 Chunyan Bai1†
Chunyan Bai1† Jiayi Ning1†Junwen Fei1
Jiayi Ning1†Junwen Fei1 Zhenbo Wang1Yu He1Jing Li1Xiaoran Zhang1
Zhenbo Wang1Yu He1Jing Li1Xiaoran Zhang1 Shuang Liang1Dali Wang2
Shuang Liang1Dali Wang2 Hao Sun1*Boxing Sun1*
Hao Sun1*Boxing Sun1*- 1College of Animal Science, Jilin University, Changchun, China
- 2Agricultural Experiment Base of Jilin University, Changchun, China
Introduction
The regulation of gene expression is influenced by multiple factors, including species origin, tissue specificity, developmental stage, and sex differences. Therefore, gene expression sequencing data obtained from various tissues of different species at different time points are of immense significance for achieving a comprehensive and clear understanding of gene functions.
Numerous pig breeds exist globally. In this report, the focus is on the Junmu No. 1 White pig (Figure 1). It is a hybrid of Belgian Seghershybrid boars and Chinese Sanjiang hybrid sows, with strong artificial selection for growth traits. The Junmu No. 1 White pig is primarily utilized as a terminal sire breed, selected specifically for superior growth rate and enhanced feed conversion efficiency (Bai et al., 2017).

Figure 1. Junmu No. 1 White boar.
Generally speaking, sense of smell is closely related to its appetite, and appetite also determines the amount of feed intake, which further affects the animal’s growth rate and the farm’s economic efficiency. Meanwhile, there is limited information available on RNA-seq of olfactory tissues in pigs, despite a single recent RNA-seq survey of porcine olfactory epithelium (Yang et al., 2024) and its conspicuous absence from the Pig RNA Atlas, comprehensive transcriptomic data for this tissue remain markedly scarce, underscoring the need for the present multi-tissue dataset. Therefore, we collected RNA sequencing to profile the transcriptomes of 13 distinct tissues from growing Junmu No. 1 White pigs, with particular focus on olfactory receptor (OR) genes. By examining the relevant databases, we discovered that, for most available transcriptome data, there are few instances where so many tissues from a single pig breed have been measured simultaneously. As a result, the data we obtained is highly valuable and rich in information, and we present this data to offer valuable insights for other researchers to investigate the functions of pig genes.
Samples collection and sequencing
The Junmu No. 1 White boars were obtained from the pig farm of Agriculture Experimental Base of Jilin University (Changchun, China). Three nursery finished healthy male piglets 70 days old with an average weight of 30 kg were randomly selected. The Spleen, ileum, brain, fat, kidney, duodenum, lung, heart, muscle, jejunum, liver, olfactory epithelium and testis tissues were collected. A small section was taken from a regular position of each tissue for each individual. Total RNA was extracted using a commercial kit (Tiangen), and sequencing was performed by Novogene Biotech (Beijing, China) using the Illumina Genome Analyzer platform.
Data quality control and variant calling
The FASTP (Chen et al., 2018) software was used to perform quality control. The clean reads were aligned to the pig reference genome (Sus scrofa 11.1) using HISAT2 (Kim et al., 2019) with default parameters. Then SAM files are converted to BAM files and sorted using the Sortsam command by GATK (Van der Auwera et al., 2013). The StringTie software (Shumate et al., 2022) was used for transcript assembly and quantification. The GFFCompare software (Pertea and Pertea, 2020) was used to annotate the transcripts. Principal component analysis (PCA) was performed using DESeq2 (Love et al., 2014).
Data description
After filtering, the average quality value of the data was significantly improved, and low-quality reads and splice sequences were effectively removed. A total of 309.8 Gb clean data was obtained. Filtered data quality values (Q30) can reach an average of 93.32%. It shows that the quality of the data has been significantly improved, providing a high-quality data base for subsequent analyses. The clean reads were aligned to the pig reference genome using HISAT2, and the results showed that the overall alignment rate was 94.61%. The results showed that most of the reads could be successfully aligned to the reference genome, indicating that the alignment process was accurate and efficient. The sequence data were deposited in the NCBI Sequence Read Achieve (SRA) and the accession number of the sequencing data was PRJNA664265. The sequencing information of each sample was shown in Supplementary Table S1.
After assembly and quantification of transcripts using StringTie, a total of 115,045 transcripts were detected with varying degrees of expression in 39 samples, these findings provided a standardised dataset for subsequent differential expression analysis. Subsequently, GFFCompare was used to annotate the transcripts, and those with a class code of “=” were specifically retained. The transcript count data obtained can be found in the figshare (https://doi.org/10.6084/m9.figshare.29192831.v1).
To elucidate the genetic distinctions between pig olfactory epithelial tissue and other tissues for future researchers, we conducted a comparison between pig olfactory epithelial tissue and the remaining 12 tissues. The results of the principal component analysis (PCA) are depicted in Figure 2. The PCA revealed that samples from the same tissue, such as liver and muscle, clustered closely together in two-dimensional space. This clustering indicates that samples from the same tissue exhibit highly similar gene expression patterns.
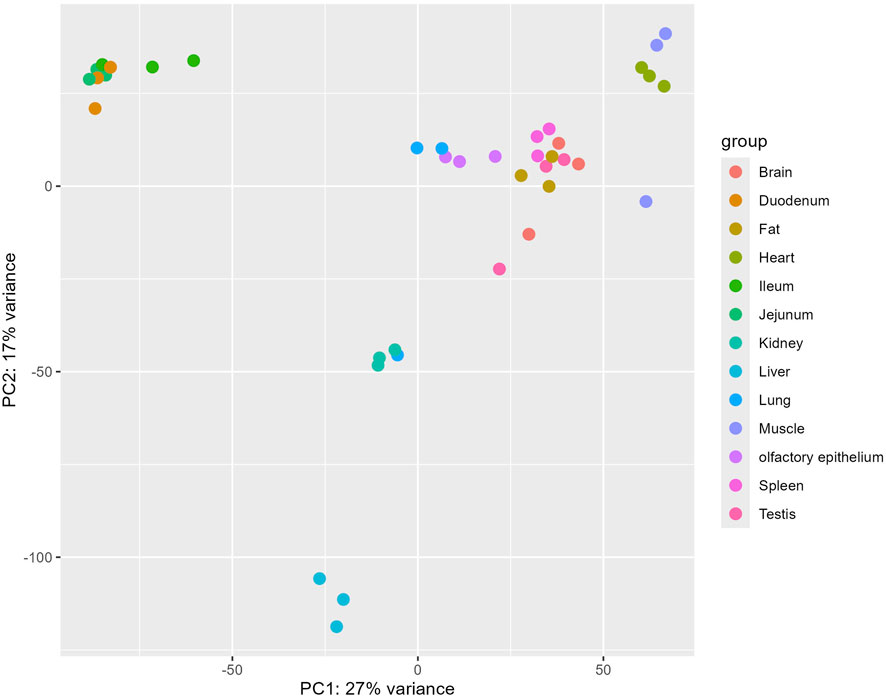
Figure 2. PCA plot of the 13 tissues based on gene expression count data.
In conclusion, this report provides 39 RNA-seq data from 13 tissues of three Junmu No. 1 White boars, which can provide candidate targets and molecular mechanism clues for further functional validation experiments (gene knockout, overexpression or single-cell sequencing) in the future. These datasets support analysis of gene function in porcine olfactory epithelium, advancing understanding of olfactory biology and its physiological impacts.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA664265.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee of Jilin University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
CB: Software, Writing – original draft, Writing – review and editing, Formal Analysis, Project administration, Resources. JN: Formal Analysis, Writing – original draft, Writing – review and editing, Methodology, Validation, Visualization. JF: Writing – review and editing, Investigation, Validation. ZW: Writing – original draft, Writing – review and editing, Investigation, Validation, Visualization. YH: Investigation, Validation, Writing – review and editing. JL: Investigation, Validation, Writing – review and editing. XZ: Investigation, Validation, Writing – review and editing. SL: Writing – review and editing, Project administration, Supervision. DW: Investigation, Writing – review and editing, Project administration, Resources, Supervision. HS: Writing – review and editing, Conceptualization, Funding acquisition, Methodology, Software, Writing – original draft. BS: Writing – review and editing, Conceptualization, Funding acquisition, Methodology, Software, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (32202628).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1641395/full#supplementary-material
References
Bai, C., Pan, Y., Wang, D., Cai, F., Yan, S., Zhao, Z., et al. (2017). Genome-wide association analysis of residual feed intake in Junmu No. 1 white pigs. Anim. Genet. 48 (6), 686–690. doi:10.1111/age.12609
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 (8), 907–915. doi:10.1038/s41587-019-0201-4
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Pertea, G., and Pertea, M. (2020). GFF utilities: Gffread and GffCompare. F1000Res 9, ISCB Comm J-304. doi:10.12688/f1000research.23297.2
Shumate, A., Wong, B., Pertea, G., and Pertea, M. (2022). Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 18 (6), e1009730. doi:10.1371/journal.pcbi.1009730
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43 (1110), 11.10.1–11.10.33. doi:10.1002/0471250953.bi1110s43
Keywords: boars, RNAseq, olfactory epithelium, piglets, mRNA
Citation: Bai C, Ning J, Fei J, Wang Z, He Y, Li J, Zhang X, Liang S, Wang D, Sun H and Sun B (2025) Data report: transcriptome profiling of 13 tissues of Junmu No. 1 boars. Front. Genet. 16:1641395. doi: 10.3389/fgene.2025.1641395
Received: 05 June 2025; Accepted: 08 August 2025;
Published: 01 September 2025.
Edited by:
Kimberly M. Davenport, Washington State University, United StatesReviewed by:
Daniela Ferreira, University of Trás-os-Montes and Alto Douro, PortugalEsma Gamze Aksel, Erciyes University, Türkiye
Copyright © 2025 Bai, Ning, Fei, Wang, He, Li, Zhang, Liang, Wang, Sun and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Sun, c3VuaGFvOTJAamx1LmVkdS5jbg==; Boxing Sun, c3VucGF0aGluZ0B2aXAuMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship