 Tingting Ge1,2†
Tingting Ge1,2† Xiaojuan Lin1,2†Xinyuan Tian3Xiaoyu Song1,2Bingbo Zhou3Ling Hui3Xiaozhuan Wang1,2Zhiqiang Zhang4
Xiaojuan Lin1,2†Xinyuan Tian3Xiaoyu Song1,2Bingbo Zhou3Ling Hui3Xiaozhuan Wang1,2Zhiqiang Zhang4 Chuan Zhang3*
Chuan Zhang3*- 1Prenatal Diagnosis Center, Gansu Provincial Maternity and Child-care Hospital, Lanzhou, China
- 2Key Laboratory of Maternal-Fetal Medicine and Reproductive Protection of Gansu Province, Lanzhou, China
- 3Center for Medical Genetics, Gansu Provincial Maternity and Child-care Hospital, Gansu Provincial Clinical Research Center for Birth Defects and Rare Diseases, Lanzhou, China
- 4Medical Imaging Center, Gansu Provincial Maternity and Child-care Hospital, Lanzhou, China
Objective: 6q terminal deletion is a rare genetic cause of prenatal brain anomalies. We evaluated five cases of cerebral dysplasia within a familial context for genetic diagnosis. Aims to analyze prenatal brain abnormalities from 6q terminal deletion of DLL1 and support prenatal diagnosis and genetic counseling.
Methods: A retrospective analysis was conducted on data from five families with fetal brain structural dysplasia, collected at Gansu Provincial Maternity and Child-care Hospital (Gansu Central Hospital) between January 2017 and April 2024. We applied copy number variation sequencing (CNV-Seq) and when negative, whole-exome sequencing (WES) to define genomic etiologies of prenatal brain anomalies.
Results: A total of 5 fetuses were included in this study. All fetuses exhibited a cerebellar diameter smaller than expected for their gestational age, as determined by US, 4/5 cases underwent MRI. In fetuses 1–4, CNV-Seq analysis identified heterozygous deletions of 1.74 Mb, 2.88 Mb, 0.72 Mb, and 21.99 Mb at the terminal region of chromosome 6q. In fetus 5, WES successfully identified the deletion that CNV-seq had missed, likely terminal coverage drop/binning limit.
Conclusion: Fetuses with reduced transverse cerebellar diameter and ventriculomegaly should be evaluated for 6q terminal deletions involving DLL1; combining CNV-seq with reflex WES reduces missed diagnoses and informs counseling.
Introduction
Disorders linked to DLL1 (OMIM: 618709) exhibit diverse neurodevelopmental symptoms, including developmental delays, intellectual disabilities, autism, attention deficits, stereotypical behaviors, and brain anomalies like hydrocephalus and dysplasia. Common symptoms also include seizures, hypotonia, joint hyperextension, ataxia, scoliosis/kyphosis, and cognitive impairment (Fischer-Zirnsak et al., 2019). Overlapping microdeletions in the 6q27 region, affecting genes like DLL1, THBS2, PHF10, and ERMARD, linked to developmental delay, intellectual disability, and brain malformations (Fischer-Zirnsak et al., 2019). During embryogenesis, DLL1, expressed in neural precursor cells, regulates differentiation through oscillatory Notch signaling, affecting brain development by inhibiting differentiation in adjacent cells (Fischer-Zirnsak et al., 2019; Louvi et al., 2006; Cao et al., 2019; Shimojo et al., 2011; Artavanis-Tsakonas et al., 1999). Research has shown that the Notch ligand DLL1 is crucial for developing the central nervous system, somites, and lymphocytes (Gray et al., 1999; Hrabĕ de Angelis et al., 1997; Marklund et al., 2010; Hiraoka et al., 2013; Dunwoodie et al., 1997; Okigawa et al., 2014; Mahler et al., 2010). Among these, DLL1 is most likely intolerant to loss of function (LoF), suggesting its deficiency may contribute to neurodevelopmental disorders (Fischer-Zirnsak et al., 2019; Karczewski et al., 2020; Peddibhotla et al., 2015). Given DLL1’s role in neurodevelopment, we investigated whether 6q terminal deletions involving DLL1 underlie prenatal brain anomalies detected by ultrasound/MRI. To delineate genomic causes, we applied CNV-seq to all cases and performed reflex WES when CNV-seq was negative.
Methods
Study design and setting and participants
This study employed a retrospective analysis of a consecutive series of fetuses referred to Gansu Provincial Maternity and Child-care Hospital (Gansu Central Hospital) between January 2017 and April 2024. Cases were initially identified based on prenatal ultrasound examination suggesting structural brain anomalies, and four cases were further assessed by prenatal magnetic resonance imaging (MRI). From this cohort, five cases with a confirmed genetic diagnosis of a 6q terminal deletion involving the DLL1 gene were selected for in-depth analysis. Comprehensive clinical, genetic, and family data were collected for all included cases. Cases were excluded for incomplete clinical or imaging data, the presence of major confounding comorbidities (e.g., aneuploidy, congenital infections), or loss to follow-up.
Sample collection
Amniocentesis was performed under sterile conditions to obtain 30 mL of amniotic fluid. Peripheral blood (2–3 mL) was collected from both parents.
CNV-seq
CNV-seq libraries were prepared using a commercial high-throughput sequencing kit (BerryGenomics) according to the manufacturer’s protocol, including DNA end repair, adapter ligation, barcoding, and purification (Zhang et al., 2018). Libraries were pooled and sequenced on the NextSeq CN500 platform—a commercially available desktop sequencer—at a minimum depth of 1× and resolution of 100 kb. Sequencing reads were aligned to the human reference genome GRCh37 (hg19), chosen for compatibility with major clinical genomics databases. Quality control metrics including average read depth, coverage uniformity, and terminal coverage behavior were evaluated. CNV pathogenicity was assessed using DECIPHER (https://www.deciphergenomics.org/browser), OMIM (https://www.omim.org/), UCSC Genome Browser (https://www.genome.ucsc.edu/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and ClinGen (https://www.clinicalgenome.org/), and interpreted according to ACMG guidelines (Riggs et al., 2020).
Detection of WES
Whole-exome sequencing (WES) was performed on the BGISEQ-2000 platform (BGI, Shenzhen, China) using the BGI Exome Capture V1 kit (BGI, Shenzhen, China). The average sequencing depth was 200×, with 98.5% of the target regions covered at ≥30×, 99% of the target regions covered at ≥20×. Variant calling for single-nucleotide polymorphisms (SNPs) and insertions/deletions (Indels) was conducted following the GATK best practices workflow. Short-read sequencing data were aligned to the reference genome GRCh37/hg19 using BWA-MEM, followed by duplicate marking, base quality score recalibration, and variant discovery using Haplotype Caller. Final variants were filtered based on recommended quality metrics. Copy number variant (CNV) analysis from exome data was applied in this study, the resolution was> 100 kb. Detected variants were annotated and interpreted according to the ACMG guidelines (Richards et al., 2015).
Ethics
Written informed consent was obtained from all participating families. This study received approval from the hospital’s Ethics Committee, with the approval number (No. 2024GSFY07).
Results
Cohort description
A total of five fetuses were included in this study. The gestational age at initial diagnosis ranged from 21.56 to 27.14 (24.45 ± 1.89) weeks by ultrasound (US). Fetuses exhibited a cerebellar diameter smaller than expected for their gestational age, as determined by US and MRI (US in all, MRI in 4/5), Figure 1.
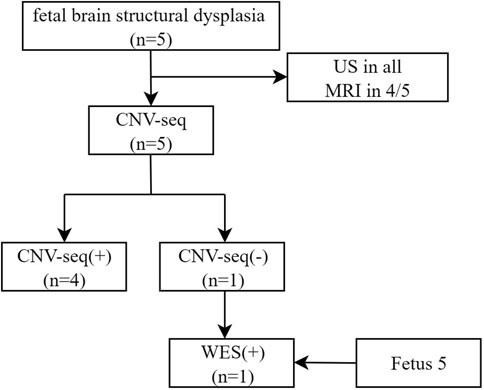
Figure 1. Workflow diagram.
Imaging findings
All fetuses exhibited a transverse cerebellar diameter smaller than expected for their gestational age, as determined by US, 4/5 cases underwent MRI (Figures 2–5). Common complications included mild lateral ventricle dilatation (5/5 cases), corpus callosum abnormalities (1/5 cases) and low positioning of the conus medullaris (2/5 cases) in a clean Table 1. Among them, fetus 4 was first found at 21 + 4 weeks of gestation with multiple system malformations, Included were fetal growth restriction, double outlet right ventricle, Nuchal fold thickened, scoliosis, hand deformities.
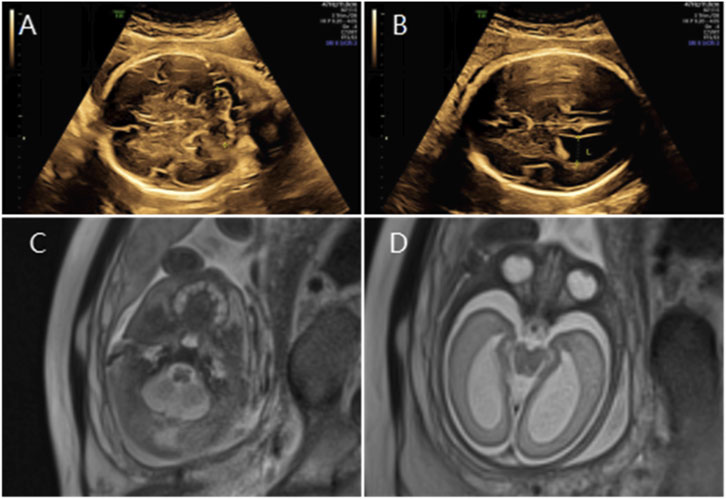
Figure 2. (A) Transverse ultrasound section showing the fetal cerebellum with a transverse cerebellar diameter of 27.8 mm (<–3.0 SD for gestational age). (B) Axial ultrasound view in thetransventricular plane demonstrating bilateral ventriculomegaly with atrial widths of 12.3 mm (left) and 12.5 mm (right). (C,D) Axial T2-weighted fetal MRI confirming reduced cerebellar size and bilateral ventriculomegaly.
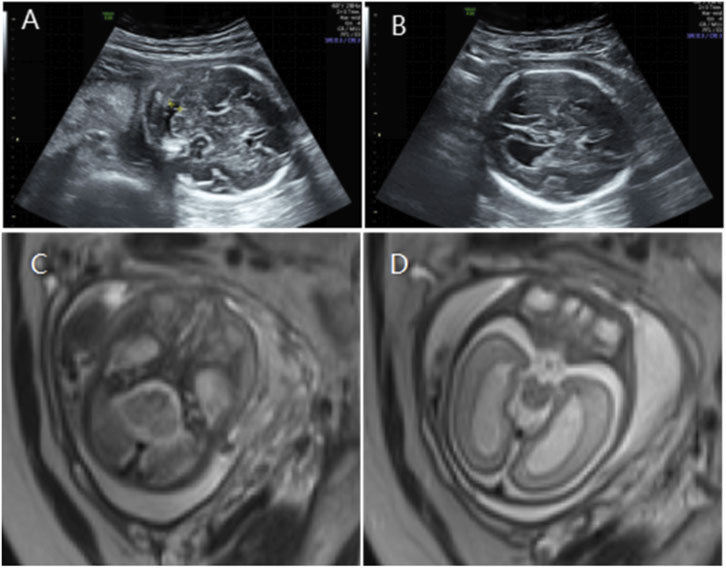
Figure 3. (A) Transverse ultrasound section showing the fetal cerebellum; transverse cerebellar diameter measured 24.8 mm (<–3.2 SD for gestational age), consistent with cerebellar hypoplasia. (B) Axial ultrasound view in the transventricular plane demonstrating bilateral ventriculomegaly, with atrial widths of 10.6 mm (left) and 11.2 mm (right). (C,D) Axial T2-weighted fetal MRI confirming cerebellar hypoplasia and bilateral ventriculomegaly.
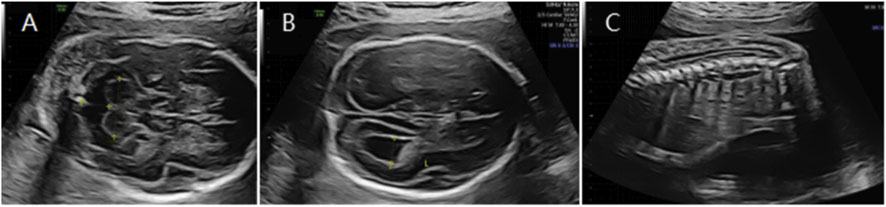
Figure 4. (A) Transverse ultrasound section showing the fetal cerebellum; transverse cerebellar diameter measured 20.2 mm (<–3.0 SD for gestational age). (B) Axial ultrasound view in the transventricular plane demonstrating bilateral ventriculomegaly, with atrial widths of approximately 8.9 mm (left) and 10.8 mm (right). (C) Sagittal ultrasound view showing a low-lying conus medullaris.
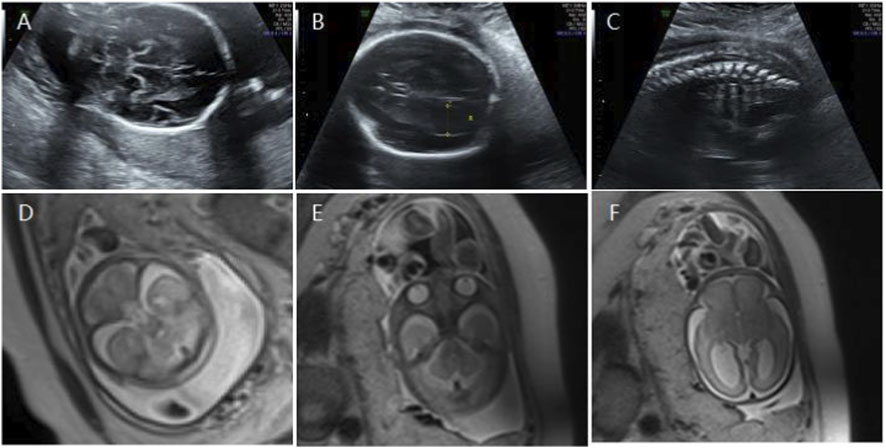
Figure 5. (A) Transverse ultrasound section showing the fetal cerebellum; transverse cerebellar diameter measured 23.4 mm (<–2.6 SD for gestational age). (B) Axial ultrasound view in the transventricular plane demonstrating bilateral ventriculomegaly, with atrial widths of 10.0 mm (left) and 13.6 mm (right). (C) Sagittal ultrasound view showing an abnormally positioned conus medullaris. (D,E) Axial T2-weighted fetal MRI confirming reduced cerebellar size. (F) Axial T2-weighted fetal MRI showing bilateral ventriculomegaly.
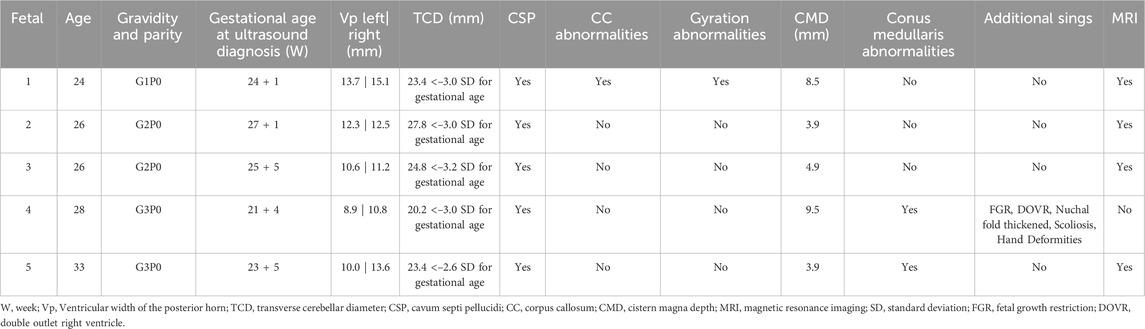
Table 1. Clinical data and prenatal ultrasound examination results of 5 fetuses.
Genetic findings
A heterozygous deletion was identified in all fetuses, with the deletion region located at the terminal end of chromosome 6q, encompassing the haploinsufficiency-sensitive gene DLL1 (2A, 1 score). The total score for fragment deletion was ≥0.99, classifying it as a pathogenic variation (Tables 1 and 2). Notably, in fetus 5, CNV-seq negative, the pregnant woman opted for further WES, this analysis revealed a heterozygous deletion of 0.14 Mb in the Chr6:g.170460966-170604541 region, likely terminal coverage drop/binning limit.
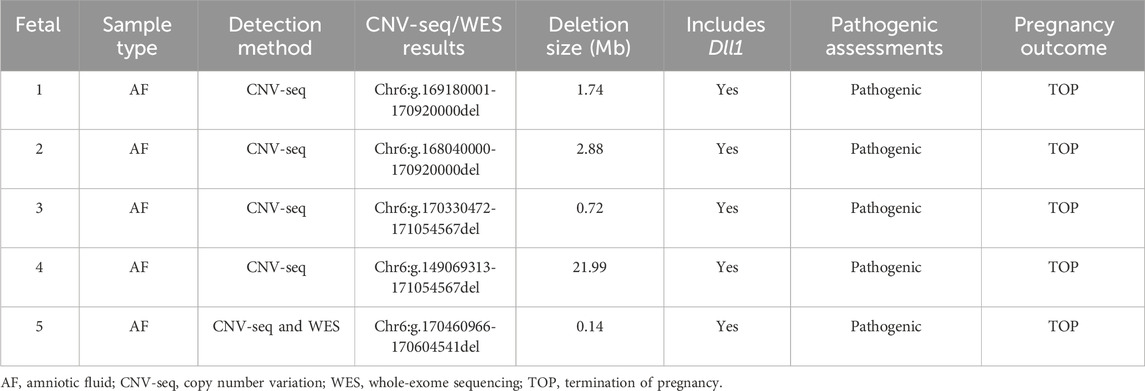
Table 2. Genetic diagnosis data of 5 fetuses.
Given that fetuses with haploinsufficiency of the DLL1 gene predominantly exhibit clinical manifestations such as intellectual disability, brain malformations, autism, epilepsy, and other related phenotypes, the parents of the five fetuses elected to terminate the pregnancy following genetic counseling.
Genotype–phenotype
DLL1 deletion exhibited altered brain structures, the most prevalent abnormalities were cerebellar dysplasia, increased ventricle width, and corpus callosum anomalies (observed in over 70% of cases) (Lesieur-Sebellin et al., 2022). When the 6q terminal deletion extends to 7.1 Mb, the majority of the patient’s clinical features can be attributed to the DLL1. Deletions exceeding 7.1 Mb result in a more severe phenotype, such as such as abnormality of the anus, either anal atresia or an ectopic anus (Engwerda et al., 2023).
Discussion
In this study, analyze five cases of cerebral dysplasia within a familial context for genetic diagnosis. All fetuses exhibited a cerebellar diameter smaller than expected for their gestational age, as determined by US, 4/5 cases underwent MRI. Common complications included mild lateral ventricle dilatation (5/5 cases), corpus callosum abnormalities (1/5 cases) and low positioning of the conus medullaris (2/5 cases). Among them, fetus 4 was first found at 21 + 4 weeks of gestation with multiple system malformations, Included were fetal growth restriction, double outlet right ventricle, Nuchal fold thickened, scoliosis, hand deformities. We applied CNV-seq to all cases. CNV-seq identified the 6q terminal deletion in fetuses 1–4, however, in fetus 5, a deletion missed by CNV-seq was detected by WES, analysis revealed a heterozygous deletion of 0.14 Mb in the Chr6:g.170460966-170604541 region, likely terminal coverage drop/binning limit.
The study of DLL1 and prenatal fetal phenotypes is relatively limited. Research by Sanlaville (Lesieur-Sebellin et al., 2022) explored the relationship between DLL1 and fetal phenotypes, revealing that fetuses with a DLL1 deletion exhibited altered brain structures. The most prevalent abnormalities were cerebellar dysplasia, increased ventricle width, and corpus callosum anomalies (observed in over 70% of cases). Gyration abnormalities were identified in 46% of the fetuses. Less common phenotypes included heterotopia cerebri, vertebral malformations, and renal abnormalities. In this study, five fetuses exhibited bilateral lateral ventricle expansion, and the cerebellar transverse diameter was smaller than expected for the gestational age, compared to typical clinical phenotypes. Two cases showed isolated chamber abnormalities.
Notably, fetus number 4 had the largest terminal deletion on chromosome 6q (21.99 Mb) and presented with the most severe phenotype. In addition to bilateral ventriculomegaly and a cerebellar transverse diameter smaller than expected for the gestational age, the condition was also associated with spinal abnormalities, including scoliosis, intrauterine growth retardation, and abnormal development of both arms. When the 6q terminal deletion extends to 7.1 Mb, the majority of the patient’s clinical features can be attributed to the DLL1. Deletions exceeding 7.1 Mb result in a more severe phenotype (Engwerda et al., 2023). DLL1 haploinsufficiency plausibly explains prenatal anomalies.
CNV-Seq analysis can detect aneuploidies as well as pathogenic/likely pathogenic CNVs, significantly improving abnormality detection rates (Zhang C et al., 2018). Currently, CNV-seq achieves a resolution limit of about 100 kb. In this study, CNV-seq successfully identified 6q terminal deletions in fetuses 1–4, but failed to detect the 0.14 Mb deletion in fetus 5, a size within its theoretical detection range. This miss may be due to insufficient coverage of the 6q terminal region during sequencing. WES has been widely used clinically for diagnosing monogenic disorders (Zhang et al., 2021), can detect single-nucleotide variants, small insertions/deletions (≤20 bp), certain exon-level CNVs, and copy number changes. Fetus 5, the CNV-seq false negative was subsequently identified by WES. Thus, in prenatal cases with findings such as hydrocephalus, ventriculomegaly, or a small cerebellar diameter, WES successfully identified the deletion that CNV-seq had missed. As of May 2024, the ClinVar database lists 78 pathogenic/likely pathogenic CNVs, 25 frameshift, 7 nonsense (across 15 variants), and 4 splicing/missense mutations related to this region. Therefore, if CNV testing is negative in fetuses with these phenotypes, further analysis for single-base changes and small indels should be pursued to exclude monogenic disorders.
This study was a retrospective study with a small sample size, no postnatal phenotyping due to pregnancy terminations, imaging variability. Future work should prioritize the establishment of prospective registries, harmonized imaging metrics, capture of postnatal outcomes when pregnancies continue.
Conclusion
Fetuses with reduced transverse cerebellar diameter, hydrocephalus, or bilateral ventriculomegaly should be evaluated for 6q terminal deletions involving DLL1. Reflex WES following negative CNV-seq reduces missed diagnoses and improves prenatal counseling.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author, email: emhhbmdjaHVhbjA0MDRAMTYzLmNvbQ==.
Ethics statement
The studies involving humans were approved by Gansu Provincial Maternity and Child-care Hospital (Gansu Central Hospital) (No. (2024) GSFY Ethics Review [07]). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
TG: Writing – original draft. XL: Writing – original draft. XT: Writing – review and editing. XS: Writing – review and editing. BZ: Writing – review and editing. LH: Writing – review and editing. XW: Writing – review and editing. ZZ: Writing – review and editing. CZ: Writing – review and editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Science and Technology Project of Gansu Province (Youth Science and Technology Fund) (23JRRA1752), National Natural Science Foundation of China (82560331), Lanzhou Science and Technology Plan Project (2022-3-15), Gansu Provincial Science and Technology Plan Project (21JR11RA171, 22YF7FA094), and Hospital Research Fund Project Support Project (GMCCH2024-2-2, GMCCH2025-2-3-08).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Artavanis-Tsakonas, S., Rand, M. D., and Lake, R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284 (5415), 770–776. doi:10.1126/science.284.5415.770
Cao, J., Spielmann, M., Qiu, X., Huang, X., Ibrahim, D. M., Hill, A. J., et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566 (7745), 496–502. doi:10.1038/s41586-019-0969-x
Dunwoodie, S. L., Henrique, D., Harrison, S. M., and Beddington, R. S. (1997). Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development 124 (16), 3065–3076. doi:10.1242/dev.124.16.3065
Engwerda, A., Kerstjens-Frederikse, W. S., Corsten-Janssen, N., Dijkhuizen, T., and van Ravenswaaij-Arts, C. M. A. (2023). The phenotypic spectrum of terminal 6q deletions based on a large cohort derived from social media and literature: a prominent role for DLL1. Orphanet J. Rare Dis. 18 (1), 59. doi:10.1186/s13023-023-02658-w
Fischer-Zirnsak, B., Segebrecht, L., Schubach, M., Charles, P., Alderman, E., Brown, K., et al. (2019). Haploinsufficiency of the notch ligand DLL1 causes variable neurodevelopmental disorders. Am. J. Hum. Genet. 105 (3), 631–639. doi:10.1016/j.ajhg.2019.07.002
Gray, G. E., Mann, R. S., Mitsiadis, E., Henrique, D., Carcangiu, M. L, Banks, A., et al. (1999). Human ligands of the Notch receptor. Am J. Pathol. 154 (3), 785–794. doi:10.1016/s0002-9440(10)65325-4
Hiraoka, Y., Komine, O., Nagaoka, M., Bai, N., Hozumi, K., and Tanaka, K. (2013). Delta-like 1 regulates Bergmann glial monolayer formation during cerebellar development. Mol. Brain 6, 25. doi:10.1186/1756-6606-6-25
Hrabĕ de Angelis, M., McIntyre, J., and Gossler, A. (1997). Maintenance of somite borders in mice requires the Delta homologue DII1. Nature 386 (6626), 717–721. doi:10.1038/386717a0
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Lesieur-Sebellin, M., Till, M., Khau Van Kien, P., Herve, B., Bourgon, N., Dupont, C., et al. (2022). Terminal 6q deletions cause brain malformations, a phenotype mimicking heterozygous DLL1 pathogenic variants: a multicenter retrospective case series. Prenat. Diagn 42 (1), 118–135. doi:10.1002/pd.6074
Louvi, A., and Artavanis-Tsakonas, S. (2006). Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 7 (2), 93–102. doi:10.1038/nrn1847
Mahler, J., Filippi, A., and Driever, W. (2010). DeltaA/DeltaD regulate multiple and temporally distinct phases of notch signaling during dopaminergic neurogenesis in zebrafish. J. Neurosci. 30 (49), 16621–16635. doi:10.1523/jneurosci.4769-10.2010
Marklund, U., Hansson, E. M., Sundström, E., de Angelis, M. H., Przemeck, G. K., Lendahl, U., et al. (2010). Domain-specific control of neurogenesis achieved through patterned regulation of Notch ligand expression. Development 137 (3), 437–445. doi:10.1242/dev.036806
Okigawa, S., Mizoguchi, T., Okano, M., Tanaka, H., Isoda, M., Jiang, Y. J., et al. (2014). Different combinations of Notch ligands and receptors regulate V2 interneuron progenitor proliferation and V2a/V2b cell fate determination. Dev. Bio. 391 (2), 196–206. doi:10.1016/j.ydbio.2014.04.011
Peddibhotla, S., Nagamani, S. C., Erez, A., Hunter, J. V., Holder, J. L., Carlin, M. E., et al. (2015). Delineation of candidate genes responsible for structural brain abnormalities in patients with terminal deletions of chromosome 6q27. Eur. J. Hum. Genet. 23 (1), 54–60. doi:10.1038/ejhg.2014.51
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Shimojo, H., Ohtsuka, T., and Kageyama, R. (2011). Dynamic expression of notch signaling genes in neural stem/progenitor cells. Front. Neurosci. 5, 78. doi:10.3389/fnins.2011.00078
Zhang, C., Hao, S. J., and Zhang, Q. H. (2018). Copy number variation analysis of abortion tissue by massively parallel sequencing. J. Reprod. Med. 27 (10), 962–965. doi:10.3969/j.issn.1004-3845.2018.10.007
Keywords: 6q, terminal, deletion, DLL1, haploinsufficiency, prenatal, brain, anomalies
Citation: Ge T, Lin X, Tian X, Song X, Zhou B, Hui L, Wang X, Zhang Z and Zhang C (2025) DLL1 haploinsufficiency in prenatal brain anomalies: a retrospective analysis of 6q terminal deletions. Front. Genet. 16:1640775. doi: 10.3389/fgene.2025.1640775
Received: 04 June 2025; Accepted: 06 October 2025;
Published: 20 October 2025.
Edited by:
Prof. Anjana Munshi, Central University of Punjab, IndiaReviewed by:
Serap Bİlge, Bursa High Specialization Training and Research Centre, TürkiyeYayun Qin, Maternal and Child Health Hospital of Hubei Province, China
Copyright © 2025 Ge, Lin, Tian, Song, Zhou, Hui, Wang, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuan Zhang, emhhbmdjaHVhbjA0MDRAMTYzLmNvbQ==
†These authors have contributed equally to this work