 Hui Tang
Hui Tang Jingjing Xiang
Jingjing Xiang Qin Zhang
Qin Zhang- Center for Reproduction and Genetics, School of Gusu, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital, Suzhou, Jiangsu, China
Yunis–Varón syndrome (YVS) is a severe autosomal recessive syndrome caused by mutations in the FIG4 gene. It is characterized by skeletal defects, including cleidocranial dysplasia and digital anomalies, and a poor prognosis due to neurological and cardiovascular involvement. In this study, we observed a Chinese family with three patients presenting thumb and hallux dysplasia. Whole-genome sequencing (WGS) identified a compound heterozygous variant in the proband: c.2097-809A>G and c.1141C>T (p.R381*). The c.2097-809A>G variant generated an aberrant splicing transcript containing a pseudoexon from intron 18, as demonstrated by further RT-PCR and splicing analysis. This is the first deep intronic variant reported in the FIG4 gene. In addition, we provided prenatal diagnoses for the family. This study expands the genetic variant spectrum, provides additional molecular and clinical information, and broadens our understanding of the molecular mechanisms involved in the disease course.
1 Introduction
Phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) is one of the seven known phosphoinositides involved in the orchestration of vesicle trafficking in mammalian cells (Martyn and Li, 2013). The FIG4 gene encodes a phosphoinositide 5-phosphatase that regulates the synthesis and turnover of PI(3,5)P2 and is involved in endolysosomal trafficking (Sbrissa et al., 2007; Martyn and Li, 2013; Lees et al., 2020). Variants in FIG4 can cause four different diseases: bilateral temporo-occipital polymicrogyria (OMIM 612691), amyotrophic lateral sclerosis 11 (OMIM 612577), Charcot–Marie–Tooth disease, type 4J (CMT4J) (OMIM 611228), and Yunis–Varón syndrome (YVS) (OMIM 216340). In an animal model, homozygous FIG4-null mice exhibit neurological features of YVS, including degeneration of the brain and peripheral ganglia and enlarged vacuoles in neurons; they also present skeletal deformities, with reduced trabecular bone volume and cortical thickness, and accumulate large vacuoles (Chow et al., 2007; Ferguson et al., 2012; Campeau et al., 2013).
Yunis–Varón syndrome is a very rare autosomal recessive syndrome that exhibits severe skeletal abnormalities (cleidocranial dysplasia, absence/hypoplasia of thumbs and halluces, pelvic bone dysplasia, and fractures), which are often lethal in infancy (Yunis and Varon, 1980; Basel-Vanagaite et al., 2008). In addition, neurological features such as hypotonia, dysphagia, global developmental delay, brain malformations, retinopathy, and facial dysmorphisms have also been attributed to it (Basel-Vanagaite et al., 2008; Campeau et al., 2013). By describing and reviewing the literature, some studies of FIG4-associated YVS have been reported (Reutter et al., 2012; Nakajima et al., 2013; Peng et al., 2021; Umair et al., 2021; Wang et al., 2023; Beauregard-Lacroix et al., 2024). However, only a few of them carried out appropriate genetic analysis and diagnosis. Variant analysis of the FIG4 gene is a valuable tool for screening and diagnosis of YVS. So far, we have found 18 variants of YVS that have been recorded: c.350C>T, c.2376 + 1G>T (Wang et al., 2023), c.573del, c.2174dup (Peng et al., 2021), c.1260_1261del, c.311G>A, c.831_838del, c.524T>C (Campeau et al., 2013), c.968A>G (Umair et al., 2021), c.1750+1del, c.2285_2286del (Nakajima et al., 2013), c.1141C>T, c.2459+1G>A, c.1294C>T, c.122T>C, c.1474C>T, c.2285_2286del, and c.1583+1G>T (Beauregard-Lacroix et al., 2024). All of them have severe clinical features of YVS, such as skeletal abnormalities, craniofacial dysmorphisms, and structural brain anomalies. We also observed prenatal phenotypes in the fetuses.
In this study, we report the identification of a novel deep intronic FIG4 variant (c.2097-809A>G) in a Chinese family with three neonates presenting hallmark features of YVS. The c.2097-809A>G variant, the first deep intronic variant reported in the FIG4 gene, was identified by whole-genome sequencing (WGS) and confirmed by Sanger sequencing. Further splicing analysis of RNA from peripheral blood mononuclear cells (PBMCs) demonstrated that c.2097-809A>G generated an aberrant transcript that destroyed the function of FIG4. Therefore, we provided prenatal diagnosis for this family. This study expands the genetic variant spectrum, provides additional molecular and clinical information, and broadens our understanding of the molecular mechanisms involved in the disease course.
2 Methods
2.1 Participants and ethical statement
This study included three children from a Chinese family who received genetic counseling at Suzhou Municipal Hospital in 2022. Written informed consent was obtained from the patients’ parents, and all procedures were conducted in accordance with the tenets of the Declaration of Helsinki. The research was performed under the approval of the Ethics Committee of Suzhou Municipal Hospital, the Affiliated Suzhou Hospital of Nanjing Medical University (approval no. 2017005).
2.2 DNA and RNA isolation
We obtained blood samples from a child, his parents, and normal controls and the amniotic fluid from the fetus. A younger afflicted child died soon after birth, and we could not collect his samples. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany), according to the standard extraction methods. Total RNA from the blood leukocytes of the patients and controls was extracted using the standard TRIzol method. cDNAs were synthesized from RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, United States).
2.3 Whole-genome sequencing
Qualified genomic DNA was randomly fragmented into proper pieces (350 bp). WGS libraries were prepared using the MGIEasy FS DNA Prep kit (BGI, Beijing, China), according to the manufacturer’s instructions. Then, they were sequenced using DNBSEQ-T7 (MGI, Beijing, China). The minimal read depth was 30-fold for WGS. Quality control for the paired-end reads was assessed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and fastp (Chen et al., 2018) was used for adapter trimming and quality filtering.
2.4 Variant analysis by direct sequencing
Direct Sanger sequencing in both directions was conducted to validate putative variants. The sequence containing the variant was amplified by polymerase chain reaction (PCR) with the primers FIG4 exon 11 (forward, 5′- GGATTTGAACCCAGGCAGTACT-3′ and reverse, 5′-TCGCTGATCTCCCAAAACTTACT-3′) and FIG4 intron 18 (forward, 5′- ACTGTTAGTGGTTTTGGTCCT-3′ and reverse, 5′-TGTTGATTTCTGCTCAGGGA-3′). PCR products were verified by agarose gel electrophoresis and subsequently sequenced in both directions using an ABI 3130 Genetic Analyzer (Applied Biosystems, United States).
2.5 Reverse transcription polymerase chain reaction and TA cloning
After extracting total RNA from the neonate and healthy controls, cDNAs were synthesized from RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, United States). DNA fragments of FIG4 intron 18 were amplified with the following primers: forward, 5′- TCCTGGGAGTTTTCCATCCCACT-3′ and reverse, 5′-CTGGCTGCCGTTTTCCGCTG-3′. The expected PCR product size was 451 bp. TA cloning was performed to isolate the aberrant transcript. PCR amplification was ligated to the T-Vector pMD19 (Simple) (TaKaRa, Japan). The positive clones were selected by blue/white screening with β-gal selection, and plasmids were extracted from the enriched white isolations using the Plasmid Mini Kit (QIAGEN, Hilden, Germany). For each sample, 10 bacterial clones were sequenced. The sequence of the amplified segment was compared with the reference sequence at UCSC.
2.6 Prenatal diagnosis
Following the identification and analysis of the FIG4 variant in the family members, prenatal genetic testing was performed for the fetus. In brief, an amniotic fluid sample was collected under ultrasonic guidance from the proband at 22 weeks of gestation. DNA was extracted using a DNA extraction kit (TIANGEN Biotech, Beijing, China), according to the manufacturer’s instructions. The FIG4 gene variant was detected using the same methods as previously described.
3 Results
3.1 Clinical features
The patient (II-2) was a 16-day-old newborn at the time of genetic consultation in 2022. He was born through cesarean section with mild asphyxia, weak crying, dysphagia, feeding difficulties, and neonatal hypotonia. He exhibited multiple facial deformities, such as a broad forehead, long philtrum, hypertelorism, depressed nasal bridge, low ear position, retrognathia, short neck, and yellow hair. His bilateral, extremely short thumbs and big toes are notable characteristics of YVS (Figure 1A). He also developed hypospadias. The echocardiography also showed an atrial septal defect and atrial septal aneurysm. His mother had an earlier child in 2019 (II-1) with similar defects. The 24-week ultrasound results showed multiple malformations, including a dilated fourth ventricle, posterior fossa cyst, low conus medullaris position with coccyx curvature abnormality, persistent left superior vena cava, slightly enlarged right heart, and a small amount of pericardial effusion (2.3 mm) (Figure 1B). After birth, he presented with abnormal skull morphology, metopic synostosis, and dyspnea. His hypoplastic thumbs and big toes were identical to those of II-2, and he died shortly thereafter. The parents denied consanguinity and any family history of related conditions (Figure 2A).
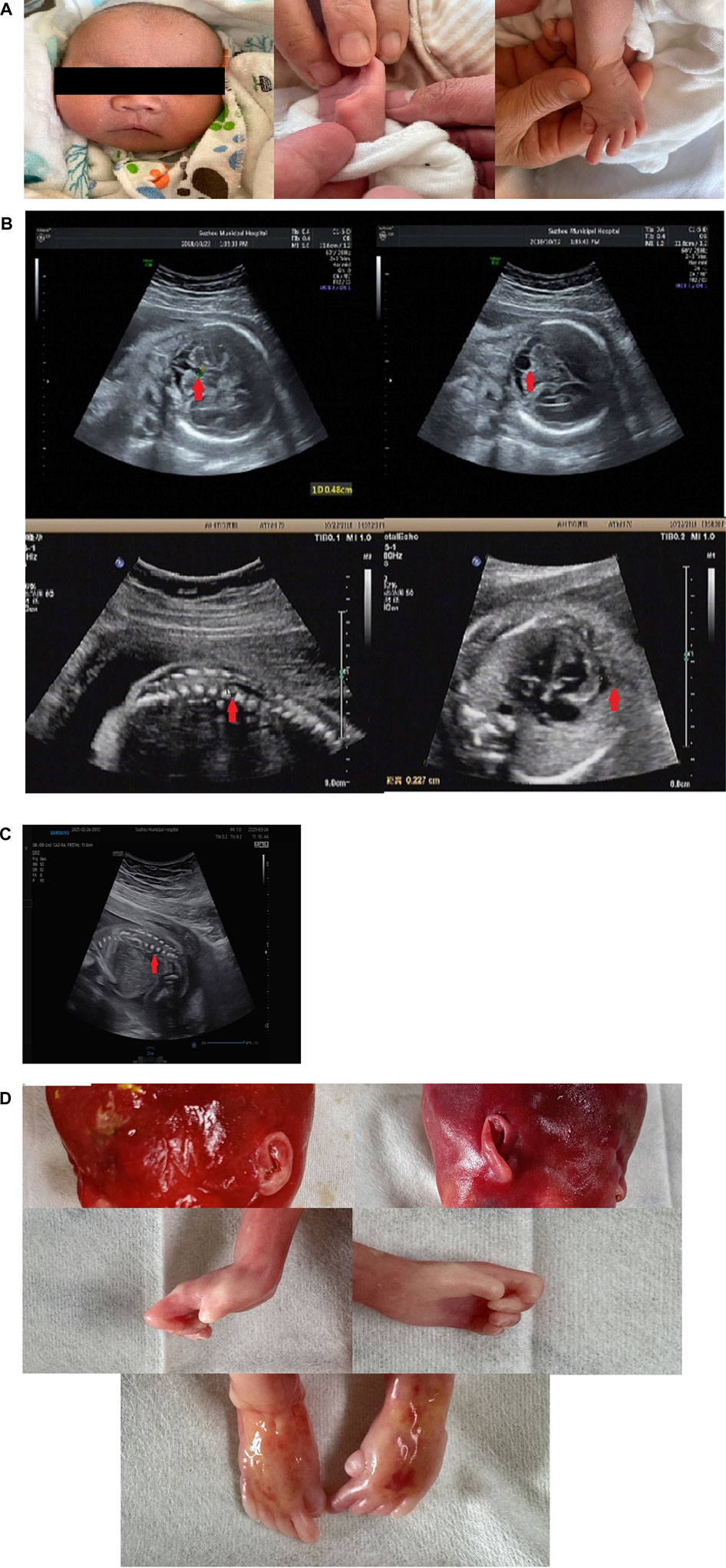
Figure 1. Phenotype of the patients: (A) facial malformation and thumb and toe anomalies of the proband. (B) Ultrasound scan of II-1 showed a dilated fourth ventricle, a posterior fossa cyst, a low conus medullaris position, and pericardial effusion. (C) Ultrasound scan of II-3 showed low conus medullaris position. (D) Low ear position and dysplasia of thumbs and halluces in II-3.
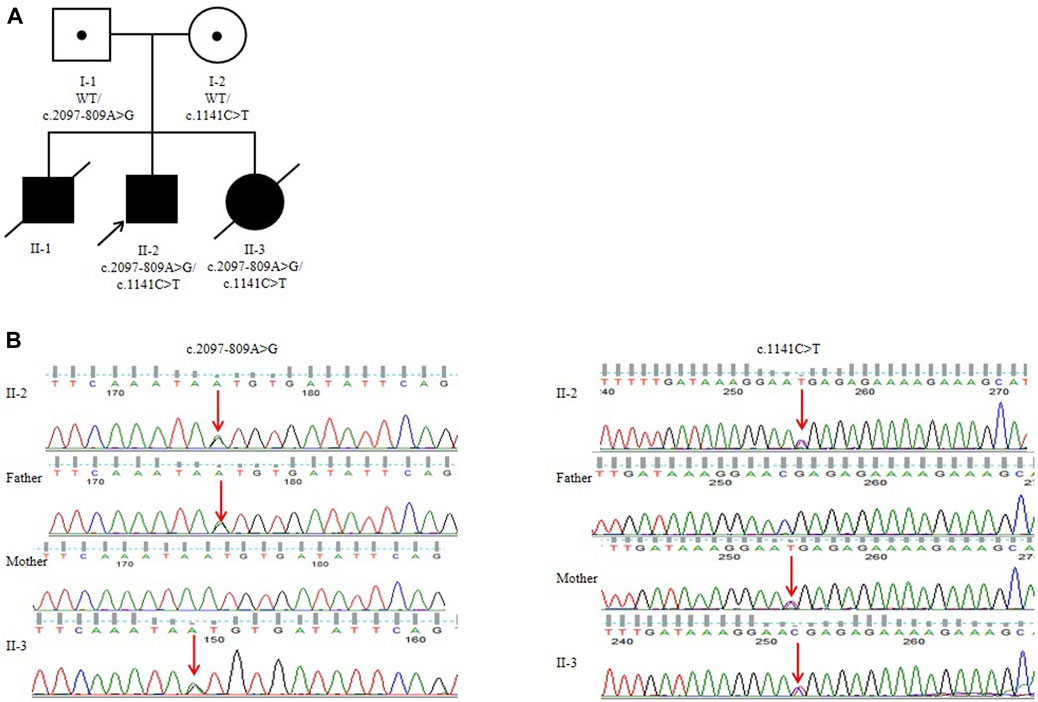
Figure 2. Pedigree analysis of the Chinese family: (A) genomic sequencing of the proband and his family. (B) I-1: Father of the proband, who carried a heterozygous variant of c.2097-809A>G; I-2: mother of the proband, who carried a heterozygous variant of c.1141C>T; II-2: the proband in this family, who carried two compound heterozygous variants of c.2097-809A>G and c.1141C>T. II-3 carried two compound heterozygous variants of c.2097-809A>G and c.1141C>T.
3.2 Identification of the variant
To examine the variant, we performed WGS using DNA from the proband and his parents’ PBMCs. The average depth of WGS was 30×, and the percentage of coverage over 10× was 96.4%, meeting the qualification for subsequent analysis. After filtering the variants using the minor allele frequency (MAF) in the 1000 Genomes Project and the Genome Aggregation Database (gnomAD) and searching for suspicious variants by the characteristic phenotype, c.1141C>T(p.R381*) in FIG4 was identified as being inherited from the mother. c.1141C>T (p.R381*) has been reported in two children with early-onset CMT4J (Nicholson et al., 2011; Zimmermann et al., 2020). In addition, the clinical findings of the two patients were highly suggestive of YVS caused by pathogenic variants in FIG4. Next, an in-depth search and analysis were performed. Then, we found c.2097-809A>G in the proband, which was inherited from the father. c.2097-809A>G was absent in various genetic variant databases or websites such as the ClinVar Database, Human Gene Variant Database (HGMD), Leiden Open Variation Database (LOVD), and PubMed. The online website SpliceAI predicted that it would destroy the acceptor sites with a score of 0.97. Subsequent family genotyping from his parents confirmed the compound heterozygous forms (Figure 2B).
3.3 RT-PCR and splicing analysis
RT-PCR of FIG4 exons 16 to 20 in mRNAs from the patient displayed an aberrant splicing transcript of 579 bp in addition to a 451-bp wild-type product (Figure 3A). Afterward, TA cloning was carried out, and cDNA sequencing results showed that the longer abnormal transcript comprised a 128-bp pseudo-exon from intron 18 (FIG4: c.2097-808_ 2097-681) (Figures 3B, C), resulting in a frameshift and generating a premature termination codon at residue 822 (p.Asp700Valfs*123).
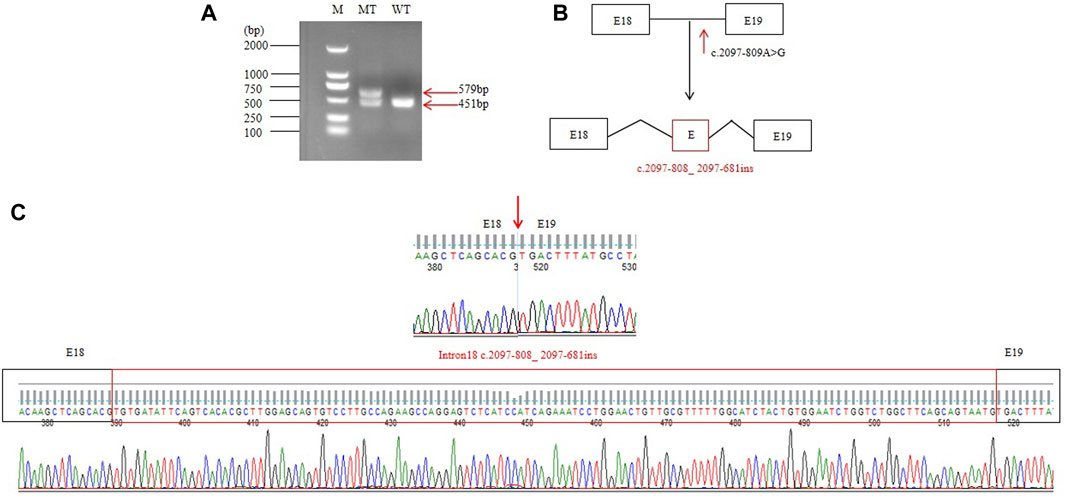
Figure 3. RT-PCR and splicing analysis of the c.2097-809A>G: (A) reverse transcription PCR analysis of mRNAs from peripheral blood leukocytes of one normal control and the patient. WT exhibited one normal splicing product (451 bp), and the patient showed a major aberrant transcript (579 bp). (B) Schematic of normal and aberrant splicing patterns. (C) Sanger sequencing of the full-length 451-bp transcript and the 579-bp aberrant transcript that included a 128-nt pseudo-exon.
3.4 Prenatal diagnosis of the FIG4 gene
In 2024, the mother was pregnant with her second child. We employed Sanger sequencing for amniotic fluid sample prenatal testing and identified that the fetus also unfortunately had compound heterozygous variants of c.2097-809A>G and c.1141C>T in the FIG4 gene (Figure 2B). Prenatal ultrasound showed a low conus medullaris position and an expanded right lateral ventricle (Figure 1C). Furthermore, SNP-array results showed that it was a female fetus. After genetic counseling, the couple chose to abort the child. After obtaining consent from the parents, we observed some phenotypes in the aborted fetus, such as low ear position and dysplasia of thumbs and halluces (Figure 1D). The parents may undergo pre-implantation genetic testing for monogenic disorders (PGT-M) for the next child.
4 Discussion
In the era of rapid sequencing technology, whole-genome sequencing has become a powerful tool for identifying candidate variants and enabling appropriate diagnoses—especially when pathogenic variants are concealed in patients with the typical phenotype and cannot be detected by routine exonic DNA sequencing methods such as Sanger sequencing or whole-exome sequencing. In this study, we detected a compound heterozygous variant in FIG4: c.1141C>T (p.R381*) and c.2097-809A>G by WGS. c.2097-809A>G is the first deep intronic variant in the FIG4 gene associated with Yunis–Varon syndrome.
Yunis–Varón syndrome is caused by complete loss-of-function of FIG4 and is characterized by severe neurological anomalies, cleidocranial dysplasia, and digital anomalies (Campeau et al., 2013). Other frequently reported malformations include a wide fontanelle with diastasis of cranial sutures, sparse scalp hairs, microcephaly, ectodermal malformations, bilateral hip dislocation, genital abnormalities, and central nervous system abnormalities such as Dandy–Walker malformation, agenesis of the corpus callosum, hydrocephalus, and hypoplastic vermis (Umair et al., 2021). Cardiovascular abnormalities such as ventricular hypertrophy and tetralogy of Fallot have also been reported (Adès et al., 1993; Abdul Wahab Siddique et al., 2019). The dysmorphic features observed in our three patients were consistent with those reported previously.
Introns are very important for eukaryotic evolution. The identification of genes with correct exon–intron architecture plays an important role in eukaryotic genome annotation (Mishra et al., 2021), and alternative splicing is crucial for the enhancement of transcriptome and proteome diversity (Keren et al., 2010). Nowadays, WGS has greatly improved the diagnosis of genetic diseases and increased the number of pathogenic deep intronic variants. These deleterious deep intronic variants usually generate pseudo-exon inclusion by the activation of atypical splice sites or changes in splicing regulatory elements (Vaz-Drago et al., 2017).
In the present study, we identified two variants in FIG4: c.1141C>T (p.R381*) inherited from the mother and c.2097-809A>G inherited from the father in the proband. His affected brother had a similar phenotype, but unfortunately, we could not obtain his sample to confirm the molecular results. c.1141C>T causes a premature termination codon at position 381 and has been reported in two children with early-onset CMT4J. c.2097-809A>G may destroy the acceptor sites predicted by SpliceAI. Subsequently, RT-PCR of FIG4 in the patient’s mRNAs displays an aberrant splicing transcript of 579 bp compared to a 451-bp wild-type product (Figure 3A) that hinted at a pseudo-exon inclusion in FIG4. We identified a 128-bp pseudo-exon from intron 18 (FIG4: c.2097-808_ 2097-681) in the mutant type by RT-PCR analysis and TA cloning. We put the full sequence of FIG4 containing the pseudo-exon into EditSeq software, and the translated protein presented a frameshift and resulted in a premature termination codon at residue 822 (p.Asp700Valfs*123). p.Asp700Valfs*123 is predicted to cause loss-of-function of the protein as a frameshift truncating mutation, resulting in an altered protein that lacks the C-terminal residues of the wild-type FIG4 protein responsible for membrane anchoring (Campeau et al., 2013). Nonsense-mediated mRNA decay is likely to occur because the variants remove more than 10% of the protein. After 1 year, the mother was pregnant again. Prenatal diagnosis showed that her second child also had compound heterozygous variants in FIG4. In addition, the aborted fetus exhibited a low conus medullaris position, an expanded right lateral ventricle, a low ear position, and dysplasia of thumbs and halluces. Unfortunately, the family had another YVS patient because of c.2097-809A>G and c.1141C>T in the FIG4 gene. According to the ACMG variant classification guideline, c.1141C>T could be classified as pathogenic (PVS1, PM3, PM2_Supporting, PP1, and PP4), and c.2097-809A>G could also be classified as pathogenic (PVS1, PM2_Supporting, PM3, PP1, and PP4). Notably, c.2097-809A>G appeared to be the first deep intronic variant in the FIG4 gene associated with YVS (Figure 4).
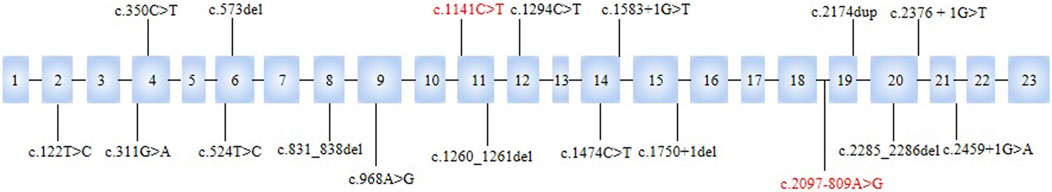
Figure 4. Locations of variants in FIG4 associated with YVS. c.2097-809A>G and c.1141C>T were identified in the present study.
To date, 18 variants of YVS have been described in previous publications and are shown in Figure 4. Of them, 61.1% (11 of 18) are loss-of-function variants, including 1 nonsense variant, 5 frameshift variants, and 5 splicing variants. c.122T>C p.(Ile41Thr), c.1474C>T p.(Arg492Cys), c.968A>G p.(Gln323Arg), c.311G>A(p.Gly104Asp), and c.524T>C alter highly conserved amino acid residues located at important domains and are predicted to affect FIG4 function (Campeau et al., 2013; Umair et al., 2021; Beauregard-Lacroix et al., 2024). In those cases, 10 patients had prenatal ultrasound manifestations (Table 1). The most frequent clinical feature in fetuses (60%, 6 of 10) is structural brain anomalies such as dilated ventricles and Dandy–Walker malformation rather than limb abnormalities (>75% of cases) (Beauregard-Lacroix et al., 2024). Only two children (20%, 2 of 10) had absent thumbs or halluces because of a low prenatal detection rate in prenatal evaluation (Vij et al., 2023). The affected individuals also presented with other manifestations, such as fetal pyelectasis, fetal hydrops, and growth retardation. It is worth noting that two patients in our study had abnormalities that have not been previously reported: a posterior fossa cyst and a low conus medullaris position with coccyx curvature abnormality. All of the cases were found in the third trimester, except one in the second trimester, which demonstrates a significant barrier to early diagnosis and intervention for patients’ families.
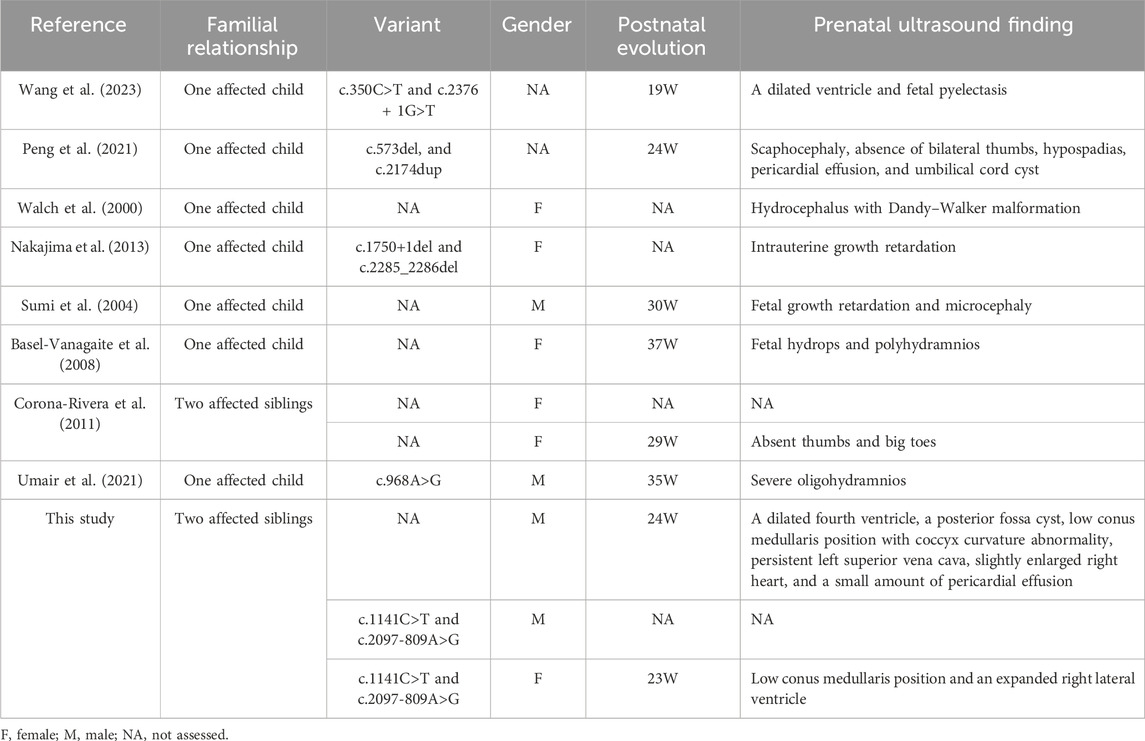
Table 1. Summary of prenatal ultrasound clinical findings in YVS.
Pathogenic deep intronic variants have been reported in more than 100 disease-associated genes. Given that the deep intronic variants identified in the present study could be detected using whole-genome sequencing and RT-PCR and splicing analysis are usually used to validate variants that have an effect on splicing in a molecular diagnostic setting (Vaz-Drago et al., 2017; Zhang et al., 2023), in this study, we used WGS to effectively detect deep intronic variants that have been widely employed in the diagnosis of genetic diseases. We detected the first deep intronic variant (c.2097-809A>G) in FIG4. Pathogenicity analysis of deep intronic variants in FIG4 can improve the genetic diagnosis rate of YVS and highlight the importance of screening of non-coding regions with functional assays to decipher the hidden variant in YVS. At the same time, in this family, spinal deformity was observed as a new phenotype in YVS. However, the small sample size is quite a limitation to our study. Therefore, additional efforts on large cohort studies are urgently needed to better clarify the genotype–phenotype correlation and provide refined genetic counseling.
5 Conclusion
We detected the first deep intronic variant in FIG4 (c.2097-809A>G) in a Chinese family with three neonates presenting the hallmark features of YVS, thereby linking this variant to the syndrome and expanding the unknown genetic variant spectrum. The splicing analysis shows how c.2097-809A>G influences the transcript and contributes to YVS. This study provides additional molecular and clinical information and broadens our understanding of the molecular mechanisms involved in the disease course.
Data availability statement
The datasets presented in this article are not readily available to assure patient confidentiality and participant privacy. Requests to access the datasets should be directed to eGlhbmdqaW5namluZzIwMTNAMTYzLmNvbQ==.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Suzhou Municipal Hospital, the Affiliated Suzhou Hospital of Nanjing Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
HT: Investigation, Writing – original draft, Conceptualization. QC: Data curation, Writing – review and editing, Resources, Formal Analysis. JX: Supervision, Writing – review and editing. QZ: Formal Analysis, Writing – review and editing, Resources, Data curation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Primary Research and Development Plan of Jiangsu Province (BE2022736) and the Jiangsu Maternal and Child Healthcare Key Discipline (FXK202142).
Acknowledgments
The authors thank the coordinators from the Chigene (Beijing) Translational Medical Research Center Co., Ltd., for sequencing technology support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdul Wahab Siddique, Z. A., Ammar Haider, H. K., and Karim, T. (2019). Yunis-varon syndrome. J. Ayub Med. Coll. Abbottabad 31 (2), 290–292.
Adès, L. C., Morris, L. L., Richardson, M., Pearson, C., and Haan, E. A. (1993). Congenital heart malformation in Yunis-Varón syndrome. J. Med. Genet. 30 (9), 788–792. doi:10.1136/jmg.30.9.788
Basel-Vanagaite, L., Kornreich, L., Schiller, O., Yacobovich, J., and Merlob, P. (2008). Yunis-Varon syndrome: further delineation of the phenotype. Am. J. Med. Genet. 146A (4), 532–537. doi:10.1002/ajmg.a.32135
Beauregard-Lacroix, É., Scott, A., Nguyen, T., Wierenga, K. J., Purcarin, G., Karstensen, A. B., et al. (2024). Exploring the phenotypic spectrum and osteopenia mechanisms in Yunis-Varón syndrome. Genet. Med. open 2, 2101837. doi:10.1016/j.gimo.2024.101837
Campeau, P. M., Lenk, G. M., Lu, J. T., Bae, Y., Burrage, L., Turnpenny, P., et al. (2013). Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am. J. Hum. Genet. 92 (5), 781–791. doi:10.1016/j.ajhg.2013.03.020
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Chow, C. Y., Zhang, Y., Dowling, J. J., Jin, N., Adamska, M., Shiga, K., et al. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448 (7149), 68–72. doi:10.1038/nature05876
Corona-Rivera, J. R., Romo-Huerta, C. O., López-Marure, E., Ramos, F. J., Estrada-Padilla, S. A., and Zepeda-Romero, L. C. (2011). New ocular findings in two sisters with Yunis-Varón syndrome and literature review. Eur J Med Genet. 54(1):76–81. doi:10.1016/j.ejmg.2010.09.013
Ferguson, C. J., Lenk, G. M., Jones, J. M., Grant, A. E., Winters, J. J., Dowling, J. J., et al. (2012). Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum. Mol. Genet. 21 (16), 3525–3534. doi:10.1093/hmg/dds179
Keren, H., Lev-Maor, G., and Ast, G. (2010). Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet. 11 (5), 345–355. doi:10.1038/nrg2776
Lees, J. A., Li, P., Kumar, N., Weisman, L. S., and Reinisch, K. M. (2020). Insights into lysosomal PI(3,5)P2 homeostasis from a structural-biochemical analysis of the PIKfyve lipid kinase complex. Mol. Cell 80 (4), 736–743.e4. doi:10.1016/j.molcel.2020.10.003
Martyn, C., and Li, J. (2013). Fig4 deficiency: a newly emerged lysosomal storage disorder. Prog. Neurobiol. 101-102, 35–45. doi:10.1016/j.pneurobio.2012.11.001
Mishra, A., Siwach, P., Misra, P., Dhiman, S., Pandey, A. K., Srivastava, P., et al. (2021). Intron exon boundary junctions in human genome have in-built unique structural and energetic signals. Nucleic Acids Res. 49 (5), 2674–2683. doi:10.1093/nar/gkab098
Nakajima, J., Okamoto, N., Shiraishi, J., Nishimura, G., Nakashima, M., Tsurusaki, Y., et al. (2013). Novel FIG4 mutations in Yunis-Varon syndrome. J. Hum. Genet. 58 (12), 822–824. doi:10.1038/jhg.2013.104
Nicholson, G., Lenk, G. M., Reddel, S. W., Grant, A. E., Towne, C. F., Ferguson, C. J., et al. (2011). Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain 134 (Pt 7), 1959–1971. doi:10.1093/brain/awr148
Peng, Y., Yang, S., Huang, X., Pang, J., Liu, J., Hu, J., et al. (2021). Whole exome sequencing analysis in fetal skeletal dysplasia detected by ultrasonography: an analysis of 38 cases. Front. Genet. 12, 12728544. doi:10.3389/fgene.2021.728544
Reutter, H., Bagci, S., Müller, A., Gembruch, U., Geipel, A., Berg, C., et al. (2012). Primary pulmonary hypertension, congenital heart defect, central nervous system malformations, hypo- and aplastic toes: another case of Yunis-Varón syndrome or report of a new entity. Eur. J. Med. Genet. 55 (1), 27–31. doi:10.1016/j.ejmg.2011.09.002
Sbrissa, D., Ikonomov, O. C., Fu, Z., Ijuin, T., Gruenberg, J., Takenawa, T., et al. (2007). Core protein machinery for mammalian phosphatidylinositol 3,5-bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel sac phosphatase joins the ArPIKfyve-PIKfyve complex. J. Biol. Chem. 282 (33), 23878–23891. doi:10.1074/jbc.M611678200
Sumi, M., Kusumoto, T., Kondoh, T., Moriuchi, H., Miyamoto, M., Masuzaki, H., et al. (2004). A case of Yunis-Varon syndrome complicated with complete cleft lip and palate. Am J Med Genet A. 125A(1):92–3. doi:10.1002/ajmg.a.20414
Umair, M., Alkharfy, T. M., Sajjad, S., and Alfadhel, M. (2021). FIG4-Associated Yunis-Varon syndrome: identification of a novel missense variant. Mol. Syndromol. 12 (6), 386–392. doi:10.1159/000516971
Vaz-Drago, R., Custódio, N., and Carmo-Fonseca, M. (2017). Deep intronic mutations and human disease. Hum. Genet. 136 (9), 1093–1111. doi:10.1007/s00439-017-1809-4
Vij, N., Goncalves, L. F., Llanes, A., Youn, S., and Belthur, M. V. (2023). Prenatal radiographic evaluation of congenital transverse limb deficiencies: a scoping review. World J. Orthop. 14 (3), 155–165. doi:10.5312/wjo.v14.i3.155
Walch, E., Schmidt, M., Brenner, R. E., Emons, D., Dame, C., Pontz, B., et al. (2000). Yunis-Varon syndrome: evidence for a lysosomal storage disease. Am J Med Genet. 95(2):157–60. doi:10.1002/1096-8628(20001113)95:2<157::aid-ajmg12>3.0.co;2-e
Wang, Y., Yin, F., Chai, Y., Jin, J., Zhang, P., Tan, Q., et al. (2023). Prenatal diagnosis of fetuses with ultrasound anomalies by whole-exome sequencing in Luoyang city, China. Front. Genet. 14, 141301439. doi:10.3389/fgene.2023.1301439
Yunis, E., and Varon, H. (1980). Cleidocranial dysostosis, severe micrognathism, bilateral absence of thumbs and first metatarsal bone, and distal aphalangia a new genetic syndrome. Am. J. Dis. Child., 134649–134653.
Zhang, C., Yan, Y., Zhou, B., Wang, Y., Tian, X., Hao, S., et al. (2023). Identification of deep intronic variants of PAH in phenylketonuria using full-length gene sequencing. Orphanet J. Rare Dis. 18 (1), 128. doi:10.1186/s13023-023-02742-1
Keywords: Yunis–Varón syndrome, FIG4 gene, deep intronic variant, whole-genome sequencing, prenatal diagnosis
Citation: Tang H, Chen Q, Xiang J and Zhang Q (2025) Identification and splicing analysis of the first deep intronic FIG4 variant causing Yunis–Varon syndrome. Front. Genet. 16:1624122. doi: 10.3389/fgene.2025.1624122
Received: 08 May 2025; Accepted: 14 July 2025;
Published: 08 August 2025.
Edited by:
Giulia Pascolini, Institute of Immaculate Dermatology (IRCCS), ItalyReviewed by:
Dora Janeth Fonseca, Rosario University, ColombiaKe Wu, Quzhou Maternity and Child Health Care Hospital, China
Copyright © 2025 Tang, Chen, Xiang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingjing Xiang, eGlhbmdqaW5namluZzIwMTNAMTYzLmNvbQ==; Qin Zhang, emhhbmdxMTEwMDA0QDE2My5jb20=