Abstract
Objective:
Hereditary spherocytosis (HS) is an inherited disorder characterized by spherical erythrocytes and abnormalities of several erythrocyte membrane proteins with extreme genotypic and phenotypic heterogeneity. HS patients were clinically diagnosed by the presence of spherical erythrocytes on the peripheral blood smear, hemolytic anemia, jaundice, and splenomegaly, with or without cholelithiasis or gallstones. To date, mutations of five genes (ANK1, EPB42, SLC4A1, SPTA1, and SPTB) have been reported to be associated with different subtypes of HS. Germline mutations of the SPTB gene cause autosomal dominant HS (Spherocytosis 2, SPH2), the rarest subtype of HS.
Methods:
In this study, we investigated a 10-year-old Chinese girl clinically diagnosed with HS and neonatal hemolytic anemia. The proband’s mother was also identified with HS and hemolytic anemia, but the proband’s father was phenotypically normal. We performed a standard G-banding karyotype to identify structural abnormalities of chromosomes in this proband. Then, we performed whole-exome sequencing and Sanger sequencing to identify the disease-causing variants in this proband. Finally, we functionally characterized the identified novel variant by performing reverse transcription polymerase chain reaction, cDNA sequencing, quantitative real-time polymerase chain reaction (PCR), and Western blot.
Results:
Whole exome sequencing identified a heterozygous novel splice-donor-site (c.647 + 1G>A) mutation in the SPTB gene in the proband. Sanger sequencing confirmed that the proband inherited this mutation from her mother, while her father was devoid of it. Reverse transcription polymerase chain reaction and cDNA sequencing showed that this novel splice-donor-site (c.647 + 1G>A) mutation causes abolition of the wild-type splice donor site, which leads to the aberrant splicing of SPTB mRNA, followed by the formation of an alternative transcript with complete loss of exon 5. The relative expression of mutated SPTB mRNA was significantly reduced in the proband and her mother compared with her father, showing normal expression of wild-type SPTB mRNA.
Conclusion:
Our present study highlighted the significance of whole-exome sequencing as the most promising path to genetic molecular diagnosis for patients with HS.
1 Introduction
Hereditary spherocytosis (HS) is an inherited disorder that manifests with spherical erythrocytes predominantly identified on a peripheral blood smear, abnormalities of several erythrocyte membrane proteins, and osmotically fragile spherocytes (Tole et al., 2020). Patients with HS are usually identified with hemolytic anemia, jaundice, and splenomegaly with or without cholelithiasis (Tole et al., 2020). The worldwide incidence of HS varies among different populations (Da Costa et al., 2013). In a Northern European population, the estimated prevalence of HS is 1 in 2000 live births, while in the Chinese population, the incidence of HS is extremely rare, affecting 1 in 100,000 live births (Da Costa et al., 2013; Perrotta et al., 2008). No significant difference in the prevalence of HS was found in male (0.18/1 million) and female (0.19/1 million) neonates (<1 year old) in the Chinese population (Wang et al., 2015). Patients with HS were usually identified with extreme phenotypic heterogeneity, which varies from asymptomatic to a severe form of the disease with skeletal abnormalities, short stature, and delayed puberty (Qin et al., 2020). HS is majorly inherited with an autosomal dominant pattern, while an autosomal recessive mode of inheritance or de novo variants have also been reported (Qin et al., 2020; Risinger and Kalfa, 2020). Germline mutations of the SPTB gene cause autosomal dominant HS (spherocytosis 2; SPH2; OMIM# 616649), the rarest form among all the HS subtypes (Qin et al., 2020; Delaunay, 2002). However, abnormalities or deficiencies of erythrocyte membrane proteins also showed genotypic and phenotypic heterogeneity among HS patients from populations of different ethnic origins (He et al., 2018; Andolfo et al., 2016).
The SPTB gene is located in the long arm of chromosome 14 (14q23.3) with a length of 100 kb. SPTB mRNA comprises 35 exons and is translated to the SPTB (β‐spectrin) protein with 2137 amino acids (Park et al., 2016). β-spectrin comprises five domains, namely, N-terminal calponin homology (CH) 1 and 2 domains (actin binding domain), spectrin domain 1 and 2 (dimerization domain), spectrin domain 3–13 and 16 (spectrin repeat domain), spectrin domain 14 and 15 (ankyrin-binding domain), and spectrin domain 17 (tetramerization domain) (Ipsaro et al., 2009). β‐spectrin is significantly involved in interacting with actin, ankyrin, and band 4.1 protein to organize and stabilize the erythrocyte plasma membrane (He et al., 2018; He et al., 2017; Boguslawska et al., 2017). Hence, a germline mutation in the SPTB gene causes the formation of partially or completely nonfunctional β‐spectrin, which may be unable to maintain the biconcave shape of human erythrocytes and causes a deficiency in erythrocyte membrane protein, finally resulting in HS (He et al., 2018; Ma et al., 2018; Fan et al., 2019). Germline mutations in the SPTB gene cause an autosomal dominant form of HS (Spherocytosis 2, SPH2), which accounts for approximately 15% of HS patients (Jang et al., 2019). To date, 57 mutations in the SPTB gene have been reported to be associated with HS. The majority of SPTB mutations are loss-of-function mutations, including frameshift, nonsense, or splice-site mutations that often lead to the formation of truncated β-spectrin (Jang et al., 2019).
In our study, we investigated a 10-year-old Han Chinese girl clinically diagnosed with HS and hemolytic anemia. The proband’s mother was also identified with HS and hemolytic anemia, but the proband’s father was phenotypically normal. Whole-exome sequencing identified a heterozygous novel splice-donor-site (c.647 + 1G>A) variant in the SPTB gene in the proband. Sanger sequencing confirmed that the proband’s mother also harbored this mutation, while her father did not carry it. Our present study not only expands the mutational spectrum of the SPTB gene associated with HS but also highlights the significance of whole-exome sequencing for genetic molecular diagnosis of HS patients.
2 Materials and methods
2.1 Participants and ethics statement
Here, we investigated a 10-year-old Chinese girl from nonconsanguineous Han Chinese parents (Figure 1A). The proband was clinically diagnosed with HS and hemolytic anemia. Peripheral blood samples were obtained from the proband and her parents at the Department of Clinical Laboratory, Shenzhen Children’s Hospital, No.7019, Yitian Road, Shenzhen, 518,038, Guangdong, China, for clinical diagnosis of the disease. Blood and serum biochemical tests were conducted in the Department of Clinical Laboratory, Shenzhen Children’s Hospital, Shenzhen, China. All participants provided their written informed consent for participating in this study. This study was formally approved by the Ethics Committee of The Shenzhen Children’s Hospital, Shenzhen, China. All procedures were performed following the approved guidelines.
FIGURE 1
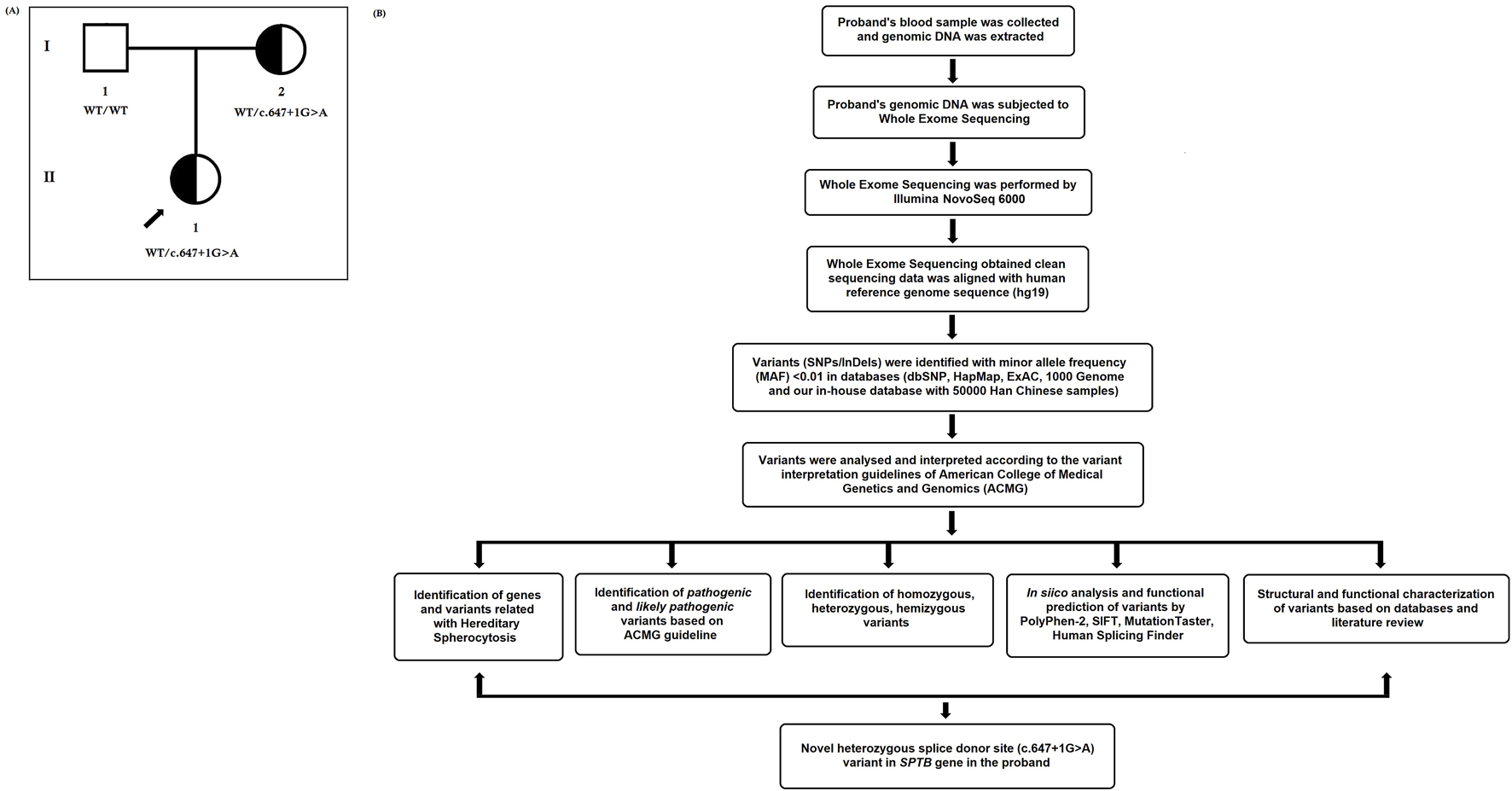
(A) Pedigree of the Chinese family with hereditary spherocytosis. Squares and circles denote males and females, respectively. Roman numerals indicate generations. Arrow indicates the proband (II-1). (B) Bioinformatics data analysis and interpretation pipeline for whole-exome sequencing.
2.2 Karyotyping analysis
Standard G-banding karyotype was performed to identify structural abnormalities of chromosomes in this proband (Zhang et al., 2020). We performed G-banding karyotype with the metaphase chromosomes obtained from temporary lymphocyte cultures of the peripheral blood of the proband according to the standard protocol (Zhang et al., 2020).
2.3 Chromosome microarray analysis (CMA)
Chromosome microarray analysis (CMA) was performed to identify copy number variations (CNV) in this proband (Zhang et al., 2020). Here, we performed CMA with the Affymetrix CytoScan HD array to identify CNVs in this proband. Affymetrix Chromosome Analysis Suite software (version 3.1) was used for CMA data analysis. Data presentation analysis has been done with reference to the NCBI37/hg19 genome assembly. Finally, the CMA data were analyzed and interpreted by using public databases [UCSC Genome Browser (https://genome.ucsc.edu/), OMIM (https://www.omim.org/), Database of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/), DECIPHER (https://www.deciphergenomics.org/), and ClinGen (https://www.clinicalgenome.org/)] with the copy number threshold of 10 kb and a marker count of ≥50. We used the UCSC Genome Browser (https://genome.ucsc.edu/) to verify the genomic content for all reported calls. OMIM (https://www.omim.org/) has been used to understand the genotype–phenotype genotype–phenotype correlation and inheritance pattern of copy number variations (CNVs). We used DGV (http://dgv.tcag.ca/dgv/) to confirm the human genomic structural variation, such as CNVs, inversions, and deletions. DECIPHER (https://www.deciphergenomics.org/) has been used to compare phenotypic and genotypic data from patients with rare diseases. We used ClinGen to understand the clinical relevance of genes and variants.
2.4 Whole exome sequencing
We performed whole exome sequencing for the proband. First, we collected the proband’s peripheral blood and extracted the genomic DNA, according to the manufacturer’s instructions (Zhang et al., 2020; Han et al., 2020). Whole-exome sequencing was performed with the proband’s genomic DNA (Zhang et al., 2020; Han et al., 2020). The sequencing library was prepared by capturing exome sequences using Agilent SureSelect version 6 (Agilent Technologies, Santa Clara, CA, United States). Then, an enriched sequencing library was subjected to whole-exome sequencing by using the Illumina NovaSeq 6000. Next, we obtained sequencing reads, and Burrows–Wheeler Aligner software (http://bio-bwa.sourceforge.net) (version 0.59) was used to align these sequencing reads with GRCh37. p10. Then, the Burrows–Wheeler aligned sequencing reads were again locally realigned by using GATK IndelRealigner (https://gatk. broadinstitute.org). After that, the base quality recalibration was performed with the Burrows–Wheeler aligned sequencing reads by using GATK Base Recalibrator (https://gatk.broadinstitute.org). Next, single-nucleotide variants (SNVs) and insertions or deletions (InDels) were identified by using GATK Unified Genotyper (https://gatk.broadinstitute.org). Then, all these identified variants (both SNVs and InDels) were annotated with the consensus coding sequence database (20,130,630) of the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi). Image analysis and base calling were performed through the Illumina pipeline (https://www.illumina.com/informatics/infrastructure-pipeline-setup.html). We designed indexed primers for data fidelity surveillance.
2.5 Bioinformatics data analysis and interpretation
All the variants obtained by whole-exome sequencing were collected. Variants were selected based on their minor allele frequency (MAF). We selected variants for bioinformatic data analysis and interpretation if their MAF was < 0.01 in the following public databases: dbSNP (https://www.ncbi.nlm.nih.gov), 1000 Genome Database (http://www.internationalgenome.org), HapMap (https://www.genome.gov), and our in-house database of 50,000 Han Chinese samples (Sherry et al., 2001; Clark et al., 2005; Auton et al., 2015). All these public databases were used to confirm the MAF of identified genetic variations in different populations. According to the MAF (cut off MAF is < 0.01), we can filter out the possible pathogenic variants for specific cases. First, we compared our identified variants with the Human Gene Mutation Database (HGMD, www.hgmd.cf.ac.uk/) (Stenson et al., 2003). HGMD is a comprehensive and updated database containing all the reported variants associated with monogenic disorders. Hence, by comparing our data with HGMD, we can interpret whether the identified genetic variants in our present study are novel variants or were previously reported to be associated with any monogenic diseases. Then, we compared our identified variants with the Online Mendelian Inheritance in Man (OMIM, https://www. omim.org) database (Hamosh et al., 2005). OMIM comprises detailed information on more than 16,000 genes and their associated Mendelian disorders, as well as their pattern of inheritance. Therefore, by comparing our identified genes and their variants with OMIM, we can understand the genotype–phenotype correlation and inheritance pattern of our identified genes and their variants. Next, we compared our identified variants with the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org) database (Karczewski et al., 2017). The ExAC database contains whole-exome sequencing data from large-scale sequencing projects from different populations globally. So, by comparing our identified genetic variations with ExAC data, we can confirm whether our identified genetic variations were already reported in some healthy individuals from different populations. After that, we compared our identified genetic variations with the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org) (Karczewski et al., 2020). The gnomAD consists of both whole-genome and whole-exome data from large-scale whole genome or whole exome sequencing projects from different populations worldwide. Therefore, by comparing our identified genetic variations with the gnomAD database, we can understand the frequency of the identified genetic variations in different populations, followed by confirming the possible pathogenicity of our identified genetic variations in this case. Then, we interpreted our identified genetic variations according to the variant interpretation guidelines of the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). Next, we used in silico webservers, namely, SIFT (https://sift.bii.a-star.edu.sg/index.html), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2), REVEL (https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=revel), Human Splicing Finder - Version 3.1 (http://www.umd.be/HSF/HSF.html), and MutationTaster (https://www.genecascade.org/MutationTaster2021/#transcript), to predict the possible pathogenic impacts of our identified genetic variations. Finally, after whole-exome sequencing data analysis and interpretation, we selected the most likely genetic variation underlying the disease phenotype in this case.
In addition, “Mutalyzer 2” software was used to confirm the expression of the genetic variant according to the HGVS rules (Lefter et al., 2021). The whole-exome sequencing quality control (QC) data of the proband is described in Table 1. The bioinformatic data analysis and interpretation are schematically presented in Figure 1B.
TABLE 1
| Total | Raw reads (All reads) | 417698473 |
| QC fail reads | 0 | |
| Raw data (Mb) | 21,102.86 | |
| Paired reads | 417698473 | |
| Mapped reads | 415569832 | |
| Fraction of mapped reads | 99.45% | |
| Mapped data (Mb) | 20,984.64 | |
| Fraction of mapped data (Mb) | 99.45% | |
| Properly paired | 405484540 | |
| Fraction of properly paired | 97.42% | |
| Read and mate paired | 414575354 | |
| Fraction of read and mate paired | 99.29% | |
| Singletons | 1148634 | |
| Read and mate map to different chromosome | 7802745.00% | |
| Read1 | 218232782 | |
| Read2 | 218232782 | |
| Read1 (rmdup) | 129342787 | |
| Read2 (rmdup) | 129312329 | |
| Forward strand reads | 217281240 | |
| Backward strand reads | 217248084 | |
| PCR duplicate reads | 186884654 | |
| Fraction of PCR duplicate reads | 43.87% | |
| Map quality cutoff value | 20 | |
| Map quality above cutoff reads | 381341984 | |
| Fraction of Map Q reads in all reads | 91.54% | |
| Fraction of Map Q reads in mapped reads | 91.84% | |
| Target | Target reads | 243693624 |
| Fraction of target reads in all reads | 56.93% | |
| Fraction of target reads in mapped reads | 57.19% | |
| Target data (Mb) | 11,846 | |
| Target data Rmdup (Mb) | 5983.84 | |
| Fraction of target data in all data | 52.42% | |
| Fraction of target data in mapped data | 52.97% | |
| Len of region | 60573987 | |
| Average depth | 183.57 | |
| Average depth (rmdup) | 98.97 | |
| Coverage (>0×) | 99.86% | |
| Coverage ( ≥ 4×) | 99.75% | |
| Coverage ( ≥ 10×) | 99.45% | |
| Coverage ( ≥ 30×) | 96.45% | |
| Coverage ( ≥ 100×) | 72.45% | |
| Target region count | 200,921 | |
| Region covered > 0× | 200,368 | |
| Fraction region covered > 0× | 99.87% | |
| Fraction region covered ≥ 4× | 99.68% | |
| Fraction region covered ≥ 10× | 99.32% | |
| Fraction region covered ≥ 30× | 96.93% | |
| Fraction region covered ≥ 100× | 72.84% | |
| Flank | Flank size | 200 |
| Len of region (not include target region) | 71847296 | |
| Average depth | 39.54 | |
| Flank reads | 77394593 | |
| Fraction of flank reads in all reads | 18.42% | |
| Fraction of flank reads in mapped reads | 18.79% | |
| Flank data (Mb) | 2793.72 | |
| Fraction of flank data in all data | 12.72% | |
| Fraction of flank data in mapped data | 12.93% | |
| Coverage (>0×) | 96.93% | |
| Coverage ( ≥ 4×) | 83.87% | |
| Coverage ( ≥ 10×) | 60.00% | |
| Coverage ( ≥ 30×) | 35.87% | |
| Coverage ( ≥ 100×) | 9.93% |
Quality control (QC) data of whole-exome sequencing of the proband.
2.6 Sanger sequencing
We performed Sanger sequencing to validate the disease-causing variant identified by whole-exome sequencing in the proband. Sanger sequencing was performed for the proband and her parents. First, we performed polymerase chain reaction (PCR). Primers were designed according to the reference genomic sequences of the Human Genome from GenBank in NCBI. Sanger sequencing was performed with the PCR products, and data were compared and analyzed.
Whole exome sequencing obtained a heterozygous novel splice-donor site variation, which was validated by Sanger sequencing. Primers used for Sanger sequencing were as follows: F′ 5′- AGCCTCTGTGTGTGGTTAGC -3’; R 5′- CAGCCCTAGCATGAAGCAGA‐3′(Product length: 534 bp; Products on intended targets: >NC_000014.9, Homo sapiens, chromosome 14, GRCh38. p14). The reference sequence NM_001024858 of SPTB was used.
2.7 Reverse transcription polymerase chain reaction (RT-PCR) and cDNA sequencing
Reverse transcription polymerase chain reaction (RT-PCR) and cDNA sequencing were performed to understand the effect of this heterozygous novel splice-donor site mutation of the SPTB gene on the splicing event of SPTB mRNA. We isolated and extracted total RNA from the peripheral blood samples of the proband and her parents. Then, we performed RT-PCR and obtained cDNA from the total RNA according to the manufacturer’s protocol (TAKARA RR037A, Shanghai, China). After that, primers were designed to encompass the coding sequence from exon 4 to exon 6. These primers were used to amplify the cDNA from the proband and her parents. After cDNA amplification, the PCR fragments were recovered and subjected to sequencing, followed by data analysis.
2.8 Quantitative real-time PCR
Quantitative real-time PCR (q-PCR) has been performed to identify the relative expression of wild type and mutated SPTB mRNA in the proband and her parents. After isolating and extracting the total RNA from the peripheral blood samples of the proband and her parents, we synthesized cDNA from total RNA by performing RT-PCR, according to the manufacturer’s protocol (TAKARA RR037A, Shanghai, China). Then, cDNAs of the proband and her parents were collected for fluorescence quantitative detection by quantitative real-time PCR (q-PCR). The q-PCR was performed with both the target gene and housekeeping gene (GAPDH) for each sample. The relative expression of both wild type and mutated SPTB mRNAs was analyzed by calculating the detected cycle threshold (Ct) value. The sequences of primers for q-PCR were as follows: SPTB- F- 5′- AAGGTCGTGAAACACGCTCA -3′, SPTB- R- 5′- TTGAATGCGTGCTCCAGGTT -3’; GAPDH- F- 5′-TCTGACTTCAACAGCGACACC-3′, GAPDH- R- 5′-CTGTTGCTGTAGCCAAATTCGT-3’.
2.9 Western blot
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using the density gradient separation method. Cells were lysed by RIPA lysis buffer (#P0013B, Beyotime, China) with added protease and phosphatase inhibitors. The lysate was centrifuged at 13,000 rpm and 4 °C for 10 min, and the cellular protein was collected. Total cellular proteins were separated on an 8% SDS-PAGE analysis and transferred to the Porablot PVDF membrane. Blocking was performed by incubating the membranes with Tris-buffered saline (TBS), pH 7.4, with 0.05% Tween (TBS-T) containing 3% bovine serum albumin (BSA). Membranes were incubated with primary antibodies, rotating at 4 °C overnight, washed three times with TBS-T, and incubated with secondary antibodies for 1 h at room temperature. Primary antibodies [the mouse anti-SPTB (1:1000 in TBS-T; Proteintech, United States of America, #26936-1-AP) and mouse anti-GAPDH (1:1000, Santa Cruz, United States of America, #sc-47724)] and secondary antibody [Goat anti-Mouse IgG HRP (1:2000, Thermos, 31,430)] were used here. Detection of immunoreactive bands was performed using the Thermo Scientific™ SuperSignal™ West Pico PLUS Chemiluminescent Substrate (#34577), according to the manufacturer’s instructions.
3 Results
3.1 Pedigree and clinical characteristics
The proband (II-1) is a 10‐year‐old Han Chinese girl from nonconsanguineous Chinese parents (Figure 1A). She is the first and only child of her parents. The proband clinically manifested HS and hemolytic anemia. Her routine blood test and reticulocyte count results showed increased neutrophil absolute value, neutrophil ratio, red blood cell variation coefficient, red blood cell distribution width, absolute value of reticulocyte, percentage of reticulocytes, reticulum ratio, immature reticulocyte ratio with decreased lymphocyte ratio, eosinophil ratio, red blood cell, hemoglobin, hematocrit, platelet distribution width, and reticulated platelet ratio (Table 2). However, glucose 6-phosphate dehydrogenase activity was found to be normal in the proband (Table 3). The red blood cell osmotic fragility test showed no abnormalities in the proband (Table 3). Hemoglobin A and Hemoglobin A2 were normal, and little elevation of Hemoglobin F was found in the proband (Table 3). Both a direct anti-human globulin test (DAT) and an indirect anti-human globulin test (IAT) were negative for the proband. An antibody screening test for the proband was also negative. The total serum protein test showed an increased level of total bilirubin (both TBIL and indirect bilirubin (IBIL) were elevated) and lactate dehydrogenase, with decreased levels of serum prealbumin, creatinine, serum bicarbonate, and iron in the proband (Table 4). Erythrocyte membrane CD55 and CD59 expressions were completely positive in the proband. In addition, an acidified glycerol dissolution test for 90 s was also positive for the proband. An abdominal ultrasound of the proband revealed diffuse enlargement of the spleen. A peripheral blood smear result showed the presence of spherical erythrocytes (10%) in the proband (Figure 2A).
TABLE 2
| Test item | Proband | Proband’s mother | Proband’s father | References | Unit |
|---|---|---|---|---|---|
| Leukocytes | 10.5 | 9.97 | 5.94 | 5–12 | 109/L |
| Neutrophil absolute value | 8.54 ↑ | 6.78 | 3.2 | 2–7 | 109/L |
| Absolute lymphocyte value | 1.56 | 2.63 | 2.22 | 0.8–4 | 109/L |
| Absolute value of monocytes | 0.35 | 0.42 | 0.41 | 0.12–1 | 109/L |
| Eosinophil absolute value | 0.02 | 0.04 | 0.08 | 0.02–0.5 | 109/L |
| Basophil absolute value | 0.03 | 0.1 | 0.03 | 0–0.1 | 109/L |
| Neutrophil ratio | 81.3 ↑ | 68 | 53.9 | 50–70 | % |
| Lymphocyte ratio | 14.9 ↓ | 26.4 | 37.4 | 20–40 | % |
| Mononuclear cell ratio | 3.3 | 4.2 | 6.9 | 3–10 | % |
| Eosinophil ratio | 0.2 ↓ | 0.4↓ | 1.3 | 0.5–5 | % |
| Ratio of basophils | 0.3 | 1 | 0.5 | 0–1 | % |
| Red blood cells | 2.99 ↓ | 3.52 | 4.45 | 3.5–5.5 | 1012/L |
| Hemoglobin | 96 ↓ | 113 | 152 | 110–160 | g/L |
| Hematocrit | 26.8 ↓ | 30.9 | 42.8 | 30–45 | % |
| Average red blood cell volume | 89.6 | 87.8 | 96.2 | 82–99 | fL |
| Average red blood cell hemoglobin content | 32.1 | 32.1 | 34.2 | 27–33 | pg |
| Mean red blood cell hemoglobin concentration | 358 | 366↑ | 355 | 320–360 | g/L |
| Red blood cell variation coefficient | 19.4 ↑ | 17.4↑ | 12.1 | 0–15 | % |
| Red blood cell distribution width | 63.1 ↑ | 54.4↑ | 42.8 | 37–50 | fL |
| Platelets | 300 | 212 | 351 | 100–300 | 109/L |
| Mean platelet volume | 8.8 | 9.2 | 8.6 | 7.4–10.4 | fL |
| Platelet packed volume | 0.25 | 0.18 | 0.28 | 0.108–0.28 | % |
| Platelet distribution width | 8.5 ↓ | 9.6 | 8.8 | 9–17 | fL |
| Large platelet ratio | 14.5 | 19.2 | 14.2 | 13–43 | % |
| Absolute value of nucleated red blood cells | 0.01 | 0 | 0 | 0–0.5 | 109/L |
| Proportion of nucleated red blood cells | 0.1 | 0 | 0 | 0–1 | /100WBC |
| Absolute value of reticulocyte | 405.4 ↑ | 287.9↑ | 70.3 | 17–70.1 | 109/L |
| Percentage of reticulocytes | 13.6 ↑ | 8.2↑ | 1.6 | 0.43–1.36 | % |
| Low fluorescence intensity reticulum ratio | 85.3↓ | 83.5↓ | 93.5 | 89.9–99.4 | % |
| Medium fluorescence intensity reticulum ratio | 11↑ | 11.4↑ | 6.3 | 1.6–9.5 | % |
| High fluorescence intensity reticulum ratio | 3.7↑ | 5.1↑ | 0.2 | 0–1.7 | % |
| Immature reticulocyte ratio | 14.7↑ | 16.5↑ | 6.5 | 1.6–10.5 | % |
| Reticulum hemoglobin content | 34.2 | 34 | 35.9 | 32.1–38.8 | pg |
| Reticulated platelet ratio | 0.7↓ | 1.4 | 1.1 | 0.8–6.3 | % |
Routine blood test results of proband, proband’s mother, and proband’s father.
TABLE 3
| Test | Result | References | Unit |
|---|---|---|---|
| Glucose 6-phosphate dehydrogenase activity | 1.5 | >1 | |
| Red blood cell osmotic fragility | 88.5 | 60–100 | % |
| Hemoglobin H | - | 0 | % |
| Hemoglobin Barts | - | 0 | % |
| Hemoglobin A | 94.9 | 94.5–96.5 | % |
| Hemoglobin F | 2.5↑ | 0.26–2.3 | % |
| Hemoglobin A2 | 2.6 | 2.5–3.5 | % |
| Hemoglobin A2+E | - | 0 | % |
| Hemoglobin N | - | 0 | % |
| Hemoglobin J | - | 0 | % |
| Hemoglobin K | - | 0 | % |
| Hemoglobin G | - | 0 | % |
| Hemoglobin D | - | 0 | % |
| Hemoglobin E | - | 0 | % |
| Hemoglobin constant spring | - | 0 | % |
Hemoglobin test results of the proband.
TABLE 4
| Test | Results | References | Unit |
|---|---|---|---|
| Total protein | 63.6 | 46–80 | g/L |
| Albumin | 41.2 | 35–55 | g/L |
| Globulin | 22.4 | 20–30 | g/L |
| Albumin–globulin ratio | 1.84 | 1.1–2.5 | |
| Serum prealbumin | 131.7↓ | 149–307 | mg/L |
| Retinol-binding protein | 15.3 | 5.9–46.6 | mg/L |
| Total bile acid | 5 | 0–15 | μmol/L |
| Total bilirubin | 31.4↑ | 0.9–17.1 | μmol/L |
| Direct bilirubin | 11.5↑ | 0–6.08 | μmol/L |
| Indirect bilirubin | 19.9↑ | 2–17 | μmol/L |
| Alanine transamination | 8 | 0–40 | IU/L |
| Aspartic acid | 35 | 0–40 | IU/L |
| Alkaline phosphatase | 115 | 40–500 | IU/L |
| γ-glutamyl transpeptidase | 8 | 0–50 | IU/L |
| Urea | 4.03 | 2.5–6.0 | mmol/L |
| Creatinine | 42.2↓ | 49–90 | μmol/L |
| Cystatin C | 0.87 | 0.6–1.55 | mg/L |
| Uric acid | 275.79 | 90–420 | μmol/L |
| Creatine kinase | 56.8 | 24–229 | IU/L |
| Creatine kinase isoenzyme | 0.3 | 0–6.8 | ng/mL |
| Lactate dehydrogenase | 318↑ | 140–280 | IU/L |
| Myoglobin | 12 | <140.2 | ng/mL |
| Troponin | 0 | <0.018 | ng/mL |
| Sodium | 134.9↓ | 135–146 | mmol/L |
| Potassium | 3.96 | 3.5–5.5 | mmol/L |
| Chlorine | 105.5 | 101–111 | mmol/L |
| Calcium | 2.23 | 2.2–2.7 | mmol/L |
| Serum bicarbonate | 17.69↓ | 20.3–30.3 | mmol/L |
| Magnesium | 0.84 | 0.7–1.15 | mmol/L |
| Iron | 6.1↓ | 9–32.6 | μmol/L |
| Inorganic phosphoric acid | 1.45 | 0.96–2.1 | mmol/L |
| Anion gap | 16 | 6–20 | mmol/L |
| Immunoglobulin G | 11.75 | 5.28–21.9 | g/L |
| Immunoglobulin M | 0.83 | 0.48–2.26 | g/L |
| Immunoglobulin A | 1.95 | 0.51–2.97 | g/L |
| Complement C3 | 0.68↓ | 0.7–2.06 | g/L |
| Complement C4 | 0.23 | 0.11–0.61 | g/L |
Total serum protein test results of the proband.
FIGURE 2
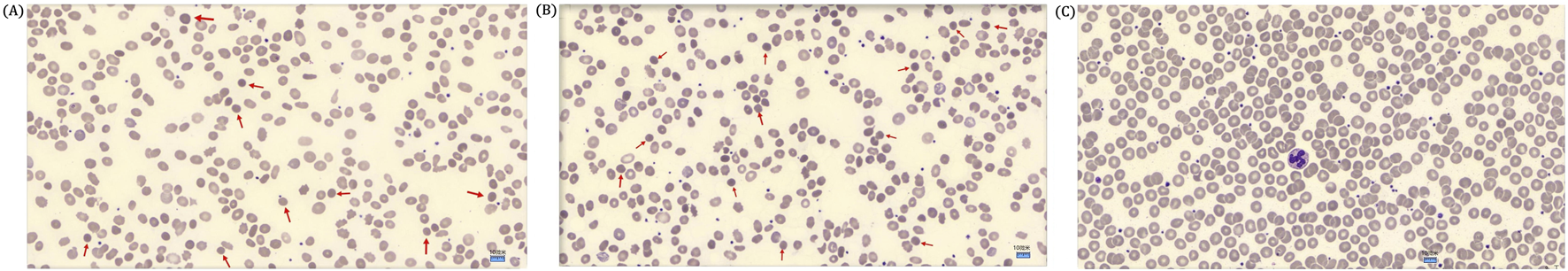
(A) Peripheral blood smear of the proband showed spherical erythrocytes. The red arrow marks a spherical erythrocyte (DI-6×0Microscope, ×1000 magnification). (B) A peripheral blood smear of the proband’s mother showed few spherical erythrocytes. A red arrow marks a spherical erythrocyte (DI-6×0Microscope, ×1000 magnification). (C) A peripheral blood smear of the proband’s father showed no spherical erythrocytes (DI-6×0Microscope, ×1000 magnification).
The proband’s mother (I-2) was a 37-year-old Han Chinese woman who had experienced HS and hemolytic anemia. Her routine blood test and reticulocyte count results showed increased mean red blood cell hemoglobin concentration, red blood cell variation coefficient, red blood cell distribution width, absolute value of reticulocyte, percentage of reticulocytes, and an immature reticulocyte ratio with a decreased eosinophil ratio (Table 2). She was also found to have a decreased level of folic acid (9.64 ng/mL, reference range: >10.6 ng/mL). The total serum protein test showed increased levels of total bilirubin (TBIL and IBIL were elevated), erythropoietin, and soluble transferrin receptor with decreased levels of serum prealbumin and alkaline phosphatase (Table 5). A peripheral blood smear result found few oval erythrocytes and very few spherical erythrocytes (Figure 2B).
TABLE 5
| Test | Result | References | Unit |
|---|---|---|---|
| Total protein | 73.8 | 46–80 | g/L |
| Albumin | 43.9 | 35–55 | g/L |
| Globulin | 29.9 | 20–30 | g/L |
| Albumin/globulin ratio | 1.47 | 1.1–2.5 | |
| Serum prealbumin | 196.7↓ | 200–400 | mg/L |
| Retinol-binding protein | 33.9 | 25–70 | mg/L |
| Total bile acid | 3 | 0–15 | μmol/L |
| Glycine | 0.68 | 0–6.7 | mg/L |
| Total bilirubin | 36.9↑ | 5–21 | μmol/L |
| Direct bilirubin | 9.9↑ | 0–6.06 | μmol/L |
| Indirect bilirubin | 27↑ | 2–17 | μmol/L |
| Alanine aminotransferase | 4 | 0–40 | IU/L |
| Aspartate aminotransferase | 11 | 0–40 | IU/L |
| Alkaline phosphatase | 34↓ | 40–500 | IU/L |
| γ-glutamyl transpeptidase | 8 | 0–50 | IU/L |
| Iron | 15.4 | 9–32.6 | μmol/L |
| Ferritin | 29.36 | 10–291 | ng/mL |
| Serum transferrin (immunoturbidimetric method) | 2.05 | 2–3.6 | g/L |
| Erythropoietin | 40.9↑ | 1.46–31.88 | mIU/L |
| Soluble transferrin receptor | 4.3↑ | 0.76–1.76 | mg/L |
Total serum protein analysis result of the proband’s mother.
The proband’s father (I-1) was a 40-year-old Han Chinese man. He is phenotypically normal. A routine blood test result of the proband’s father showed no abnormalities (Table 2). A peripheral blood smear result found no spherical erythrocytes (Figure 2C).
According to biochemical tests and the peripheral blood smear experiment, the proband was clinically diagnosed with HS with hemolytic anemia according to the diagnostic thresholds for HS in practice guidelines.
3.2 Karyotype and chromosomal microarray analyses
We identified no chromosomal structural abnormalities in the proband by karyotype analysis (46, XX) (Figure 3A). We have not found any pathogenic copy number variations (CNVs) in the chromosomes of the proband by CMA (Figure 3B).
FIGURE 3
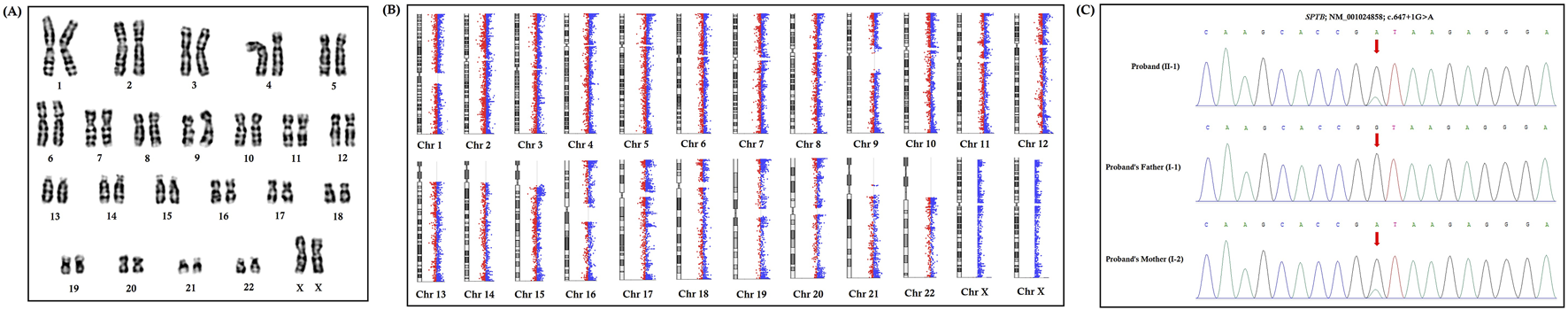
Karyotype analysis, chromosome microarray analysis, and Sanger sequencing. (A) Karyotype analysis identified no chromosomal structural abnormalities in the proband (46, XX). (B) Chromosome microarray analysis identified no pathogenic copy number variations (CNVs) in the proband. (C) Partial DNA sequences in the SPTB gene were obtained by Sanger sequencing of the family. The reference sequence NM_001024858 of the SPTB gene was used.
3.3 Identification of a splice-donor site mutation in the SPTB gene
Whole-exome sequencing identified a novel heterozygous splice-donor site mutation (c.647 + 1G>A) in the first base of intron 5 of the SPTB gene in this proband (Figure 3C). Sanger sequencing also confirmed that this heterozygous novel mutation was also present in the proband’s mother, while the proband’s father did not harbor this mutation (Figure 3C). Therefore, the proband inherited this heterozygous novel mutation from her mother.
This heterozygous novel mutation (c.647 + 1G>A) has not been found in 100 ethnically matched normal healthy controls. This mutation was also not found in the Human Gene Mutation database (HGMD, www.hgmd.cf.ac.uk/), Online Mendelian Inheritance in Man (MIM, (https://www.omim.org), Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org), dbSNP (https://www.ncbi.nlm.nih.gov), Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org), International Genome Sample Resource (1000 Genome Database, http://www.internationalgenome.org), or our in-house database which consists of ∼50,000 Chinese Han samples.
According to the variant interpretation guidelines of the American College of Medical Genetics and Genomics (ACMG), this novel heterozygous splice-donor site mutation (c.647 + 1G>A) was classified as [PVS1, PS3, PM2, PP1] “likely pathogenic” variants (Richards et al., 2015).
In addition, this novel heterozygous splice-donor site mutation (c.647 + 1G>A) was predicted to be a “Broken Wild Type (WT) Donor Site: Alternation of the Wild Type (WT) Donor site,” most probably affecting splicing, by Human Splicing Finder Pro (https://www.genomnis.com/hsf).
This novel heterozygous splice-donor site mutation (c.647 + 1G>A) in the first base of intron 5 of the SPTB gene was co-segregated well with the disease phenotype in this family with an autosomal dominant mode of inheritance.
3.4 Functional characterization of the splice-donor site mutation
The novel heterozygous splice-donor site (c.647 + 1G>A) mutation in the first base of intron 5 of the SPTB gene disrupts the wild type SPTB exon 5 splice donor site. RT-PCR and cDNA sequencing showed normal splicing of exon 4 to exon 6 in the proband’s father, while a complete loss of exon 5 (81 bp) was identified in the proband and her mother (Figures 4A–C). The normal and aberrant splicing of SPTB mRNA upon this splice-donor-site mutation is schematically presented in Figures 4D,E.
FIGURE 4
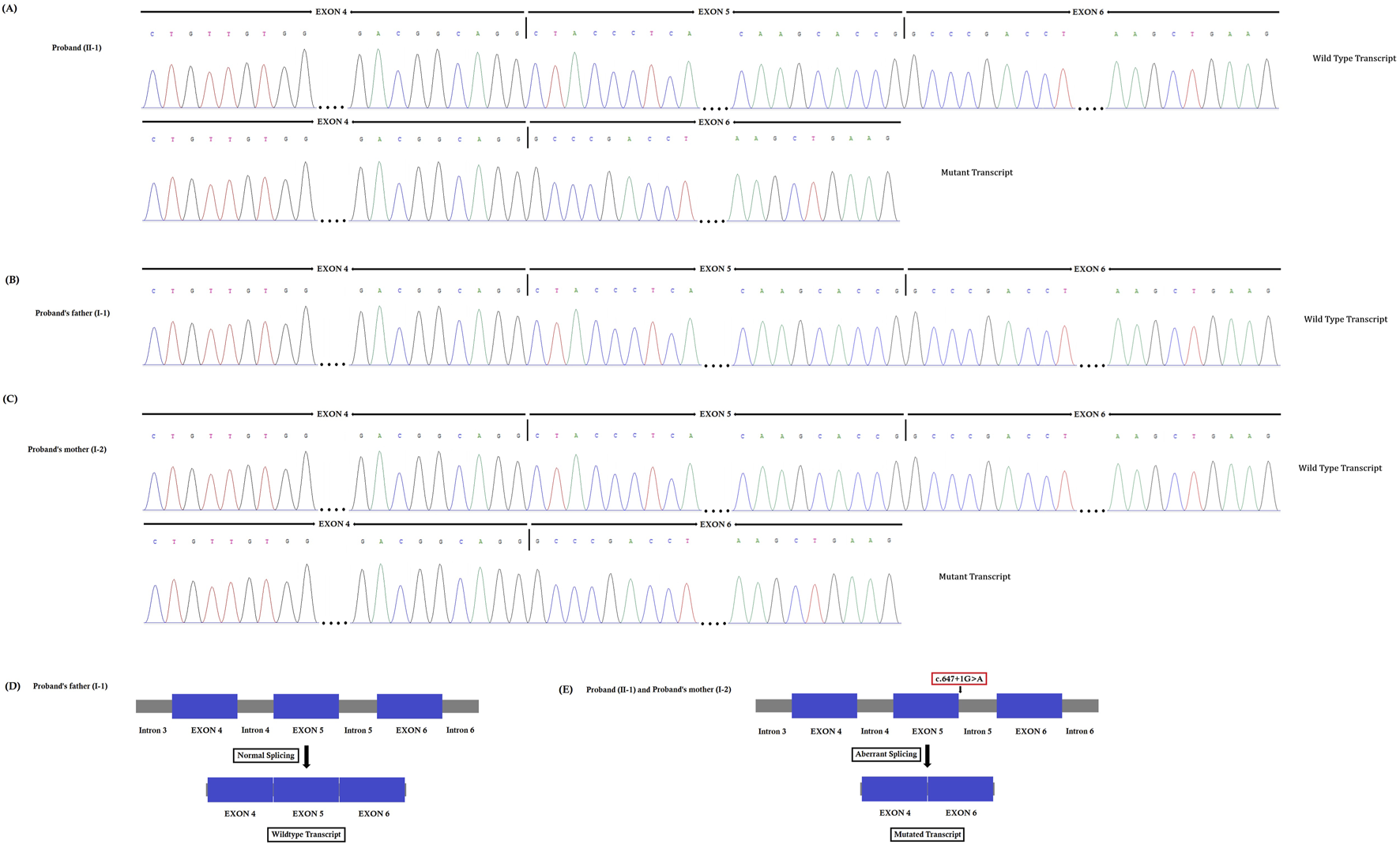
(A–C). Reverse transcription polymerase chain reaction (RT-PCR) and cDNA sequencing. This novel heterozygous splice-donor site (c.647 + 1G>A) mutation in the first base of intron 5 of the SPTB gene disrupts the wild type SPTB exon 5 splice donor site. RT-PCR and Sanger sequencing of SPTB cDNA showed normal splicing of exon 4 to exon 6 in the proband’s father, (B) while a complete loss of exon 5 (81 bp) was identified in the proband (A) and her mother (C). (D–E). Schematic presentation of normal and aberrant splicing of SPTB mRNA.
3.5 Relative expression of SPTB mRNA by quantitative real-time PCR
Relative expression of SPTB mRNA revealed a significantly decreased mutated SPTB transcript level in the proband and her mother, while the proband’s father showed normal expression of wild-type SPTB mRNA (Figure 5A). Our result also suggested that the mutant SPTB transcript was present at a detectable level in the proband and her mother and was not probably degraded by the nonsense-mediated mRNA decay pathway.
FIGURE 5
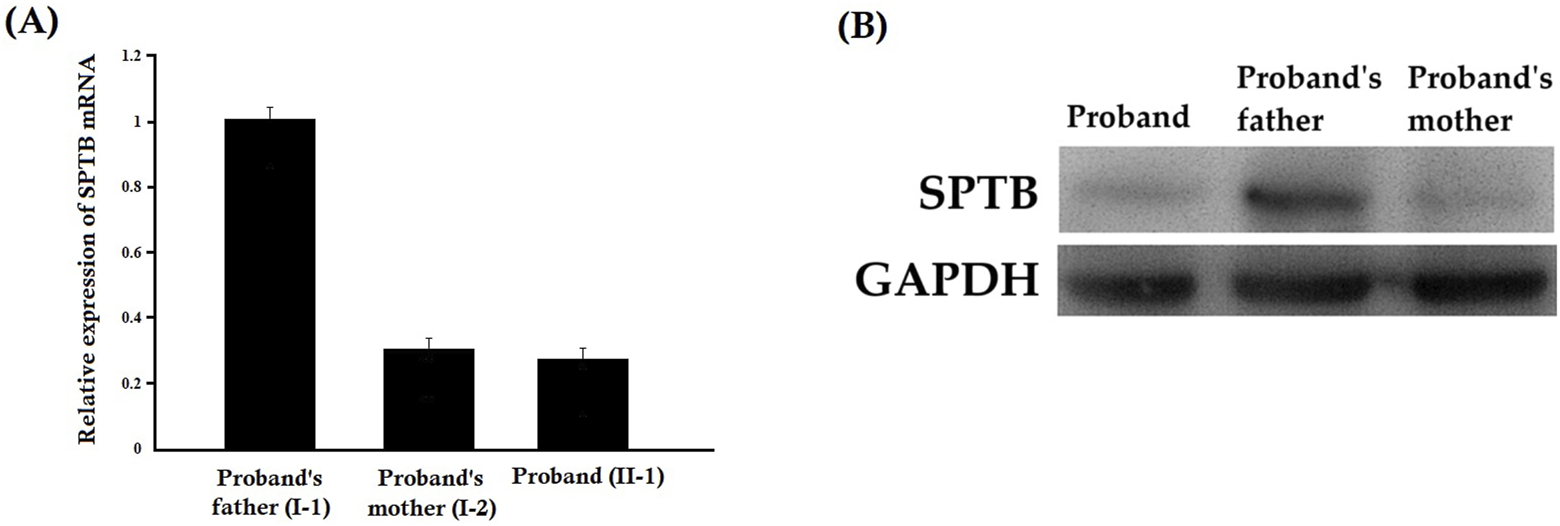
(A) Relative expression of SPTB mRNA by quantitative real-time PCR (q-PCR). Relative expression of SPTB mRNA revealed a significantly decreased SPTB transcript level in the proband and her mother, while the proband’s father showed normal expression of SPTB mRNA. Data analysis was performed by using the comparative threshold cycle (2-△△CT) method. (B) Relative expression of SPTB protein by Western blot. The relative expression of the SPTB protein showed a significantly decreased mutated SPTB protein level in the proband and her mother, while the proband’s father showed normal expression of wild type SPTB protein.
3.6 Relative expression of the SPTB protein by Western blot
The relative expression of the SPTB protein showed a significantly decreased mutated SPTB protein level in the proband and her mother, while the proband’s father showed normal expression of wild-type SPTB protein (Figure 5B). Notably, a truncated protein product corresponding to the predicted size of the mutant protein (lacking 27 amino acids) was not detected. This suggests that the mutant protein is likely unstable and degraded, and that the clinical phenotype results from haploinsufficiency of the wild-type SPTB allele (Supplementary Figure S1).
4 Discussion
Here, we investigated a 10-year-old Chinese girl with hereditary spherocytosis and hemolytic anemia. Whole-exome sequencing and Sanger sequencing identified a novel heterozygous splice-donor site mutation (c.647 + 1G>A) in intron 5 of the SPTB gene in this proband. The proband inherited this variant from her mother, while the proband’s father did not carry it.
We know that any splice-site mutation exerts its effect by causing aberrant splicing of mutated mRNA, followed by the formation of an alternative transcript with partial or complete retention of intron or skipping of exons. In order to understand the effect of this heterozygous novel splice-donor site mutation (c.647 + 1G>A), we performed RT-PCR and subsequent cDNA sequencing of both wild type and mutated SPTB mRNA from the proband and her parents. Our results showed that this novel spice site mutation causes aberrant splicing of SPTB mRNA, followed by the formation of an alternative transcript with complete loss of exon 5. The exon 5 of SPTB mRNA consists of 81 bp, translating into 27 amino acids, constituting part of calponin homology (CH) 2 domains significantly involved in actin binding. Hence, this mutation causes partial loss of the CH2 domain and may reduce the actin-binding ability of the mutated β-spectrin protein. The wild-type β-spectrin protein and the mutated β-spectrin protein are schematically presented in Figures 6 A,B.
FIGURE 6
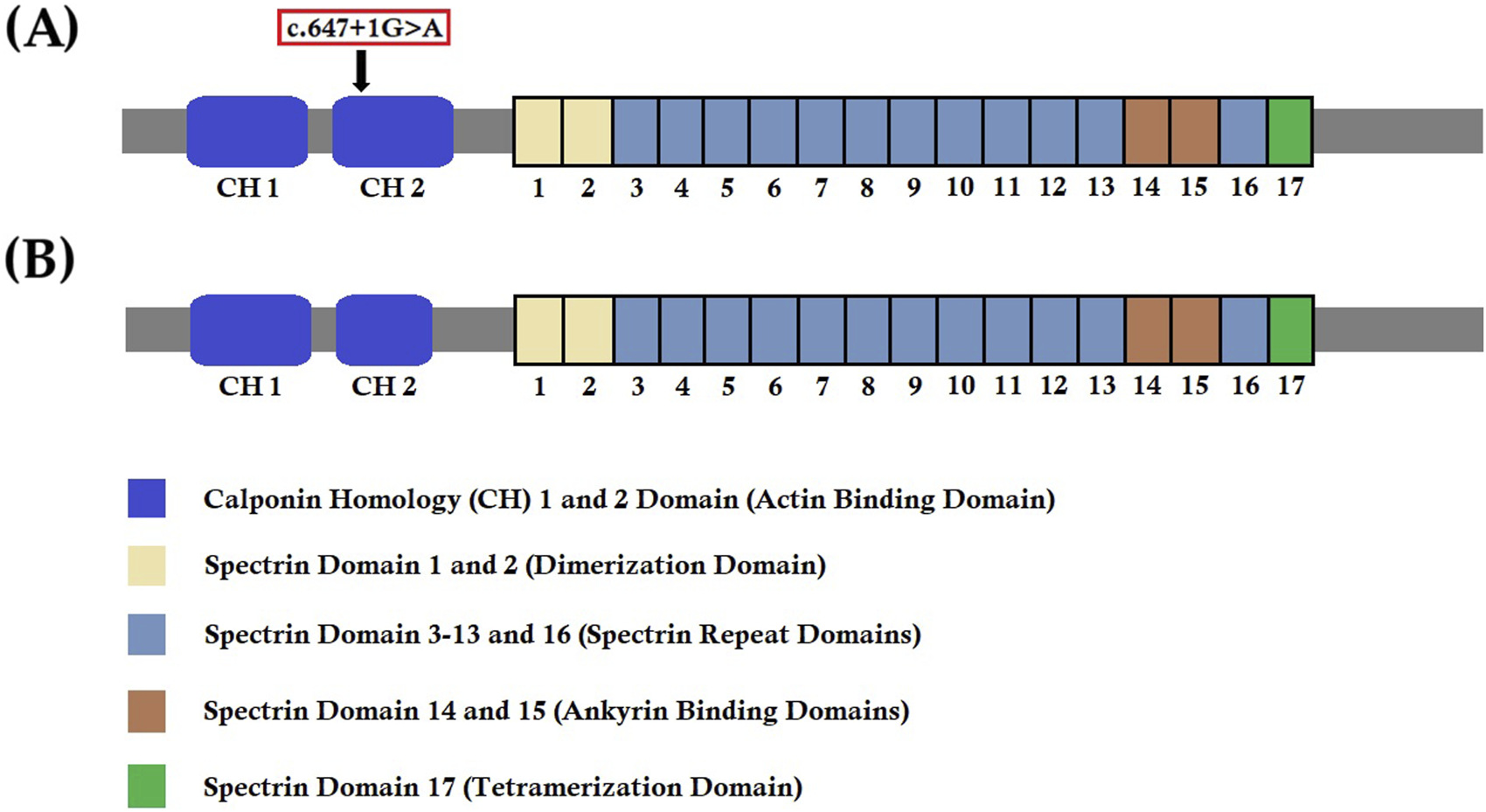
Wild-type and mutant SPTB protein structures. (A) Wild-type structure of the SPTB protein showing the location of the identified mutation (c.647G>A) in our study. (B) Mutated structure of the SPTB protein after complete loss of exon 5 (81 bp).
Our results also showed the presence of both the wild-type and mutated SPTB transcripts in the proband and her mother, whereas only the wild-type SPTB transcript was found in the proband’s father, suggesting that mutant mRNA was present in detectable levels and was probably not degraded by the nonsense-mediated mRNA decay pathway. In addition, we also found that the relative expressions of mutated SPTB mRNA of the proband and her mother were significantly reduced compared with the wild-type SPTB mRNA of her father. Therefore, this splice-site mutation identified in our study exerts its dominant negative effect as well as causes β-spectrin haploinsufficiency underlying the disease phenotype in this family.
The clinical diagnosis of HS patients is usually performed based on the presence of spherical erythrocytes on the peripheral blood smear, extravascular hemolysis, and physical findings with a positive family history. However, the clinical diagnosis of HS patients is becoming a great challenge due to phenotypic heterogeneity, complex pathophysiology, lack of familial history, and co-occurrence of HS together with other types of hereditary anemias (Fan et al., 2019; Wu et al., 2021; Yang et al., 2019; Aggarwal et al., 2020; Zou et al., 2020). In addition, the prevalence and molecular profile of HS are significantly different in different populations worldwide (Xue et al., 2019; Bogusławska et al., 2021). However, in the Chinese population, a higher rate of misdiagnosis has been reported among HS patients due to mild or moderate clinical manifestations, the presence of other diseases (β-thalassemia or Gilbert syndrome) along with HS, and inconsistent HS genotype and phenotype (He et al., 2018; Andolfo et al., 2016; Roy et al., 2016). Therefore, molecular genetic diagnostics through the application of genomic sequencing technologies will provide proper clinical diagnosis, disease management, and genetic consultation to reduce the risk of disease occurrence in the next generation (Qin et al., 2020; He et al., 2017; Del Orbe Barreto et al., 2016; Schafer et al., 2009; Koboldt et al., 2013; Russo et al., 2018; Xu et al., 2022).
Previous studies showed that the identification of disease-causing mutations in HS patients is usually done by targeted gene panel-based next-generation sequencing or whole-exome sequencing (Tole et al., 2020; Qin et al., 2020; Wu et al., 2021; Yang et al., 2019; Aggarwal et al., 2020; Bogusławska et al., 2021; Xu et al., 2022). However, to date, only two studies have been performed with the application of whole genome sequencing (Nieminen et al., 2021; Du et al., 2021). Whole-exome sequencing provides more sequencing coverage and depth as well as a cost-effective way of identifying disease-causing variants in HS patients more efficiently than targeted gene panel-based next-generation sequencing and whole-exome sequencing. Hence, presently, whole exome sequencing is widely used to identify candidate genes and disease-causing variants in HS patients. Additionally, identification of the mutation will also help us classify the HS and further enable us to provide the clinical or disease management for the patients as well as genetic counseling of the family (Fan et al., 2019; Bolton-Maggs et al., 2012). Genetic counseling and prenatal diagnosis could play a significant role in prenatal and postnatal care of HS patients inheriting diseases causing germline mutation in the SPTB gene (Ribeiro et al., 2000). In this study, we also performed whole exome sequencing to identify the mutational characteristics of a Chinese family with HS and rapidly discovered the causative genotype in this family, thus providing genetic information for reproductive risk consultation.
HS patients with germline mutations in the SPTB gene usually present with extreme phenotypic heterogeneity. We recommend treatment or therapeutic interventions for HS patients according to their clinical symptoms. HS patients clinically manifest with an enlarged spleen, jaundice, gallstones, and hemolytic anemia. We recommend phototherapy or light treatment for a newborn with jaundice and recommend blood transfusion for treating anemia. Splenectomy or surgical removal of the spleen is also recommended for HS patients. In addition, surgical removal of the gallbladder, or cholecystectomy, has been recommended for HS patients. We also recommend iron chelation therapy for removing excess iron in HS patients with iron overload due to regular blood transfusions. These are the recommended disease management practices for HS patients. Here, we recommend a blood transfusion for the proband if her hemoglobin is lower than 60 g/L. We usually performed blood transfusions for the proband and her mother 1–2 times a year.
β-spectrin protein interacts with the α-spectrin protein to form a heterotetramer (α2β2), which finally results in the formation of a dense erythrocyte-membrane-skeleton network and connects it to the lipid bilayer for maintaining the normal structure of the erythrocyte (Maddala et al., 2016; Bennett and Lorenzo, 2016; Meng et al., 2019). β-spectrin is a rate-limiting protein and plays a key role in the formation of the α2β2-heterotetrameric network. The C-terminal region of β-spectrin attaches to the α2β2-heterotetrameric network with the lipid bilayer (Meng et al., 2019). β-spectrin also binds with actin, ankyrin, and band 4.1 protein and is significantly involved in the formation of the cytoskeletal superstructure of the erythrocyte cell membrane, as well as stabilizing it (Maddala et al., 2016; Bennett and Lorenzo, 2016). β-spectrin binds with ankyrin to anchor the cytoplasmic face of the cell membrane (Bennett and Lorenzo, 2016; Ipsaro and Mondragon, 2010). Hence, β-spectrin helps to maintain the deformability of erythrocytes in capillaries through membrane skeleton cohesion (Meng et al., 2019). β-spectrin also maintains the biconcave shape of human erythrocytes, regulates the plasma membrane components, and maintains the lipid asymmetry of the plasma membrane (Pishesha et al., 2014; Narla and Mohandas, 2017; Machnicka et al., 2014). Hence, germline mutation in the SPTB gene results in a decrease in both the cohesion of the membrane skeleton and the surface area of the membrane, leading to the formation of spherical erythrocyte, which finally causes decreased deformability of erythrocytes and premature destruction of erythrocytes in the spleen (He et al., 2018; Meng et al., 2019; Shin et al., 2018; Christensen et al., 2014).
Here, we investigated all the previous reports and found that the SPTB gene mutation-associated HS accounts for 20%–25% of all HS cases in Europe and the United States, while it showed a potentially higher rate in China (Perrotta et al., 2008; Wang et al., 2018; Hassoun et al., 1997). In China, germline heterozygous mutations in the SPTB gene have been identified in 45% of HS patients (Qin et al., 2020; Wang et al., 2018; Wang et al., 2020). These studies suggested that HS patients showed different geographical distribution of mutations in the SPTB gene and, most interestingly, SPTB is the major gene among HS patients in the Chinese population. We also investigated the SPTB gene mutation databases (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk; Leiden Open Variation Database, LOVD v.3.0, https://www.lovd.nl/) and found that HS-associated SPTB mutations include nonsense, frameshift, and splice-site mutations, most often exerting a dominant negative effect on the splicing event of SPTB mRNA, which finally results in the formation of truncated β-spectrin (Maciag et al., 2009; Li et al., 2025). However, we found no common SPTB mutations or mutation hotspots in the SPTB gene. So far, all the identified SPTB mutations associated with HS are unique and occur in individual families. Hence, we could not predict the severity of the disease based on the type or location of the SPTB mutations (Tole et al., 2020; Agarwal et al., 2023; Wang et al., 2024; Qin et al., 2025). However, to date, genotype–phenotype association studies have been rarely reported in HS patients and require further investigation.
5 Conclusion
In conclusion, in the present study, we investigated a Chinese girl with hereditary spherocytosis and hemolytic anemia. Whole-exome sequencing identified a heterozygous novel splice-site mutation (c.647 + 1G>A) in the SPTB gene in the proband. Sanger sequencing confirmed that the proband inherited this heterozygous novel splice-donor site mutation from her mother, while her father did not carry this mutation. Our study confirmed the functional impact of this identified mutation at the molecular level and expanded the spectrum of SPTB mutations associated with HS. Our results also significantly highlighted the application of whole-exome sequencing for clinical diagnosis of patients with HS, which may contribute to the clinical management and genetic counseling of HS patients.
Databases and Web servers used:
1000 Genome Database: http://www.internationalgenome.org
Burrows–Wheeler Aligner software: http://bio-bwa.sourceforge.net, version 0.59
ClinGen: https://www.clinicalgenome.org/
dbSNP: https://www.ncbi.nlm.nih.gov
Database of Genomic Variants (DGV): http://dgv.tcag.ca/dgv/
DECIPHER: https://www.deciphergenomics.org/
Exome Aggregation Consortium (ExAC): http://exac.broadinstitute.org
GATK Base Recalibrator: https://gatk.broadinstitute.org
GATK IndelRealigner: https://gatk.broadinstitute.org
GATK Unified Genotyper: https://gatk.broadinstitute.org
Genome Aggregation Database (gnomAD): https://gnomad.broadinstitute.org
HapMap: https://www.genome.gov
Human Gene Mutation Database (HGMD): www.hgmd.cf
Human Splicing Finder - Version 3.1: http://www.umd.be/HSF/HSF.html
Human Splicing Finder Pro: https://www.genomnis.com/hsf
Illumina pipeline: https://www.illumina.com/informatics/infrastructure-pipeline-setup.html
Leiden Open Variation Database, LOVD v.3.0: https://www.lovd.nl/
MutationTaster: https://www.genecascade.org/MutationTaster2021/#transcript
National Center for Biotechnology Information (NCBI): https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi
Online Mendelian Inheritance in Man (OMIM): https://www.omim.org
Polyphen-2: http://genetics.bwh.harvard.edu/pph2
REVEL: https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=revel
UCSC Genome Browser: https://genome.ucsc.edu/)
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://bigd.big.ac.cn/gsa-human/browse/HRA010244,HRA010244.
Ethics statement
The studies involving humans were approved by the Ethics Committee of The Shenzhen Children’s Hospital, Shenzhen, China. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. The animal studies were approved by the Ethics Committee of The Shenzhen Children’s Hospital, Shenzhen, China. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
KC: Investigation, Writing – original draft, Formal Analysis, Data curation, Methodology, Writing – review and editing. XL: Writing – review and editing, Formal Analysis, Data curation, Investigation, Methodology, Writing – original draft. LL: Data curation, Methodology, Writing – review and editing, Writing – original draft, Formal Analysis. XM: Writing – original draft, Methodology, Formal Analysis, Writing – review and editing. RL: Writing – original draft, Writing – review and editing, Methodology, Formal Analysis. YC: Supervision, Project administration, Writing – review and editing, Funding acquisition, Writing – original draft, Investigation, Resources, Data curation, Conceptualization. SB: Funding acquisition, Project administration, Supervision, Investigation, Writing – review and editing, Writing – original draft, Conceptualization, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The study was supported by the Guangdong Basic and Applied Basic Research Foundation (2023A1515220156, 2022A1515220033) and the Shenzhen Science and Technology Program (JCYJ20220530155811025, JCYJ20230807093820041, JCYJ20240813112420027).
Acknowledgments
The authors are grateful to the proband and her family for their participation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1626155/full#supplementary-material
References
1
Agarwal A. M. McMurty V. Clayton A. L. Bolia A. Reading N. S. Mani C. et al (2023). Clinical utility of targeted next-generation sequencing panel in routine diagnosis of hereditary hemolytic anemia: a national reference laboratory experience. Eur. J. Haematol.110 (6), 688–695. 10.1111/ejh.13951
2
Aggarwal A. Jamwal M. Sharma P. Sachdeva M. U. S. Bansal D. Malhotra P. et al (2020). Deciphering molecular heterogeneity of Indian families with hereditary spherocytosis using targeted next-generation sequencing: first South Asian study. Br. J. Haematol.188 (5), 784–795. 10.1111/bjh.16244
3
Andolfo I. Russo R. Gambale A. Iolascon A. (2016). New insights on hereditary erythrocyte membrane defects. Haematologica101 (11), 1284–1294. 10.3324/haematol.2016.142463
4
Auton A. Brooks L. D. Durbin R. M. Garrison E. P. Kang H. M. Korbel J. O. et al (2015). A global reference for human genetic variation. Nature526 (7571), 68–74. 10.1038/nature15393
5
Bennett V. Lorenzo D. N. (2016). An adaptable spectrin/ankyrin‐Based mechanism for long‐range organization of plasma membranes in vertebrate tissues. Curr. Top. Membr.77, 143–184. 10.1016/bs.ctm.2015.10.001
6
Boguslawska D. M. Heger E. Machnicka B. Skulski M. Kuliczkowski K. Sikorski A. F. (2017). A new frameshift mutation of the β-spectrin gene associated with hereditary spherocytosis. Ann. Hematol.96 (1), 163–165. 10.1007/s00277-016-2838-0
7
Bogusławska D. M. Skulski M. Machnicka B. Potoczek S. Kraszewski S. Kuliczkowski K. et al (2021). Identification of a novel mutation of beta-Spectrin in hereditary spherocytosis using whole exome sequencing. Int. J. Mol. Sci.22 (20), 11007. 10.3390/ijms222011007
8
Bolton-Maggs P. H. Langer J. C. Iolascon A. Tittensor P. King M. J. General Haematology Task Force of the British Committee for Standards in Haematology (2012). Guidelines for the diagnosis and management of hereditary spherocytosis-2011 update. Br. J. Haematol.156 (1), 37–49. 10.1111/j.1365-2141.2011.08921.x
9
Christensen R. D. Nussenzveig R. H. Reading N. S. Agarwal A. M. Prchal J. T. Yaish H. M. (2014). Variations in both α-spectrin (SPTA1) and β-spectrin (SPTB) in a neonate with prolonged jaundice in a family where nine individuals had hereditary elliptocytosis. Neonatology105 (1), 1–4. 10.1159/000354884
10
Clark A. G. Hubisz M. J. Bustamante C. D. Williamson S. H. Nielsen R. (2005). Ascertainment bias in studies of human genome-wide polymorphism. Res.15 (11), 1496–1502. 10.1101/gr.4107905
11
Da Costa L. Galimand J. Fenneteau O. Mohandas N. (2013). Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Rev.27 (4), 167–178. 10.1016/j.blre.2013.04.003
12
Del Orbe Barreto R. Arrizabalaga B. De la Hoz A. B. García-Orad Á. Tejada M. I. Garcia-Ruiz J. C. et al (2016). Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. Int. J. Lab. Hematol.38 (6), 629–638. 10.1111/ijlh.12551
13
Delaunay J. (2002). Molecular basis of red cell membrane disorders. Acta. Haematol.108 (4), 210–218. 10.1159/000065657
14
Du Z. Luo G. Wang K. Bing Z. Pan S. (2021). Identification of a novel heterozygous SPTB mutation by whole genome sequencing in a Chinese patient with hereditary spherocytosis and atrial septal defect: a case report. Bmc. Pediatr.21 (1), 291. 10.1186/s12887-021-02771-4
15
Fan L. L. Liu J. S. Huang H. Du R. Xiang R. (2019). Whole exome sequencing identified a novel mutation (p.Ala1884Pro) of β-spectrin in a Chinese family with hereditary spherocytosis. J. Gene. Med.21 (2-3), e3073. 10.1002/jgm.3073
16
Hamosh A. Scott A. F. Amberger J. S. Bocchini C. A. McKusick V. A. (2005). Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic. Acids. Res.33 (Database issue), D514–D517. 10.1093/nar/gki033
17
Han P. Wei G. Cai K. Xiang X. Deng W. P. Li Y. B. et al (2020). Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J. Cell. Mol. Med.24 (2), 1286–1299. 10.1111/jcmm.14768
18
Hassoun H. Vassiliadis J. N. Murray J. Njolstad P. R. Rogus J. J. Ballas S. K. et al (1997). Characterization of the underlying molecular defect in hereditary spherocytosis associated with spectrin deficiency. Blood90 (1), 398–406.
19
He Y. Jia S. Dewan R. K. Liao N. (2017). Novel mutations in patients with hereditary red blood cell membrane disorders using next-generation sequencing. Gene627, 556–562. 10.1016/j.gene.2017.07.009
20
He B. J. Liao L. Deng Z. F. Tao Y. F. Xu Y. C. Lin F. Q. (2018). Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta. Haematol.139 (1), 60–66. 10.1159/000486229
21
Ipsaro J. J. Mondragon A. (2010). Structural basis for spectrin recognition by ankyrin. Blood115 (20), 4093–4101. 10.1182/blood-2009-11-255604
22
Ipsaro J. J. Huang L. Mondragon A. (2009). Structures of the spectrin ankyrin interaction binding domains. Blood113 (22), 5385–5393. 10.1182/blood-2008-10-184358
23
Jang W. Kim J. Chae H. Kim M. Koh K. M. Park C. J. et al (2019). Hereditary spherocytosis caused by copy number variation in SPTB gene identified through targeted next-generation sequencing. Int. J. Hematol.110 (2), 250–254. 10.1007/s12185-019-02630-0
24
Karczewski K. J. Weisburd B. Thomas B. Solomonson M. Ruderfer D. M. Kavanagh D. et al (2017). The ExAC browser: displaying reference data information from over 60000 exomes. Nucleic. Acids. Res.45 (D1), D840–D845. 10.1093/nar/gkw971
25
Karczewski K. J. Francioli L. C. Tiao G. Cummings B. B. Alföldi J. Wang Q. et al (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature581 (7809), 434–443. 10.1038/s41586-020-2308-7
26
Koboldt D. C. Steinberg K. M. Larson D. E. Wilson R. K. Mardis E. R. (2013). The next-generation sequencing revolution and its impact on genomics. Cell155 (1), 27–38. 10.1016/j.cell.2013.09.006
27
Lefter M. Vis J. K. Vermaat M. den Dunnen J. T. Taschner P. E. M. Laros J. F. J. (2021). Mutalyzer 2: next generation HGVS nomenclature checker. Bioinformatics37 (18), 2811–2817. 10.1093/bioinformatics/btab051
28
Li X. Zhang T. Li X. Wang L. Li Q. Liu Q. et al (2025). Identification of a novel SPTB gene splicing mutation in hereditary spherocytosis: a case report and diagnostic insights. Front. Genet.15, 1522204. 10.3389/fgene.2024.1522204
29
Ma S. Qin J. Wei A. Li X. Qin Y. Liao L. et al (2018). Novel compound heterozygous SPTA1 mutations in a patient with hereditary elliptocytosis. Mol. Med. Rep.17 (4), 5903–5911. 10.3892/mmr.2018.8632
30
Machnicka B. Czogalla A. Hryniewicz-Jankowska A. Bogusławska D. M. Grochowalska R. Heger E. et al (2014). Spectrins: a structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim. Biophys. Acta.1838 (2), 620–634. 10.1016/j.bbamem.2013.05.002
31
Maciag M. Płochocka D. Adamowicz-Salach A. Burzyńska B. (2009). Novel beta-spectrin mutations in hereditary spherocytosis associated with decreased levels of mRNA. Br. J. Haematol.146 (3), 326–332. 10.1111/j.1365-2141.2009.07759.x
32
Maddala R. Walters M. Brophy P. J. Bennett V. Rao P. V. (2016). Ankyrin‐B directs membrane tethering of periaxin and is required for maintenance of lens fiber cell hexagonal shape and mechanics. Am. J. Physiol. Cell. Physiol.310 (2), C115–C126. 10.1152/ajpcell.00111.2015
33
Meng L. L. Yuan S. M. Tu C. F. Lin G. Lu G. X. Tan Y. Q. (2019). Next‐generation sequencing identified a novel SPTB frameshift insertion causing hereditary spherocytosis in China. Ann. Hematol.98 (1), 223–226. 10.1007/s00277-018-3417-3
34
Narla J. Mohandas N. (2017). Red cell membrane disorders. Int. J. Lab. Hematol.39 (Suppl. 1), 47–52. 10.1111/ijlh.12657
35
Nieminen T. T. Liyanarachchi S. Comiskey D. F. Jr Wang Y. Li W. Hendrickson I. V. et al (2021). A novel essential splice site variant in SPTB in a large hereditary spherocytosis family. Mol. Genet. Genomic Med.9 (5), e1641. 10.1002/mgg3.1641
36
Park J. Jeong D. C. Yoo J. Jang W. Chae H. Kim J. et al (2016). Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin. Genet.90 (1), 69–78. 10.1111/cge.12749
37
Perrotta S. Gallagher P. G. Mohandas N. (2008). Hereditary spherocytosis. Lancet372 (9647), 1411–1426. 10.1016/S0140-6736(08)61588-3
38
Pishesha N. Thiru P. Shi J. Eng J. C. Sankaran V. G. Lodish H. F. (2014). Transcriptional divergence and conservation of human and mouse erythropoiesis. Proc. Natl. Acad. Sci. USA.111 (11), 4103–4108. 10.1073/pnas.1401598111
39
Qin L. Nie Y. Zhang H. Chen L. Zhang D. Lin Y. et al (2020). Identification of new mutations in patients with hereditary spherocytosis by next-generation sequencing. J. Hum. Genet.65 (4), 427–434. 10.1038/s10038-020-0724-z
40
Qin L. Jia Y. Wang H. Feng Y. Zou J. Zhou J. et al (2025). Identification of novel variants in hereditary spherocytosis patients by whole-exome sequencing. Clin. Chim. Acta.565, 119989. 10.1016/j.cca.2024.119989
41
Ribeiro M. L. Alloisio N. Almeida H. Gomes C. Texier P. Lemos C. et al (2000). Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood96 (4), 1602–1604.
42
Richards S. Aziz N. Bale S. Bick D. Das S. Gastier-Foster J. et al (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med.17 (5), 405–424. 10.1038/gim.2015.30
43
Risinger M. Kalfa T. A. (2020). Red cell membrane disorders: structure meets function. Blood136 (11), 1250–1261. 10.1182/blood.2019000946
44
Roy N. B. A. Wilson E. A. Henderson S. Wray K. Babbs C. Okoli S. et al (2016). A novel 33-Gene targeted resequencing panel provides accurate, clinical-grade diagnosis and improves patient management for rare inherited anaemias. Br. J. Haematol.175 (2), 318–330. 10.1111/bjh.14221
45
Russo R. Andolfo I. Manna F. Gambale A. Marra R. Rosato B. E. et al (2018). Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am. J. Hematol.93 (5), 672–682. 10.1002/ajh.25058
46
Schafer A. Fraccarollo D. Widder J. Eigenthaler M. Ertl G. Bauersachs J. (2009). Inhibition of platelet activation in rats with severe congestive heart failure by a novel endothelial nitric oxide synthase transcription enhancer. Eur. J. Heart. Fail.11 (4), 336–341. 10.1093/eurjhf/hfp005
47
Sherry S. T. Ward M. H. Kholodov M. Baker J. Phan L. Smigielski E. M. et al (2001). dbSNP: the NCBI database of genetic variation. Nucleic. Acids. Res.29 (1), 308–311. 10.1093/nar/29.1.308
48
Shin S. Jang W. Kim M. Kim Y. Park S. Y. Park J. et al (2018). Targeted next‐generation sequencing identifies a novel nonsense mutation in SPTB for hereditary spherocytosis: a case report of a Korean family. Med. Baltim.97 (3), e9677. 10.1097/MD.0000000000009677
49
Stenson P. D. Ball E. V. Mort M. Phillips A. D. Shiel J. A. Thomas N. S. T. et al (2003). Human gene mutation database (HGMD): 2003 update. Hum. Mutat.21 (6), 577–581. 10.1002/humu.10212
50
Tole S. Dhir P. Pugi J. Drury L. J. Butchart S. Fantauzzi M. et al (2020). Genotype-phenotype correlation in children with hereditary spherocytosis. Br. J. Haematol.191 (3), 486–496. 10.1111/bjh.16750
51
Wang C. Cui Y. Li Y. Liu X. Han J. (2015). A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable. Rare. Dis. Res.4 (2), 76–81. 10.5582/irdr.2015.01002
52
Wang R. Yang S. Xu M. Huang J. Liu H. Gu W. et al (2018). Exome sequencing confirms molecular diagnoses in 38 Chinese families with hereditary spherocytosis. Sci. China. Life. Sci.61 (8), 947–953. 10.1007/s11427-017-9232-6
53
Wang X. Zhang A. Huang M. Chen L. Hu Q. Lu Y. et al (2020). Genetic and clinical characteristics of patients with hereditary spherocytosis in hubei province of China. Front. Genet.11, 953. 10.3389/fgene.2020.00953
54
Wang Y. Liu T. Jia C. Xiao L. Wang W. Zhang Y. et al (2024). A novel variant in the SPTB gene underlying hereditary spherocytosis and a literature review of previous variants. Bmc. Med. Genomics.17 (1), 206. 10.1186/s12920-024-01973-w
55
Wu Y. Liao L. Lin F. (2021). The diagnostic protocol for hereditary spherocytosis-2021 update. J. Clin. Lab. Anal.35 (12), e24034. 10.1002/jcla.24034
56
Xu C. Wu Y. Wang D. Zhang X. Wang N. (2022). Novel SPTB frameshift mutation in a Chinese neonatal case of hereditary spherocytosis type 2: a case report. Exp. Ther. Med.24 (3), 600. 10.3892/etm.2022.11537
57
Xue J. He Q. Xie X. Su A. Cao S. (2019). Clinical utility of targeted gene enrichment and sequencing technique in the diagnosis of adult hereditary spherocytosis. Ann. Transl. Med.7 (20), 527. 10.21037/atm.2019.09.163
58
Yang K. Ren Q. Wu Y. Zhou Y. Yin X. (2019). A case of hereditary spherocytosis caused by a novel homozygous mutation in the SPTB gene misdiagnosed as β-Thalassemia intermedia due to a KLF1 gene mutation. Hemoglobin43 (2), 140–144. 10.1080/03630269.2019.1620764
59
Zhang R. Chen S. Han P. Chen F. Kuang S. Meng Z. et al (2020). Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes meckel syndrome. J. Cell. Mol. Med.24 (2), 1906–1916. 10.1111/jcmm.14887
60
Zou W. Zhang Z. Tan Y. Zhang L. (2020). Hereditary spherocytosis associated with Gilbert syndrome diagnosed with liver biopsy examination and exome sequencing. J. Coll. Physicians. Surg. Pak.30 (2), 213–215. 10.29271/jcpsp.2020.02.213
Summary
Keywords
hereditary spherocytosis, novel mutation, SPTB gene, hemolytic anemia, splice-donor site mutation
Citation
Cao K, Luo X, Liu L, Mao X, Liu R, Chen Y and Banerjee S (2025) Case Report: Identification and functional characterization of a novel heterozygous splice-donor (c.647+1G>A) site mutation in the SPTB gene that causes hereditary spherocytosis with hemolytic anemia. Front. Genet. 16:1626155. doi: 10.3389/fgene.2025.1626155
Received
10 May 2025
Revised
10 September 2025
Accepted
10 October 2025
Published
14 November 2025
Volume
16 - 2025
Edited by
Mara Marongiu, National Research Council (CNR), Italy
Reviewed by
Prabhakar S Kedar, Indian Council of Medical Research, India
Kyril Turpaev, Center for Theoretical Problems of Physicochemical Pharmacology (RAS), Russia
Updates
Copyright
© 2025 Cao, Luo, Liu, Mao, Liu, Chen and Banerjee.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Santasree Banerjee, santasree.banerjee@yahoo.com; Yunsheng Chen, chenyunsheng66@163.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.