as nuclease-free water, were used as the negative and blank controls, respectively. The DNA marker was included in the kit, and an amplification fragment of 1.7 kb indicated the presence of the internal control sequence.
Structural prediction and visualization
The hemoglobin structures of the wild-type and mutant forms, including Hb O-Arab and Hb D-Punjab, were predicted using the ColabFold v1.5.5 (RRID: SCR_025453, https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb), an online platform for AlphaFold2. The predicted local distance difference test (pLDDT) score above 70 was considered with high confidence. All models were visualized using PyMOL (RRID: SCR_000305), with hydrogen bonds displayed using the default settings.
Retrospective analysis of similar cases reported in the literature
The literature search was performed to summarize the hematological features and clinical symptoms of individuals with the rare variants, Hb O-Arab and Hb D-Punjab. The literature search was conducted in the PubMed, Web of Science, and National Center for Biotechnology Information (NCBI) databases through keywords “hemoglobin O Arab”, “Hemoglobin O Arab”, “Hb O-Arab”, “hemoglobin D-Punjab”, “Hemoglobin D-Punjab”, “Hb D-Punjab”. Titles and abstracts selected from the initial search were first scanned, and the full papers of potentially eligible studies were reviewed. Articles were excluded for the following reasons: 1) the articles were not in English; 2) the full version of the articles were not available; 3) the articles did not report the hematological characteristics and clinical manifestations.
Results
A total of 11,019 participants from throughout Guangxi were recruited via the genetic screening program for hemoglobinopathies, with ages ranged from the neonatal period to 86 years old, including 2,673 children and adolescents, and 8,346 adults. Among these participants, there were 5,310 males and 5,709 females. Totally, 165 hemoglobin variants were identified, of which 83 were variants in the HBB gene. In two unrelated families, the hemoglobin variants, Hb O-Arab and Hb D-Punjab at the same genetic locus, were identified with an extremely low frequency of 0.0045% [1/(11,019*2), respectively] among the screened population.
A rare compound heterozygous variant Hb O-Arab and β0-thalassemia in family 1
The proband in Family 1, an 18-year-old Chinese man, visited our hospital with complaints of increased bilirubin, icteric sclera, and skin, but without subcutaneous bleeding, which indicated possible hemolytic anemia. The SMRT sequencing detected substitutions at codons 17 [NM_000518.5(HBB): c.52A>T p.(Lys18Ter)] and 122 [NM_000518.5(HBB): c.364G>A p.(Glu122Lys)] of the HBB gene simultaneously (Figure 1A). These two variants were located in trans and presented in a compound heterozygous pattern. Family analysis showed that the variant c.52A>T was from the father and c.364G>A was from the mother (Figures 1B,C, 2C). Additionally, a hotspot heterozygous -α3.7 deletion (chr16: g.34164_37967del3804) was identified in the father (Figure 1B). The variants c. 52A>T and c. 364G>A were further confirmed by Sanger sequencing (Figure 2A) and the -α3.7 was validated by Gap-PCR (Figure 2B).
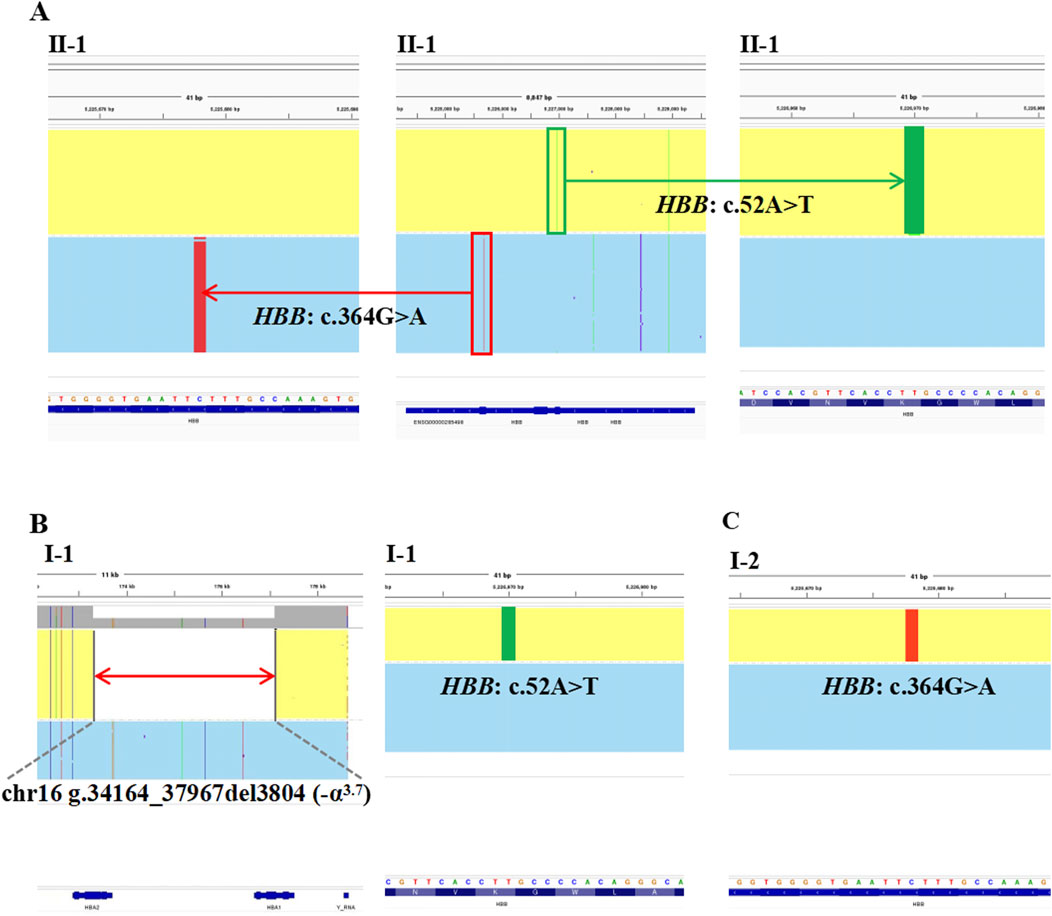
Figure 1. The variants identified by SMRT in Family 1. The integrative genomics viewer plots of the variants detected by SMRT in Family 1. (A) The variants, HBB: c.364G>A, co-occurring with HBB: c.52A>T, in II-1. (B) The deletion, chr16: g.34164_37967del3804 (−α3.7) and the variant, HBB: c.52A>T, in I-1. (C) The variant, HBB: c.364G>A, in I-2.
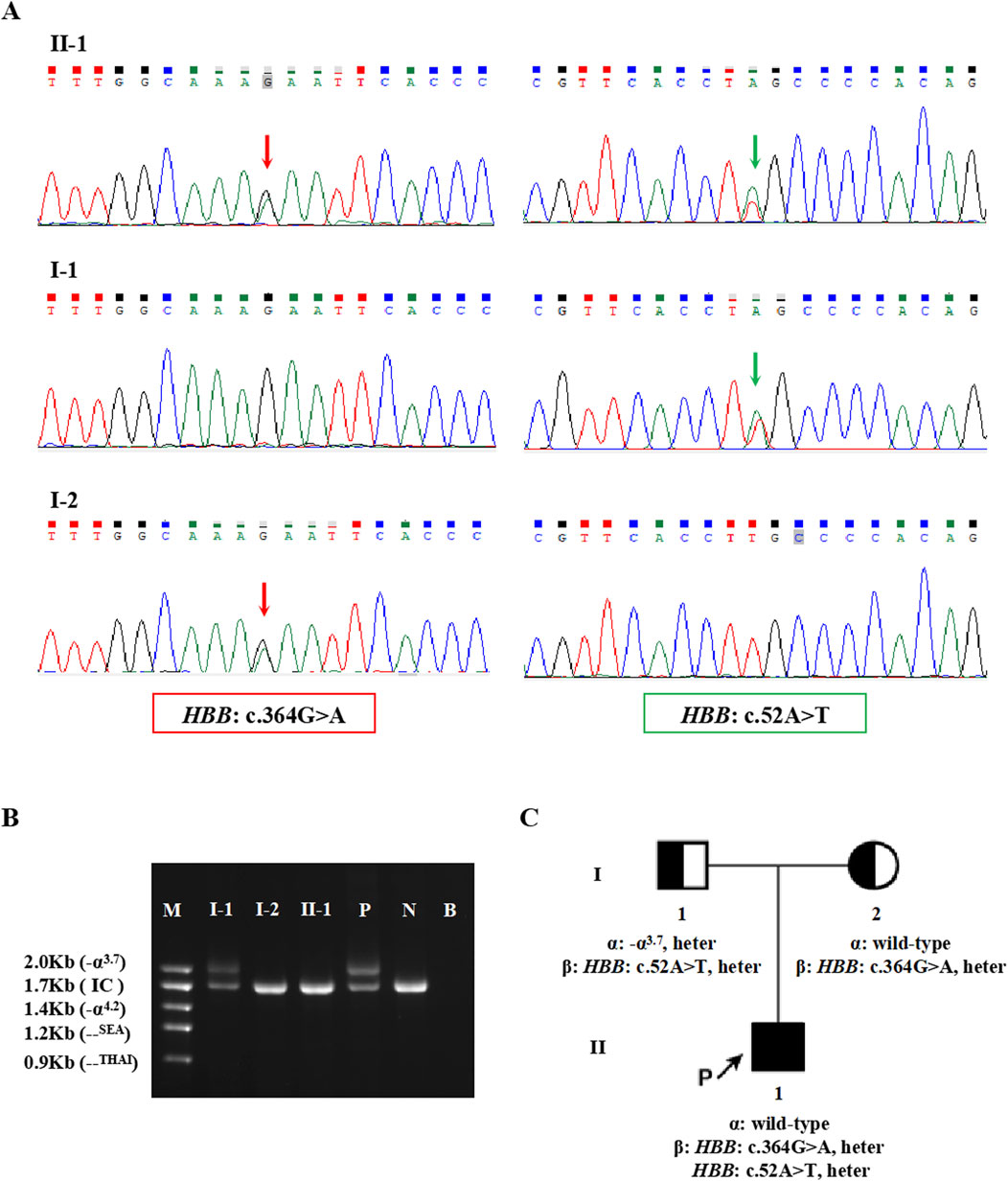
Figure 2. The variants validated by conventional approaches in Family 1. (A) The results of Sanger sequencing in Family 1. The red arrows indicate the variant, HBB: c.364G>A, and the green arrows indicate the variant, HBB: c.52A>T. (B) Agarose gel electrophoresis analysis for the Gap-PCR products in Family 1. M: DNA marker, P: positive control, N: negative control, B: blank control, IC: internal control. (C) Pedigree chart of Family 1.
Among the two variants, c.52A>T also called CD17 (A>T) could cause β0-thalassemia, and c.364G>A also known as Hb O-Arab could alter the structure of hemoglobin. Thus, the heterozygosity of these two variants may explain the proband’s phenotype. This was confirmed by the routine hematological analysis, which showed significantly decreased MCV and MCH indicating microcytic hypochromic anemia (Table 1), together with abnormal hemoglobin presenting as an overlapping peak of Hb A2 and Hb O-Arab (92.5%), an Hb F peak (6.4%) and an uncharacterized Hb X peak (1.1%), but without the Hb A peak (Figure 3A). The parents also exhibited relatively low MCV and MCH levels (Table 1). An abnormal overlapping peak of Hb A2 and Hb O-Arab was also observed in his mother, while a mild increase in Hb A2 (5.4%) was detected in his father (Figures 3B,C).
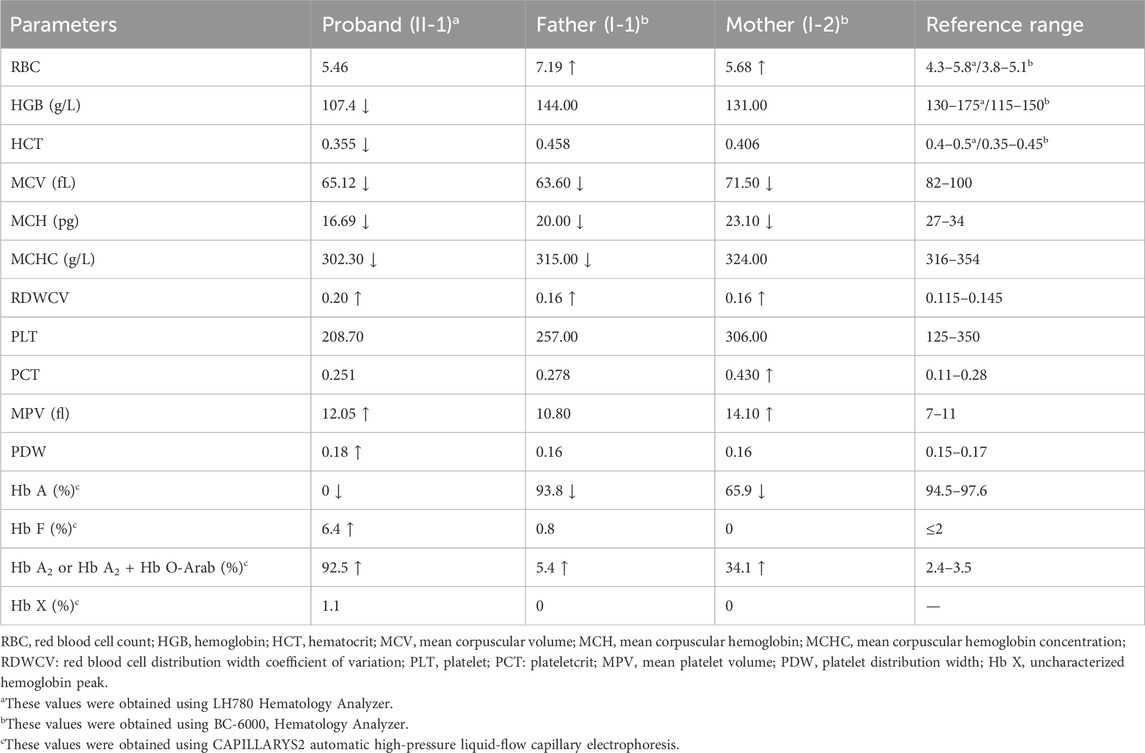
Table 1. Hematological and electrophoretic characteristics in Family 1.
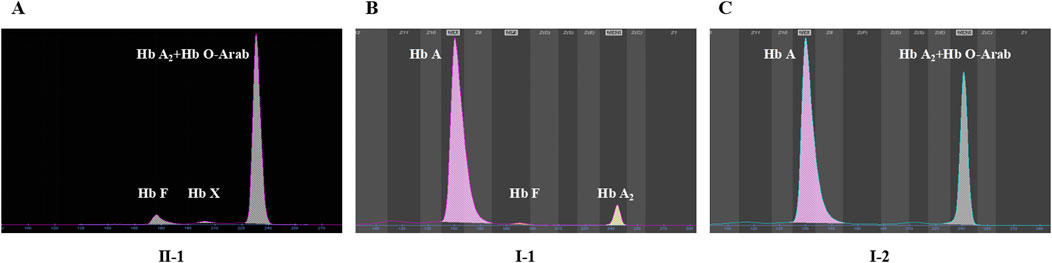
Figure 3. The capillary electrophoresis analysis of hemoglobin (Hb) in Family 1. (A–C) show the electrophoresis results of the proband (II-1), and his father (I-1) and mother (I-2) in Family 1, respectively. Specific hemoglobin peaks for Hb A, Hb A2, Hb F, and Hb O-Arab are displayed. Hb X: uncharacterized hemoglobin peak.
A rare heterozygous variant Hb D-Punjab in family 2
The proband in Family 2 was a 4 years old boy and participated in the genetic screening program for hemoglobinopathies. A heterozygous variant, NM_000518.5(HBB): c.364G>C p.(Glu122Gln), also called Hb D-Punjab, was identified in the proband by the SMRT sequencing (Figure 4A) and was validated by Sanger sequencing (Figure 4B). It was a rare Hb variant at the same genetic locus as the Hb O-Arab. Unfortunately, the hematological results of the proband were unavailable and the family refused to blood resampling for further analysis and validation.
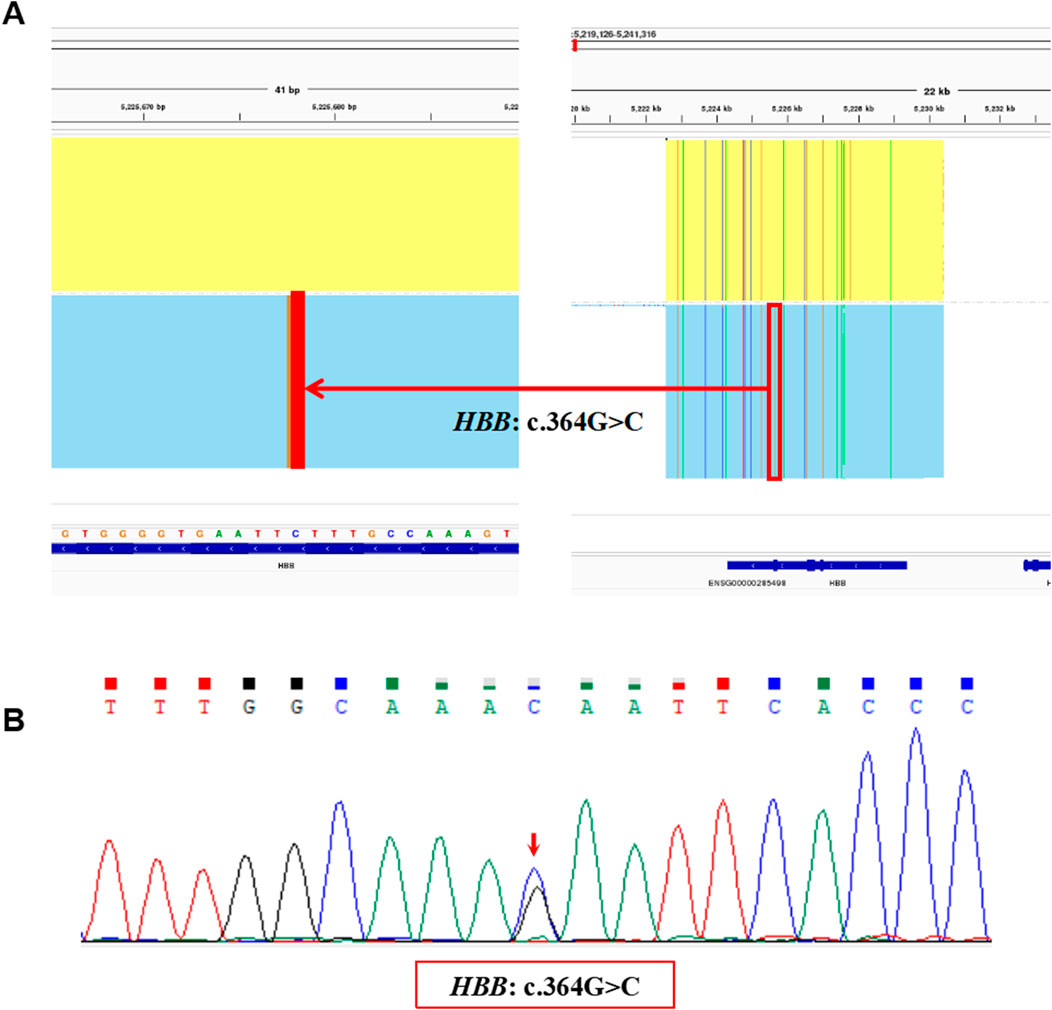
Figure 4. The variants identified by SMRT and validated by Sanger sequencing in Family 2. (A) The integrative genomics viewer plot of the variant, HBB: c.364G>C, detected by SMRT in Family 2. (B) Sequentially, Sanger sequencing confirmed the presence of the variant, HBB: c.364G>C indicated by the red arrow.
Structural prediction and alterations of hydrogen bonds
The pLDDT scores of all the structures were above 70, which indicated that the predictions were highly confident. In wild-type hemoglobin, the glutamic acid at residue 122 showed hydrogen bonds with lysine 18 (2.6Å), phenylalanine 119 (3.1Å), and threonine 124 (3.0Å), respectively (Figure 5A). However, in Hb O-Arab, the substitution of glutamic acid by lysine at residue 122 decreased the number of hydrogen bonds with the surrounding residues, only interacting with phenylalanine 119 (3.2Å) and threonine 124 (3.0Å) (Figure 5B). The loss of hydrogen bonds may alter the stability of the protein, potentially resulting in the clinical manifestation of anemia. By contrast, in Hb D-Punjab, the substitution of glutamic acid by glutamine at residue 122 maintains interactions with lysine 18 (2.6Å), phenylalanine 119 (3.1Å), and threonine 124 (3.0Å) (Figure 5C). Both the number and length of these hydrogen bonds remained unchanged, which indicated Hb D-Punjab was unlikely to significantly alter the protein structure, further suggesting its effect on the phenotype may be minimal.
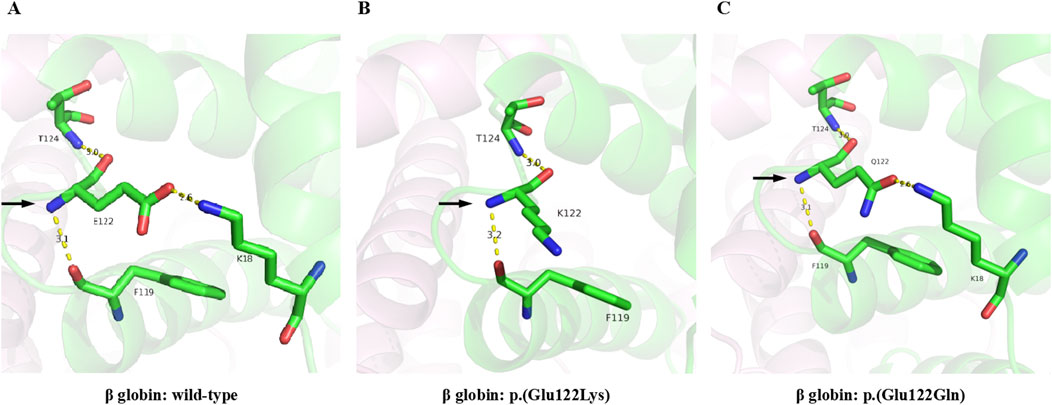
Figure 5. 3D-structure of Hb O-Arab and Hb D-Punjab compared with wild-type β globin. The 3D-structures of the wild-type β globin (A), Hb O-Arab (B), and Hb D-Punjab (C) were predicted by Alphafold2. The carbon, nitrogen, and oxygen atoms are colored green, blue, and red, respectively. The hydrogen bonds are displayed as yellow dashed lines.
Hematological characteristics and clinical manifestations of individuals carrying the variants
Individuals with Hb O-Arab showed various hematological features and clinical symptoms across different situations. Although the subjects with homozygous Hb O-Arab typically had mild to moderate anemia with a lower hemoglobin levels, most of them were asymptomatic, and only a few of them exhibited mild clinical symptoms such as lassitude, jaundice and splenomegaly (Dror, 2013; Nagel et al., 1999; Efremov et al., 1977; Heard et al., 1991). When the variant co-existed with Hb S, the corresponding cases usually presented with mild to moderate anemia, jaundice, and splenomegaly, as well as clinical characteristics similar to those with sickle cell disease (SCD), such as acute chest syndrome, recurrent vaso-occlusive painful events, dactylitis, hemolytic and so on (Nagel et al., 1999; Rachmilewitz et al., 1985; Zimmerman et al., 1999). Individuals carrying compound heterozygous Hb O-Arab and β-thalassemia variants typically manifested mild to moderate microcytic hypochromic anemia with reduced levels of MCV and MCH as well as an elevated level of Hb A2, similar to β-thalassemia traits. They were typically asymptomatic, and only a few individuals exhibited mild jaundice and splenomegaly. As expected, the hematological characteristics and clinical manifestations of individuals carrying compound heterozygous Hb O-Arab and β0-thalassemia variants were more severe than those of individuals with Hb O-Arab and β+-thalassemia variants (Rachmilewitz et al., 1985; Lacerra et al., 1993; Kalai et al., 2024; Morlé et al., 1984; Moumni et al., 2011; Nikolov et al., 1989). Detailed information of the reported cases was provided in Table 2.
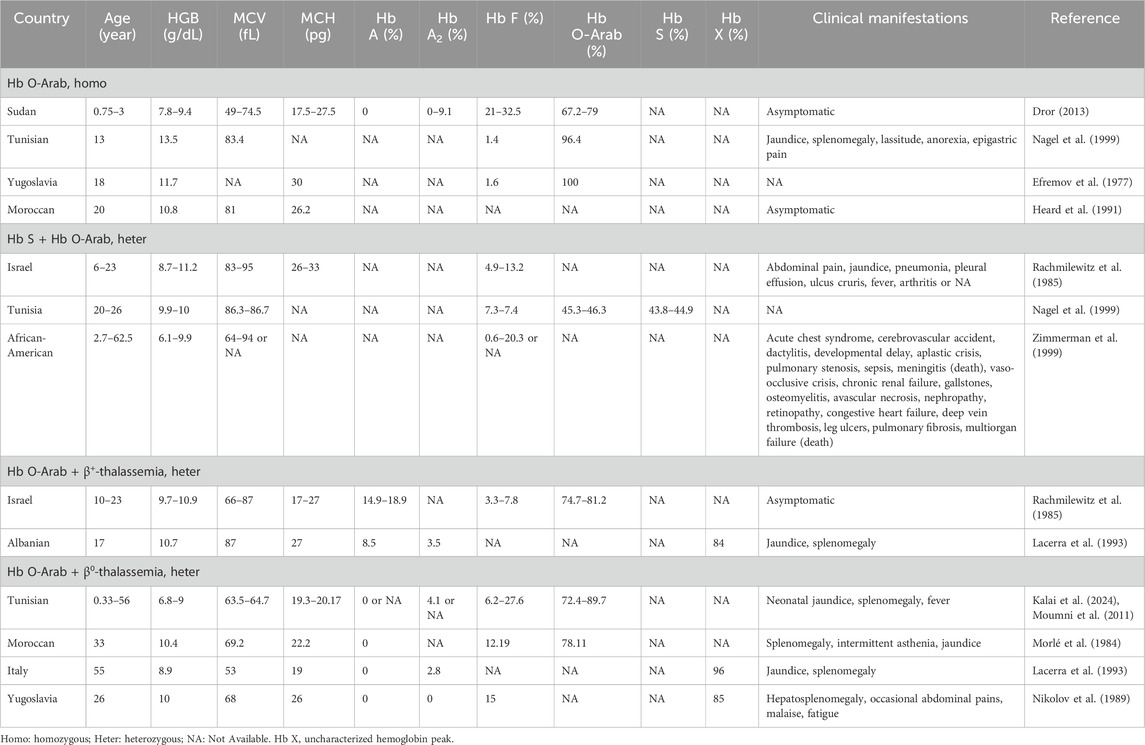
Table 2. Hematological characteristics and clinical manifestations of cases with Hb O-Arab.
The Hb D-Punjab mainly existed in three forms: Hb D-Punjab homozygous, Hb S combined with Hb D-Punjab, Hb D-Punjab combined with β-thalassemia. Individuals with homozygous Hb D-Punjab were typically asymptomatic with normal hematological characteristics (el-Kalla and Mathews, 1997; Silva-Pinto et al., 2014), although a few of them developed mild to moderate anemia and led to pallor and fatigability (Singh et al., 2023; Spandana et al., 2022). The association of this variant with Hb S or thalassemia also occurred. Usually, the compound heterozygous Hb D-Punjab and β-thalassemia caused mild microcytic and hypochromic anemia with reductions in MCV and MCH and elevated Hb A2, but showed no clinical changes (el-Kalla and Mathews, 1997; Perea et al., 1999; Panyasai et al., 2017; Fucharoen et al., 2002). Occasionally, individuals with this profile experienced weakness, hepatosplenomegaly and jaundice (Shekhda et al., 2017). The compound heterozygosity for Hb S and Hb D-Punjab resulted in moderately severe anemia with a reduction of Hb levels, and in addition to jaundice and hepatosplenomegaly, these individuals also presented clinical symptoms similar to those of sickle cell disease (SCD). Pain due to vaso-occlusive crisis was one of the most common complications, and acute chest syndrome as well as acute splenic sequestration (Torres et al., 2016; Adekile et al., 2010), also occurred in cases of this form. Detailed information was provided in Table 3.
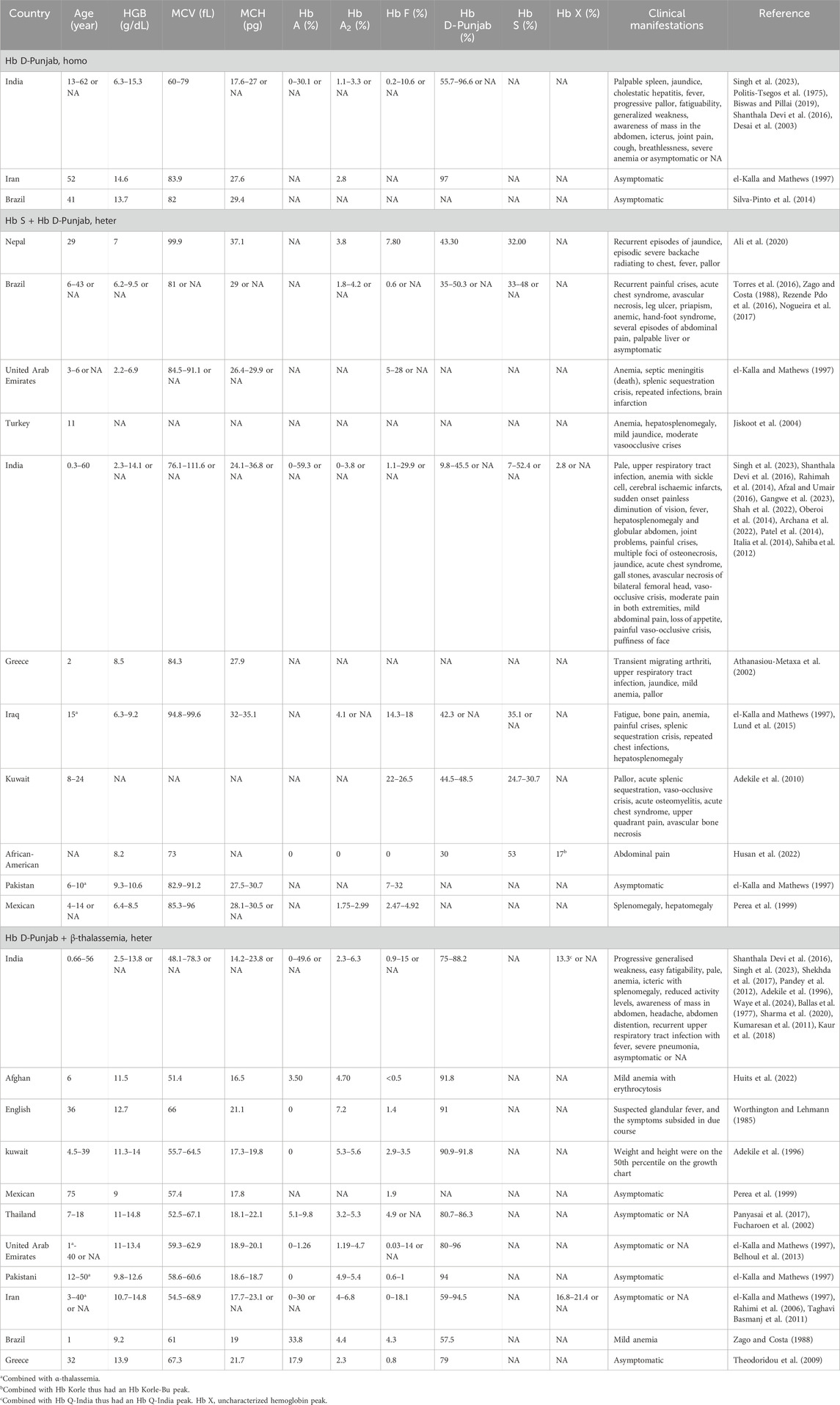
Table 3. Hematological characteristics and clinical manifestations of cases with Hb D-Punjab.
Discussion
In this study, the SMRT sequencing was employed to conduct a population-based genetic screening for hemoglobinopathies among large-scale individuals all over Guangxi, China. The hemoglobin variants, Hb O-Arab and Hb D-Punjab at the same genetic locus, were identified in two unrelated families, with an extremely low frequency among the screened population. Furthermore, we predicted the impact of these two rare variants on the structure of hemoglobin, and conducted a literature review to analyze their origins and genotype-phenotype correlations.
Hb O-Arab was first described in 1960 (Ramot et al., 1960), and was then discovered to cause the substitution of glutamic acid by lysine at residue 122 (Baglioni and Lehmann, 1962). Worldwide, Hb O-Arab was most common in the Pomak village in Greece with an allele frequency of 4.4% (Kuchenbaecker et al., 2022). Its frequency increased due to high genetic drift within the Pomak population, leading to its dispersion throughout the Mediterranean basin and the Middle East, with minor variations in its haplotypic pattern (Papadopoul et al., 2005). To date, Hb O-Arab had been reported in Israeli Arabs (Rachmilewitz et al., 1985), Tunisia (Kalai et al., 2024; Moumni et al., 2011; Nagel et al., 1999), Morocco (Morlé et al., 1984), and Bulgaria (Kantchev et al., 1975). However, to the best of our knowledge, this was the first reported family with the Hb O-Arab variant in the Chinese population, among whom it was expected to have an extremely low occurrence. It was believed that the Hb O-Arab variant originated from the Ottoman Empire, as the high-incidence regions for Hb O-Arab were consistent with the areas colonized by the Ottoman Empire (Elbashir and Elsayed Yousif, 2023). Few clues could be found to support the origin of the variant in this Chinese family. Despite this, the identification of the variant expanded the mutational spectrum of the HBB gene in the Chinese population. It should be declared that this variant had not been included in the routine screening panels in most clinical settings in China, and therefore the true frequency of the variant in the population may have been higher than anticipated. Additionally, heterozygous carriers of the Hb O-Arab variant were clinically asymptomatic and could have been easily neglected in routine screening, resulting in an unusually low detection rate.
By reviewing the reported cases in the literature, we found highly variable phenotypes among individuals carrying the Hb O-Arab variant. The characteristics of Hb O-Arab homozygotes varied from asymptomatic with only mild anemia to mild symptoms, including jaundice, splenomegaly, lassitude, anorexia, and epigastric pain (Efremov et al., 1977; Dror, 2013). The phenotypes of compound heterozygotes for Hb O-Arab and Hb S were similar to those of homozygotes for Hb S disease, presenting with hemolytic anemia, jaundice, and sickle cell disease characteristics (Rachmilewitz et al., 1985). The compound heterozygotes for Hb O-Arab and the β-thalassemia variant typically manifested mild to moderate anemia, and the hematological characteristics and clinical manifestations of individuals with Hb O-Arab combined with β0-thalassemia individuals were considerably more severe than those of individuals with Hb O-Arab combined with β+-thalassemia individuals (Moumni et al., 2011; Morlé et al., 1984; Kantchev et al., 1975; Rachmilewitz et al., 1985; Kalai et al., 2024), indicating that β-thalassemia variants were the main contributors to the phenotypic variability in these individuals. A previous study reported a four-month-old infant with compound heterozygous Hb O-Arab and β0-thalassemia variant (HBB: c.92 + 1G>A), presenting severe manifestations, including neonatal hemolytic anemia and an enlarged spleen (Kalai et al., 2024). However, the proband in Family 1 that we reported did not present with microcytic hypochromic anemia combined with abnormal hemoglobin content, increased bilirubin, icteric sclera, and skin discoloration until the age of 18, suggesting variable severity and onset age of the manifestations in these cases.
Hb D-Punjab, also known as Hb D-Los Angeles, was one of the most common hemoglobin variants worldwide, following Hb S and Hb C. It was most prevalent in India, and also had been found in other countries, including Italy, Spain, Thailand, and so on (Torres et al., 2015). In China, Hb D-Punjab was most common in Xinjiang province, accounting for 55.6% of total abnormal hemoglobin variants (Li et al., 1986). The Hb D-Punjab variant had been reported in both heterozygous and homozygous states as well as in combination with other abnormal hemoglobins such as thalassemia or Hb S. Hb D-Punjab heterozygotes and homozygotes, the rarest form of inheritance, presented no clinical or hematological alterations, but occasionally manifested mild to moderate hemolytic anemia. Usually, the interaction between Hb D-Punjab and β-thalassemia led to mild microcytic and hypochromic anemia, but did not present relevant clinical or hematological changes. However, the co-inheritance of Hb S and D-Punjab resulted in moderate to severe clinical manifestations similar to those of homozygous Hb S. In this study, we observed a heterozygous Hb D-Punjab in the proband from Family 2, who was expected to be asymptomatic. However, the genetic counseling and carrier screening for his partner in the future are still necessary due to the increased risk of having offspring with compound heterozygosity for Hb S and Hb D-Punjab.
The selection of strategies for diagnosing hemoglobinopathies is largely determined by the variant spectrum and prevalence of the variants in the local population. In China, the conventional methods of genetic testing for thalassemia, such as Gap-PCR and PCR-RDB (Polymerase Chain Reaction-Reverse Dot Blot), typically detect only the 24 hotspot variants commonly found in the Chinese population, which cover approximately 95%–98% of α- and β-thalassemia carriers (Li and Ye, 2024). However, various rare and complex variants are booming with global migration. Population-specific assays can not fully satisfy the needs of hemoglobinopathies controlling and prevention, thus optimizing diagnostic strategies and improving testing rates are essential. Compared with conventional methods, the SMRT approach provides the ability to uncover new variations and complex structural rearrangements such as triplications of the α-globin genes which worsen β-thalassemia phenotypes with high efficiency benefiting from its free of PCR amplification during sequencing and use of ultra-long reads (Xu et al., 2024). Furthermore, it can distinguish whether the identified variants are in cis or trans configuration without pedigree analysis (Xu et al., 2020). Recently, the SMRT sequencing has emerged as a reliable method for preconception screening and prenatal testing of hemoglobinopathies (Huang et al., 2024; Liang et al., 2023). Integrating this approach into screening programs for newborns in high-incidence areas who are at high risk of hemoglobinopathies could enhance early diagnosis, enable personalized treatment, support informed decision-making, and ultimately improve public health outcomes for these conditions.
Conclusion
Two rare hemoglobin variants, Hb O-Arab and Hb D-Punjab, were identified in the population-based genetic screening throughout Guangxi, China, using the SMRT sequencing. The first report of Hb O-Arab enriches the spectrum of hemoglobin variants in the Chinese population. Analyzing the frequency, origin, and genotype-phenotype correlation of these variants could pave the way for clinical management and genetic counseling for hemoglobinopathies, including thalassemia. Due to the limited number of subjects enrolled and the complex genotype-phenotype correlation involved, further research based on a larger volume of participants in specific populations is still required to summarize the key hematological and clinical features of these rare variants. This study also verified the SMRT sequencing-based assay as a valuable and comprehensive method for the detection of rare hemoglobin variants.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Second Affiliated Hospital of Guangxi Medical University (approval number No. 2022KY-0476). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
CG: Writing – original draft, Data curation, Funding acquisition, Methodology, Writing – review and editing, Investigation. ZC: Writing – review and editing, Writing – original draft, Investigation, Data curation. YC: Data curation, Investigation, Writing – review and editing, Resources. YM: Investigation, Writing – review and editing, Validation. HC: Data curation, Investigation, Writing – review and editing. WW: Validation, Writing – review and editing, Methodology. XW: Data curation, Writing – review and editing, Investigation. JL: Validation, Methodology, Writing – review and editing. XZ: Investigation, Writing – review and editing. QD: Writing – review and editing, Methodology, Data curation. YL: Supervision, Investigation, Writing – review and editing, Resources. BG: Supervision, Conceptualization, Writing – review and editing, Data curation, Methodology, Funding acquisition, Investigation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported in part by the Guangxi Major Research Programme (AB22035013 to BG), the Joint Project on Regional High-Incidence Diseases Research of Guangxi Natural Science Foundation (2023GXNSFBA026124 to CG), the National High-Level Hospital Clinical Research Funding (2022-PUMCH-C-033 to BG as team member), the Guangxi Medical University Training Program for Distinguished Young Scholars (to BG as team member), the Science Foundation for Young Scholars of Guangxi Medical University (GXMUYSF202115 to CG), the Special Scientific Research Fund of Guangxi Ten-Hundred-Thousand Talents Project (2021186 to BG), and the Initial Scientific Research Fund for Advanced Talents from the Second Affiliated Hospital of Guangxi Medical University (2019112 to BG).
Acknowledgments
We would like to thank the participants and their families for their contributions to this work.
Conflict of interest
Author QD was employed by Berry Genomics Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adekile, A. D., Kazanetz, E. G., Leonova, J. Y., Marouf, R., Khmis, A., and Huisman, T. H. (1996). Co-inheritance of Hb D-Punjab (codon 121; GAA--CAA) and beta (0) -thalassemia (IVS-II-1; G--A). J. Pediatr. Hematology/Oncology 18 (2), 151–153. doi:10.1097/00043426-199605000-00010
Adekile, A., Mullah-Ali, A., and Akar, N. A. (2010). Does elevated hemoglobin F modulate the phenotype in Hb SD-Los Angeles? Acta Haematol. 123 (3), 135–139. doi:10.1159/000276998
Afzal, H., and Umair, S. F. (2016). Haemoglobin sickle D disease: a presentation with ischaemic stroke. JPMA J. Pak. Med. Assoc. 66 (3), 348–350.
Ali, W., Jain, M., Agarwal, S., and Kumar, A. (2020). A case of hemoglobin sickle-D Punjab. Indian J. Hematol. Blood Transfus. 36 (1), 205–207. doi:10.1007/s12288-019-01179-6
Archana, R., Vidya, C., Sumithra, N., Jyothi, M., and Sanil, R. (2022). Arab-Indian -530 ß-distal promoter haplotype and sickle/Hb D heterozygosis in Badagas of Nilgiris: is it suggestive of Harappan origin? J. Genet. 101, 17. doi:10.1007/s12041-021-01348-5
Athanasiou-Metaxa, M., Economou, M., Tsatra, I., Pratsidou, P., and Tsantali, C. (2002). Co-inheritance of hemoglobin D-Punjab and hemoglobin S: case report. J. Pediatr. Hematology/Oncology 24 (5), 421. doi:10.1097/00043426-200206000-00022
Baglioni, C., and Lehmann, H. (1962). Chemical heterogeneity of haemoglobin O. Nature 196, 229–232. doi:10.1038/196229a0
Ballas, S. K., Atwater, J., and Norris, D. G. (1977). The interaction of betaO-thalassemia with hemoglobin D Punjab: a study of globin chain synthesis in an Indian family. Int. J. Hemoglobin Res. 1 (7), 697–701. doi:10.3109/03630267708999176
Belhoul, K., Bakir, M. L., and Abdulrahman, M. J. H. (2013). Misdiagnosis of Hb D-Punjab/β-Thalassemia is a potential pitfall in hemoglobinopathy screening programs: a case report. Hemoglobin 37, 119–123. doi:10.3109/03630269.2013.769174
Biswas, T. K., and Pillai, A. (2019). Hemoglobin-D Punjab—rare hemolytic anemia in the elderly: a case report. MGM J. Med. Sci. 6 (3), 148–151. doi:10.4103/mgmj.mgmj_12_20
Desai, D. V., Dhanani, H., Shah, M. A., Dayal, N., Kapoor, A., and Yeluri, S. (2003). Homozygous hemoglobin D disease: a case report. Internet J. Pathology 3. doi:10.5580/1a8b
Dror, S. (2013). Clinical and hematological features of homozygous hemoglobin O-Arab [beta 121 Glu → Lys]. Pediatr. Blood Cancer 60 (3), 506–507. doi:10.1002/pbc.24414
Efremov, G. D., Sadikario, A., Stojancov, A., Dojcinov, D., and Huisman, T. H. (1977). Homozygous hemoglobin O Arab in a gypsy family in Yugoslavia. Hemoglobin 1 (4), 389–394. doi:10.3109/03630267708996897
el-Kalla, S., and Mathews, A. R. (1997). Hb D-Punjab in the United Arab Emirates. Hemoglobin 21 (4), 369–375. doi:10.3109/03630269709000669
Elbashir, I., and Elsayed Yousif, T. Y. (2023). Molecular detection of hemoglobin O-Arab in the Sudanese population. Int. J. General Med. 16, 3323–3330. doi:10.2147/IJGM.S421140
Fucharoen, S., Changtrakun, Y., Surapot, S., Fucharoen, G., and Sanchaisuriya, K. (2002). Molecular characterization of Hb D-Punjab [beta121(GH4)Glu--Gln] in Thailand. Hemoglobin 26 (3), 261–269. doi:10.1081/hem-120015030
Gangwe, A. B., Singh, A., Parchand, S. M., Agrawal, D., Ekumankama, C. B., and Azad, R. (2023). Asymmetric sickle cell retinopathy in a patient with sickle cell hemoglobin D disease: a case report. Indian J. Ophthalmol. 3 (3), 760–761. doi:10.4103/ijo.ijo_981_23
Giardine, B. M., Joly, P., Pissard, S., Wajcman, H., Chui, D. H. K., Hardison, R. C., et al. (2021). Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 49 (D1), D1192–D1196. doi:10.1093/nar/gkaa959
Harteveld, C. L., Achour, A., Arkesteijn, S. J. G., Ter Huurne, J., Verschuren, M., Bhagwandien-Bisoen, S., et al. (2022). The hemoglobinopathies, molecular disease mechanisms and diagnostics. Int. J. Lab. Hematol. 44 (Suppl. 1), 28–36. doi:10.1111/ijlh.13885
Heard, S. E., Westwood, N. B., Pearson, T. C., and Stephens, A. D. (1991). Homozygous haemoglobin O-Arab in pregnancy. Clin. Laboratory Haematol. 13 (3), 319–320. doi:10.1111/j.1365-2257.1991.tb00289.x
Huang, R., Liu, Y., Xu, J., Lin, D., Mao, A., Yang, L., et al. (2024). Back-to-Back comparison of third-generation sequencing and next-generation sequencing in carrier screening of thalassemia. Archives Pathology Laboratory Med. 148 (7), 797–804. doi:10.5858/arpa.2022-0168-OA
Huits, R., Feyens, A. M., Lonneville, N., Peyrassol, X., Adam, A.-S., Gulbis, B., et al. (2022). Diagnosis and clinical relevance of co-inheritance of haemoglobin D-Punjab/β+-thalassemia traits in an immigrant Afghan family. J. Clin. Pathol. 75, 861–864. doi:10.1136/jclinpath-2021-208009
Husan, A., Amarasinghe, S. N., Fontenot, A., and Khan, M. W. (2022). Rare combinational hemoglobinopathies. Cureus 14 (12), e32327. doi:10.7759/cureus.32327
Italia, K., Upadhye, D., Dabke, P., Kangane, H., Colaco, S., Sawant, P., et al. (2014). Clinical and hematological presentation among Indian patients with common hemoglobin variants. Clin. Chim. Acta 431, 46–51. doi:10.1016/j.cca.2014.01.028
Jiskoot, P. M., Halsey, C., Rivers, R., Bain, B. J., and Wilkins, B. S. (2004). Unusual splenic sinusoidal iron overload in sickle cell/haemoglobin D-Punjab disease. J. Clin. Pathology 57 (5), 539–540. doi:10.1136/jcp.2002.004481
Kalai, M., Moumni, I., Ouragini, H., Chaouechi, D., Boudriga, I., and Menif, S. (2024). Coinheritance of HbO Arab/β0-thalassemia with severe manifestation in newborn. Am. J. Perinatology 41 (5), 594–597. doi:10.1055/s-0042-1743185
Kantchev, K., Tcholakov, B., Casey, R., Lehmann, H., and El Hazmi, M. J. H. (1975). Twelve families with Hb O Arab in the Burgas district of Bulgaria. Observations on sixteen examples of Hb O Arab-beta (0) thalassaemia. Humangenetik 26, 93–97. doi:10.1007/BF00278434
Kaur, M., Bodal, V. K., Sibia, R. P. S., Gupta, D., and Murarka, S. (2018). Education m. A rare presentation of haemoglobin D-thalassemia. J. Res. Med. Edu 18 (1). doi:10.56412/GMCP.2018.1.1.13
Kohne, E. (2011). Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Dtsch. Arzteblatt Int. 108 (31-32), 532–540. doi:10.3238/arztebl.2011.0532
Kountouris, P., Lederer, C. W., Fanis, P., Feleki, X., Old, J., and Kleanthous, M. (2014). IthaGenes: an interactive database for haemoglobin variations and epidemiology. PLoS One 9 (7), e103020. doi:10.1371/journal.pone.0103020
Kuchenbaecker, K., Gilly, A., Suveges, D., Southam, L., Giannakopoulou, O., Kilian, B., et al. (2022). Insights into the genetic architecture of haematological traits from deep phenotyping and whole-genome sequencing for two Mediterranean isolated populations. Sci. Rep. 12 (1), 1131. doi:10.1038/s41598-021-04436-9
Kumaresan, K., Gupta, K., Kalra, N., and Das, R. (2011). A rare association of giant adrenal myelolipoma in a young female double heterozygous for HbD Punjab and β-thalassemia trait. Indian J. Pathol. Microbiol. 54 (3), 635–637. doi:10.4103/0377-4929.85126
Lacerra, G., Fioretti, G., Hani, A., Duka, D., De Angioletti, M., Pagano, L., et al. (1993). Hb O-Arab [beta 121(GH4)Glu--Lys]: association with DNA polymorphisms of African ancestry in two Mediterranean families. Hemoglobin 17 (6), 523–535. doi:10.3109/03630269309043492
Lai, K., Huang, G., Su, L., and He, Y. (2017). The prevalence of thalassemia in mainland China: evidence from epidemiological surveys. Sci. Rep. 7 (1), 920. doi:10.1038/s41598-017-00967-2
Li, W., and Ye, Y. (2024). Application of third-generation sequencing technology in the genetic testing of thalassemia. Mol. Cytogenet. 17 (1), 32. doi:10.1186/s13039-024-00701-4
Li, H. J., Liu, D. X., Li, L., Liu, Z. G., Lo, S. L., Zhao, J., et al. (1986). A note about the incidence and origin of Hb D-Punjab in Xinjiang, People's Republic of China. Hemoglobin 10 (6), 667–671. doi:10.3109/03630268609036571
Liang, Q., Gu, W., Chen, P., Li, Y., Liu, Y., Tian, M., et al. (2021). A more universal approach to comprehensive analysis of thalassemia alleles (CATSA). J. Mol. Diagnostics JMD. 23 (9), 1195–1204. doi:10.1016/j.jmoldx.2021.06.008
Liang, Q., He, J., Li, Q., Zhou, Y., Liu, Y., Li, Y., et al. (2023). Evaluating the clinical utility of a long-read sequencing-based approach in prenatal diagnosis of thalassemia. Clin. Chem. 69 (3), 239–250. doi:10.1093/clinchem/hvac200
Lund, K., Chakravorty, S., Toma, S., and Bain, B. J. (2015). Compound heterozygosity for hemoglobins S and D. Am. J. Hematol. 90 (9), 842. doi:10.1002/ajh.24095
Modell, B., and Darlison, M. (2008). Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 86 (6), 480–487. doi:10.2471/blt.06.036673
Morlé, F., Morlé, L., Baklouti, F., Dorléac, E., Baudonnet, C., Delaunay, J., et al. (1984). The Hb F composition in a Moroccan family with β°-thalassaemia and Hb O-Arab. Scand. J. Haematol. 33 (3), 281–287. doi:10.1111/j.1600-0609.1984.tb02229.x
Moumni, I., Yalaoui, S., Ghrairi, N., Hamzaoui, A., Zoraï, A., and Abbes, S. (2011). Molecular characterization of a discrete hemoglobinopathy upon investigation for a lung hydatic cyst in an old Tunisian patient. Ann. Biol. Clin. Paris. 69 (3), 353–356. doi:10.1684/abc.2011.0582
Nagel, R. L., Krishnamoorthy, R., Fattoum, S., Elion, J., Genard, N., Romero, J., et al. (1999). The erythrocyte effects of haemoglobin O(ARAB). Br. J. Haematol. 107 (3), 516–521. doi:10.1046/j.1365-2141.1999.01755.x
Nikolov, N., Andreeva, M., Janković, L., and Efremov, G. D. (1989). Hemoglobin O Arab in interaction with beta 0-thalassemia. Lijec. Vjesn. 111 (1-2), 27–30.
Nogueira, R. N., Leite, CMBT, Souza, L. X., and Barbosa, A. A. L. (2017). Clinical and laboratory repercussions in patient with hemoglobin SD-Punjab disease: a case report. J. Bras. Patol. Med. Lab. 53 (5). doi:10.5935/1676-2444.20170049
Oberoi, S., Das, R., Trehan, A., Ahluwalia, J., Bansal, D., Malhotra, P., et al. (2014). HbSD-Punjab: clinical and hematological profile of a rare hemoglobinopathy. J. Pediatr. Hematology/Oncology 36 (3), e140–e144. doi:10.1097/MPH.0000000000000049
Pandey, S., Ranjan, R., Mishra, R. M., Pandey, S., and Saxena, R. (2012). Interaction of - α 3.7, ß thalassemia mutation IVS 1-5 and HbD Punjab in a family: a case report. Indian J. Clin. Biochem. IJCB 27 (3), 314–317. doi:10.1007/s12291-012-0189-8
Panyasai, S., Rahad, S., and Pornprasert, S. (2017). Coinheritance of hemoglobin D-Punjab and β(0)-thalassemia 3.4 kb deletion in a Thai girl. Asian J. Transfus. Sci. 11 (2), 199–202. doi:10.4103/ajts.AJTS_117_16
Papadopoulos, V., Dermitzakis, E., Konstantinidou, D., Petridis, D., Xanthopoulidis, G., and Loukopoulos, D. (2005). HbO-Arab mutation originated in the Pomak population of Greek Thrace. Haematologica 90 (2), 255–257.
Patel, S., Purohit, P., Mashon, R. S., Dehury, S., Meher, S., Sahoo, S., et al. (2014). The effect of hydroxyurea on compound heterozygotes for sickle cell-hemoglobin D-Punjab--a single centre experience in eastern India. Pediatr. Blood Cancer 61 (8), 1341–1346. doi:10.1002/pbc.25004
Perea, F. J., Casas-Castañeda, M., Villalobos-Arámbula, A. R., Barajas, H., Alvarez, F., Camacho, A., et al. (1999). Hb D-Los Angeles associated with Hb S or beta-thalassemia in four Mexican Mestizo families. Hemoglobin 23 (3), 231–237. doi:10.3109/03630269909005703
Politis-Tsegos, C., Kynoch, P., Lang, A., Lehmann, H., Lorkin, P. A., Stathopoulou, R., et al. (1975). Homozygous haemoglobin D Punjab. J. Med. Genet. 12 (3), 269–274. doi:10.1136/jmg.12.3.269
Rachmilewitz, E. A., Tamari, H., Liff, F., Ueda, Y., and Nagel, R (1985). The interaction of hemoglobin O Arab with Hb S and beta+ thalassemia among Israeli Arabs. Hum. Genet. 70, 119–125. doi:10.1007/BF00273069
Rahimah, A., Syahira Lazira, O., Siti Hida, H. M., Faidatul Syazlin, A. H., Nur Aisyah, A., Nik Hafidzah, N. M., et al. (2014). Haemoglobin sickle d Punjab: - a case report. Med. J. Malays. 69 (1), 42–43.
Rahimi, Z., Akramipour, R., Korani, S., and Nagel, R. L. (2006). Hb D-Punjab [beta 121 (GH4) Glu--Gln]/beta0-thalassemia [IVSII.1(G--A)] in two cases from an Iranian family: first report. Am. J. Hematol. 81 (4), 302–303. doi:10.1002/ajh.20537
Ramot, B., Fisher, S., Remez, D., Schneerson, R., Kahane, D., Ager, J. A., et al. (1960). Haemoglobin O in an Arab family. Br. Med. J. 2 (5208), 1262–1264. doi:10.1136/bmj.2.5208.1262
Rezende Pdo, V., Costa, K. S., Domingues, J. C., Silveira, P. B., Belisário, A. R., Silva, C. M., et al. (2016). Clinical, hematological and genetic data of a cohort of children with hemoglobin SD. Rev. Bras. Hematol. Hemoter. 38 (3), 240–246. doi:10.1016/j.bjhh.2016.05.002
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sahiba, K., Amandeep, K., and Namrata, C. (2012). Hemoglobin SD disease - a case report. Biochem. 6 (7), 223–225.
Shah, V., Shah, M., Jhaveri, P., and Shah, F. (2022). Double heterozygosity of Hbs and Hbd Punjab in two siblings with Covid-19 infection: a case report. Indian J. Appl. Basic Med. Sci. 24, 18–23. doi:10.48165/ijabms.2022.243803
Shanthala Devi, A. M., Rameshkumar, K., and Sitalakshmi, S. (2016). Hb D: a not so rare hemoglobinopathy. Indian J. Hematol. Blood Transfus. 32 (Suppl. 1), 294–298. doi:10.1007/s12288-013-0319-3
Sharma, P., Jandial, A., Rajasekaran, S., Das, R., Chhabra, S., Hira, J. K., et al. (2020). Missing Hb Q-India peak in a triple-heterozygous patient with Hb D-Punjab/Hb Q-India/β-Thalassemia trait. Hemoglobin 44 (3), 211–213. doi:10.1080/03630269.2020.1767128
Shekhda, K. M., Leuva, A. C., Mannari, J. G., Ponda, A. V., and Amin, A. (2017). Co-inheritance of haemoglobin D-Punjab and beta thalassemia - a rare variant. J. Clin. Diagn. Res. 11 (6), OD21–OD22. doi:10.7860/JCDR/2017/27816.10114
Silva-Pinto, A. C., Silva, T. J., Moretto, E. L., Ottoboni, M., Rodrigues, E. S., and Covas, D. T. (2014). Blood donor homozygous for Hb D Los Angeles. Transfus. Apher. Sci. 51 (2), 219–220. doi:10.1016/j.transci.2014.08.021
Singh, N., Seth, T., and Tyagi, S. (2023). Review of clinical and hematological profile of hemoglobin D cases in a single centre. J. Mar. Med. Soc. 25, S74–S79. doi:10.4103/jmms.jmms_165_22
Spandana, R., Panneerselvam, K., Mani, S., and Krishnamoorthy, N. (2022). An interesting and rare case of hemoglobin D-Punjab variant in Tamil Nadu. Cureus 14 (2), e22668. doi:10.7759/cureus.22668
Taghavi Basmanj, M., Karimipoor, M., Amirian, A., Jafarinejad, M., Katouzian, L., Valaei, A., et al. (2011). Co-inheritance of hemoglobin D and β-thalassemia traits in three Iranian families: clinical relevance. Arch. Iran. Med. 14 (1), 61–63.
Theodoridou, S., Alemayechou, M., Perperidou, P., Sinopoulou, C., Karafoulidou, T., and Kiriakopoulou, G. (2009). Compound heterozygosity for Hb D-Punjab/β-thalassemia and blood donation: case report. Turk J. Hematol. 26 (2), 100–101.
Torres, L. S., Okumura, J. V., Silva, D. G., and Bonini-Domingos, C. R. (2015). Hemoglobin D-Punjab: origin, distribution and laboratory diagnosis. Rev. Bras. Hematol. Hemoter. 37 (2), 120–126. doi:10.1016/j.bjhh.2015.02.007
Torres, L. S., Okumura, J. V., Belini-Júnior, É., Oliveira, R. G., Nascimento, P. P., Silva, D. G., et al. (2016). Phenotypic diversity of sickle cell disease in patients with a double heterozygosity for Hb S and Hb D-Punjab. Hemoglobin 40 (5), 356–358. doi:10.1080/03630269.2016.1222295
van Gammeren, A. J., Pelkmans, L., Endschot, C., Roelofsen-de Beer, R., and Harteveld, C. L. (2020). An unusual compound heterozygosity for Hb O-Arab (HBB: c.364G>A) and Hb D-Los Angeles (HBB: c.364G>C). Hemoglobin 44 (1), 61–63. doi:10.1080/03630269.2019.1710530
Waye, J. S., Hanna, M., Hohenadel, B. A., Nakamura, L., Walker, L., Eng, B., et al. (2024). β(0)-Thalassemia caused by a novel nonsense mutation [HBB: c.199A > T]. Hemoglobin 48 (1), 69–70. doi:10.1080/03630269.2024.2322518
Weatherall, D. J. (2010). The inherited diseases of hemoglobin are an emerging global health burden. Blood 115 (22), 4331–4336. doi:10.1182/blood-2010-01-251348
Worthington, S., and Lehmann, H. (1985). The first observation of Hb D Punjab beta zero thalassaemia in an English family with 22 cases of unsuspected beta zero thalassaemia minor among its members. J. Med. Genet. 22 (5), 377–381. doi:10.1136/jmg.22.5.377
Xu, L., Mao, A., Liu, H., Gui, B., Choy, K. W., Huang, H., et al. (2020). Long-molecule sequencing: a new approach for identification of clinically significant DNA variants in α-thalassemia and β-thalassemia carriers. J. Mol. Diagnostics JMD. 22 (8), 1087–1095. doi:10.1016/j.jmoldx.2020.05.004
Xu, A., Ye, Y., Huang, Y., Huang, Y., Guo, H., and Ji, L. (2024). Identification of Hb Lepore, Hb anti-Lepore, and α-globin gene triplications by long-read single-molecule real-time sequencing. Am. J. Clin. Pathology 161 (4), 411–417. doi:10.1093/ajcp/aqad155
Zago, M. A., and Costa, F. F. (1988). Hb D-Los Angeles in Brazil: simple heterozygotes and associations with beta-thalassemia and with Hb S. Hemoglobin 12 (4), 399–403. doi:10.3109/03630268808998040
Keywords: Hb O-Arab, Hb D-Punjab, hemoglobinopathy, genetic screening, Chinese population, single-molecule real-time sequencing, genotype-phenotype correlation
Citation: Gui C, Cheng Z, Chen Y, Ma Y, Chen H, Wei W, Wei X, Liu J, Zhou X, Du Q, Lai Y and Gui B (2025) The rare hemoglobin variants Hb O-Arab and Hb D-Punjab identified in population-based genetic screening throughout Guangxi, China. Front. Genet. 16:1622391. doi: 10.3389/fgene.2025.1622391
Received: 03 May 2025; Accepted: 30 July 2025;
Published: 14 August 2025.
Edited by:
Evandra Strazza Rodrigues, Hemocentro Foundation of Ribeirão Preto, BrazilReviewed by:
Oyesola Ojewunmi, Queen Mary University of London, United KingdomHarsha Garadi Suresh, St. Jude Children’s Research Hospital, United States
Copyright © 2025 Gui, Cheng, Chen, Ma, Chen, Wei, Wei, Liu, Zhou, Du, Lai and Gui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoheng Gui, YmFvaGVuZ2d1aUB5ZWFoLm5ldA==; Yinghui Lai, eWluZ2h1aWxhaUBzaW5hLmNvbQ==
†These authors have contributed equally to this work