 Chang Liu1,2†
Chang Liu1,2† Yanlin Huang1,2†
Yanlin Huang1,2† Anpeng Fu1,2Yunan Wang1,3Jing Wu1,3Yan Zhang1,2Li Du1,3Hongke Ding1,2
Anpeng Fu1,2Yunan Wang1,3Jing Wu1,3Yan Zhang1,2Li Du1,3Hongke Ding1,2 Lihua Yu1,2Fake Li1,2
Lihua Yu1,2Fake Li1,2 Yiming Qi1,2Yuan Liu1,2Xingwang Wang1,2Yukun Zeng1,2Ling Liu1,2Ying Xiong1,3Yuanling Liu1,3Xin Zhao1,3Liyuan Fang1,3Jiayi Jian1,3
Yiming Qi1,2Yuan Liu1,2Xingwang Wang1,2Yukun Zeng1,2Ling Liu1,2Ying Xiong1,3Yuanling Liu1,3Xin Zhao1,3Liyuan Fang1,3Jiayi Jian1,3 Aihua Yin1,3*Yanqin You4*
Aihua Yin1,3*Yanqin You4*- 1Medical Genetic Center, Guangdong Women and Children Hospital, Guangzhou, Guangdong, China
- 2Guangzhou Key Laboratory of Prenatal Screening and Diagnosis, Guangdong Women and Children Hospital, Guangzhou, Guangdong, China
- 3Prenatal Diagnosis Center, Guangdong Women and Children Hospital, Guangzhou, Guangdong, China
- 4Department of Obstetrics and Gynecology, First Medical Center of Chinese PLA General Hospital, Beijing, China
Background: Hearing loss (HL) is a prevalent sensorineural disorder with a highly heterogeneous etiology. Next-generation sequencing (NGS) has revolutionized the genetic testing landscape for diseases characterized by high genetic and allelic heterogeneity, enabling the simultaneous screening of hundreds of genes.
Methods: One hundred and seventy-one unrelated patients with non-syndromic or syndromic HL were enrolled in this study. Exome sequencing (ES) was applied to explore molecular etiology in the cohort, and clinical reports were provided by geneticists and genetic counselors. Multidisciplinary team forums were conducted to ensure accurate diagnoses and improved patient management.
Results: The molecular cause of HL was determined in 78 of 171 probands (45.6%): 54 with an autosomal recessive (AR) inheritance pattern, 23 with an autosomal dominant (AD) pattern, and 1 with both AR/AD inheritance patterns. Candidate variants in 33 genes were identified in the study cohort: 14 with an AR inheritance pattern, 18 with an AD pattern, and 1 with both AR/AD inheritance patterns. Twenty-eight of the variants identified in the study were novel.
Conclusion: Exome sequencing facilitates genetic diagnosis and improves the management of patients with HL in clinical practice. Identifying the etiology of HL may improve patient care, refine genetic counseling, and facilitate the estimation of recurrence risk.
1 Introduction
According to estimates from the World Health Organization, approximately 466 million individuals globally are affected by disabling hearing loss (HL), including 34 million children under the age of fifteen years (Wilson et al., 2017). HL is a prevalent sensorineural disorder with implications that extend far beyond sensory impairment (World Health Organization, 2018). Insufficient auditory stimulation or inadequate language exposure during early childhood may alter brain connectivity and processing; thus, children who have not acquired adequate auditory stimulation may encounter significant challenges in their subsequent linguistic acquisition, cognitive development, and psychosocial functioning (World Health Organization, 2018; Kral and O’Donoghue, 2010; Lieu et al., 2020). HL can manifest at any stage of life, impairing communication, affecting social interactions, and creating difficulties in professional settings (World Health Organization, 2018).
The complex etiology of HL, coupled with the highly variable and often overlapping presentations of different types of HL, poses challenges for traditional clinical diagnosis (Alford et al., 2014). It is estimated that up to 60% of educationally significant congenital and early-onset HL is associated with genetic factors (Alford et al., 2014; Schimmenti et al., 2004). Although the role of genetic factors in adult-onset HL remains less clear, an increasing number of susceptibility loci have been identified, and a substantial proportion of cases have been attributed to genetic causes (Alford et al., 2014; Michels et al., 2019). Approximately 120 non-syndromic loci have been identified in humans (hereditaryhearingloss.org), and over 400 genetic syndromes include HL as a characteristic feature (Alford et al., 2014; García-García et al., 2020). Recent recommendations advocate genetic testing as the initial diagnostic step for children with bilateral sensorineural hearing loss (SNHL), following cytomegalovirus (CMV) testing. Identifying the genetic etiology of HL can provide several potential benefits for patients and their families, including improved clinical management, informed planning for future medical and educational needs, and more accurate estimation of recurrence risk (Alford et al., 2014). Furthermore, given the rapid advancement of SNHL gene therapy research, understanding the genetic etiology of HL is crucial for assessing the eligibility for imminent clinical trials.
Traditional molecular diagnostic tests for HL primarily involved genotyping or DNA sequencing to identify specific HL variants or screen a limited number of genes associated with it. In the last decade, next-generation sequencing (NGS) has revolutionized the genetic testing landscape for diseases characterized by high genetic and allelic heterogeneity, such as HL, enabling the simultaneous screening of hundreds of genes (García-García et al., 2020). In the present study, exome sequencing (ES) was utilized to facilitate genetic diagnosis and improve the management of patients with HL in clinical practice.
2 Materials and methods
2.1 Ethics statement
The study was approved by the Medical Ethics Committee of Guangdong Women and Children Hospital. Written informed consent was obtained from all participants and their parents or legal guardians (in the case of children under 18). The authors had access to identifiable patient information, which was anonymized prior to submission. All the procedures conducted in the study adhered to the Declaration of Helsinki, as previously described (Liu et al., 2022).
2.2 Patients and samples
A total of 171 unrelated patients diagnosed with non-syndromic or syndromic HL were enrolled from January 2017 to September 2024 under an institutional review board-approved protocol of informed consent. A comprehensive history and physical examination were obtained for each participant, including clinical history, severity of HL, age and cause of onset, infection history, exposure to aminoglycoside antibiotics, and genetic factors associated with hearing impairment (Yin et al., 2014). The recruited patients exhibited sensorineural or mixed HL. The severity of HL was established as mild (hearing thresholds 26 dB–40 dB), moderate (hearing thresholds 41 dB–60 dB), severe (hearing thresholds 61 dB–80 dB), or profound (hearing thresholds more than 80 dB). A stepwise approach was provided as an alternative (Yin et al., 2014), and four patients in the study cohort opted for this approach and were pre-screened for the GJB2, SLC26A4, and MT-RNR1 gene variants prior to ES.
Peripheral blood samples were collected from the patients and their first-degree relatives if available. Genomic DNA was extracted using the SolPure Blood DNA Kit (Magen, Shanghai, China), following the manufacturer’s instructions, which included optional RNase treatment. The DNA was then fragmented using a Q800R Sonicator (Qsonica, CT, United States). The paired-end libraries were prepared following the Illumina library preparation protocol.
2.3 Exome sequencing
All cases underwent genetic evaluations using either whole-exome sequencing (WES) or clinical exome sequencing (CES). Custom-designed NimbleGen SeqCap probes (Roche NimbleGen, Madison, WI) were used for in-solution hybridization to enrich target sequences for WES or, in the case of clinical ES, the target sequences included ∼5,000 genes that were potentially associated with known Mendelian genetic diseases (AmCare Genomic Laboratory, Guangzhou, China). Low-quality reads (Phred score < Q20) were removed before demultiplexing. Sequences were aligned to the hg19 reference genome using NextGENe software (SoftGenetics, State College, PA) with standard parameters for single-nucleotide variant (SNV) and insertion/deletion (indel) detection.
2.4 Data analysis
The resulting sequencing data were analyzed using NextGENe software with reference to the human assembly GRCh37/hg19. This software program performs the alignment, variant calling, and annotation of the variants. The annotated variants were filtered according to a minor allele frequency (MAF) value >0.01, and the frequency of the variants was explored in the gnomAD v4.1.0 database (https://gnomad.broadinstitute.org/). To classify the variants, we considered their annotation in dbSNP (www.ncbi.nlm.nih.gov/SNP/); their description in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), VarSome (https://Varsome.com/), and HGMD (http://www.hgmd.cf.ac.uk/); and the variant type, as previously described (García-García et al., 2020). The detailed process and quality control system for identifying the candidate variants were applied as previously described (Liu et al., 2022). Variant classification adhered to the 2015 ACMG/AMP guidelines (Richards et al., 2015; Tavtigian et al., 2020). Copy number variation (CNV) identification utilized a CNVkit (Talevich et al., 2016). AnnotSV annotated the detected CNVs in each tested sample for clinical interpretation (Geoffroy et al., 2018). Candidate CNV clinical significance was evaluated following the ACMG/ClinGen criteria (Riggs et al., 2020). Diagnostic variants were validated by Sanger sequencing or quantitative polymerase chain reaction (qPCR) when applicable.
2.5 Reanalysis and reclassification
The purpose of variant reinterpretation was to reassess the pathogenicity of the variants. All the variants were reinterpreted according to the standards and guidelines recommended by the ACMG/AMP. Negative and uncertain results were reanalyzed from the raw sequencing data stored as compressed fastq files (Nambot et al., 2018). All variants of the final analysis file were interpreted. The reclassification focused on variants that were previously and newly reported as pathogenic/probably pathogenic (P/LP) in updated public databases of clinical interest (OMIM, https://omim.org/; ClinVar, http://www.ncbi.nlm.nih.gov/clinvar; DECIPHER, https://www.deciphergenomics.org/; HGMD, http://www.hgmd.cf.ac.uk/) or as affecting the well-established human disease genes (Nambot et al., 2018). The interpretation was then extended to all other variants, namely, those not meeting the diagnostic interpretation criteria (Nambot et al., 2018). Variants that remained undiagnosed were subject to periodic re-interpretation.
3 Results
3.1 Cohort description
A total of 171 unrelated patients were enrolled in the study, and the characteristics of the study cohort are summarized in Table 1. One hundred and thirty-four patients were affected with non-syndromic hearing loss (NSHL), and 37 were affected with syndromic hearing loss (SHL). The degree of HL in the patients varied, and severe-to-profound HL was observed in the majority (78.4%, 134/171). In addition, pre-lingual HL was detected in 80.6% (145/171) of the patients.
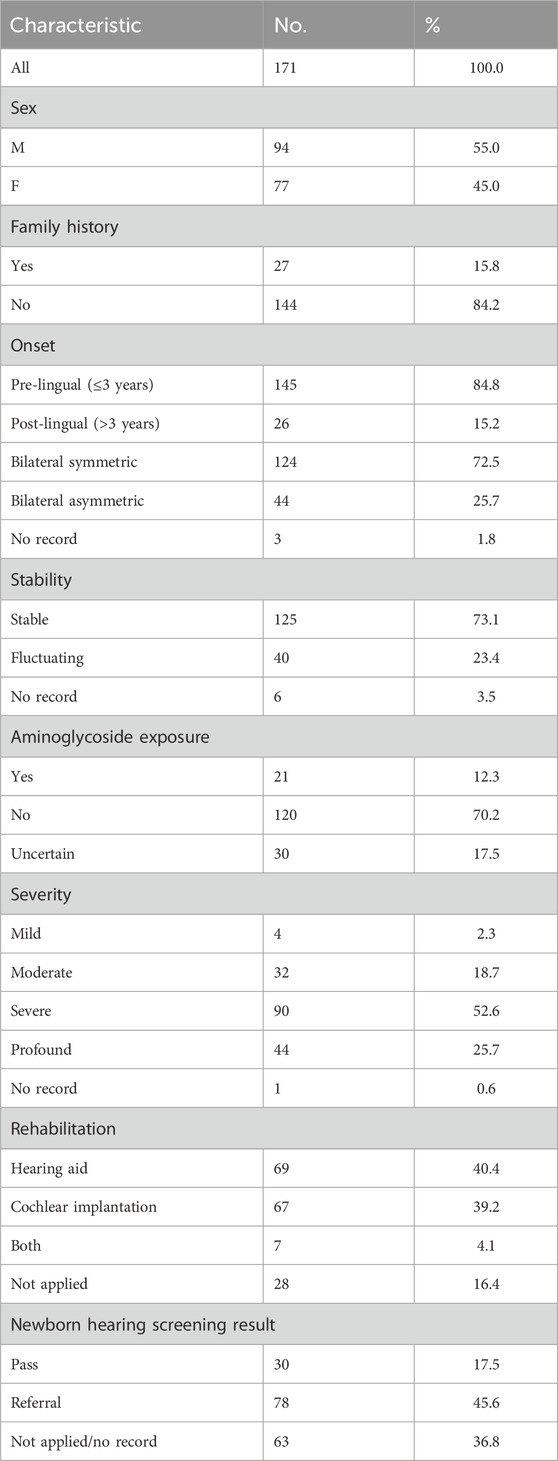
Table 1. Characteristics of the study cohort.
3.2 Diagnostic findings
The etiologic diagnosis for HL has been identified for 78 out of 171 probands (45.6%), among which 55 had an autosomal recessive (AR) inheritance pattern and 23 had an autosomal dominant (AD) pattern. Table 2 contains the clinical data and genetic evaluations of the diagnosed patients. Candidate variants in 33 genes were identified in the study cohort, with 14 having an AR inheritance pattern and 19 having an AD pattern (Figure 1). A total of 91 candidate variants were identified, including 33 missense, 21 frameshift, 16 nonsense, 9 canonical splicing, 4 intronic, 3 CNVs, 2 in-frame, 2 synonymous, and 1 multi-nucleotide variants. Of the 91 variants, 28 were novel (Table 4).

Table 2. Clinical data and genetic evaluations of the diagnosed patients.
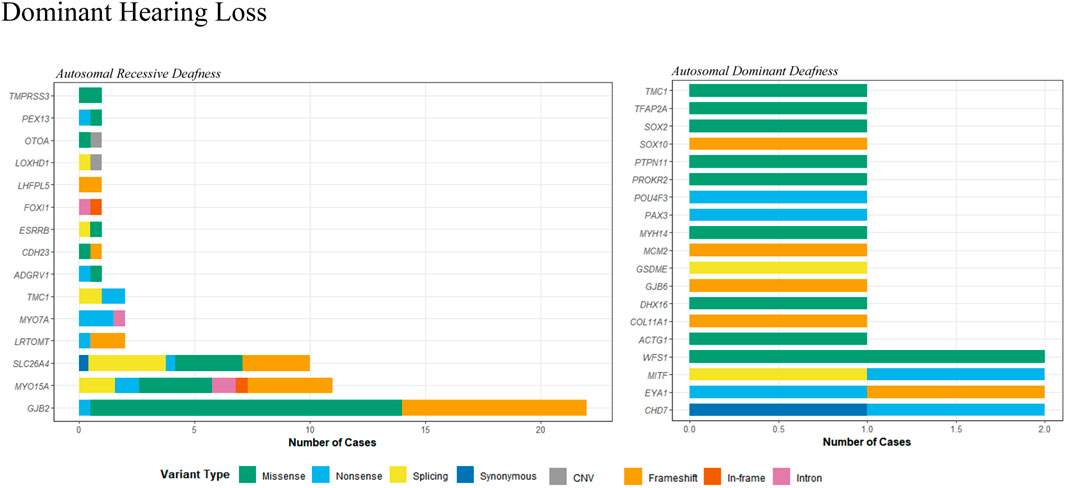
Figure 1. Distribution of variant types in genes associated with autosomal recessive and dominant hearing loss.
Fifty-four probands carried bi-allelic pathogenic or likely pathogenic variants associated with an AR pattern of inheritance (Table 2A). Fifty-one cases presented with NSHL. These were linked to GJB2 (22 cases), SLC26A4 (10 cases), MYO15A (10 cases), MYO7A (2 cases), LRTOMT (2 cases), TMC1 (2 cases), and 1 case in each of the ADGRV1, CDH23, TMPRSS3, LOXHD1, ESRRB, FOXI1, PEX13, OTOA, and LHFPL5 genes (Figure 1). Of these, three cases may have had more than one gene that contributed to the HL. In patient 18, a homozygous c.109G>A (p.Val37Ile) variant in GJB2 was identified, in addition to two P/LP variants in the PEX13 gene. Similarly, a homozygous c.109G>A (p.Val37Ile) variant in GJB2 was identified in patient 23, in addition to two LP/VUS variants in the OTOA gene. Another patient (P49) with Usher syndrome was found to carry LP variants in the ADGRV1 gene, in addition to two LP/VUS variants in the MYO15A gene.
Twenty-three patients were identified with autosomal dominant HL (Table 2B). Eight cases presented with NSHL, and 15 patients presented with SHL. They were linked to CHD7 (two cases), EYA1 (two cases), MITF (two cases), WFS1 (two cases), and one case in each of the ACTG1, TMC1, GJB6, DHX16, COL1A1, PTPN11, TFAP2A, SOX2, SOX10, MYH14, POU4F3, PROKR2, PAX3, and GSDME genes (Figure 1). Moreover, one patient (P39) may have had HL contributions from both a gene associated with AD inheritance and a gene associated with AR inheritance. A VUS variant, c.993dup, in the MCM2 gene was identified, in addition to two LP/VUS variants in the MYO15A gene.
Identifying the genetic causes of HL enabled the recommendation of personalized treatment options, such as cochlear implants and hearing aids. Of the 78 cases with positive genetic diagnoses, 73 patients (93.6%) adopted rehabilitation approaches and achieved favorable therapeutic outcomes. Moreover, after genetic counseling, 27 couples utilized preimplantation genetic diagnosis or prenatal diagnosis after identifying the pathogenic variants, enabling informed reproductive choices.
3.3 Undiagnosed probands by ES
Reanalysis was performed for patients with a non-positive result, and the reclassification was carried out for all variants in cases with positive or uncertain results to reassess the pathogenicity of the variants. A total of 93 probands still remained undiagnosed after follow-up studies (Table 3), with 9.7% (9/93) showing inconclusive findings due to limited variant pathogenic evidence. For instance, patient 91 harbored a loss-of-function variant, c.2953delG (p.Glu985SerfsTer6), in the MACF1 gene with an AD inheritance pattern. The variant was not found in gnomAD exomes/genomes and was predicted as damaging by multiple computational algorithms in VarSome, but further studies are needed to determine its pathogenicity. A similar situation was observed for patients 81, 88, and 95. Patient 90 harbored a homozygous c.6070G>A (p.Ala2024Thr) variant in the PTPRQ gene. The variant was not found in gnomAD exomes/genomes, but further studies are needed to determine its pathogenicity. Patient 92 carried two VUS variants in the DIAPH3 gene with an AD inheritance pattern. The missense variant c.1124C>T (p.Pro375Leu) was absent from gnomAD exomes/genomes and was predicted to be deleterious by multiple computational algorithms in VarSome. The c.180 + 5G>T variant was also absent in gnomAD exomes/genomes and scored 0.867 by delta score of donor loss in spliceAI. It was predicted to affect mRNA transcription and splicing, but additional studies are needed to determine its pathogenicity.

Table 3. Undiagnosed probands by exome sequencing.
Moreover, diagnostic proband-only ES revealed the presence of a single pathogenic or likely pathogenic variant in AR inheritance-pattern genes in 10 patients (10.8%). For example, patient 85 harbored the c.1251 + 5G>A variant in the CEP78 gene. Studies have shown that this variant results in skipping of exon 10 and introduces a premature termination codon (Fu et al., 2017). It has also been reported in individual(s) with CEP78-related conditions (Fu et al., 2017). However, in the absence of another causal variant, the case remains inconclusive. Patient 87 harbored the c.439G>A (p.Glu147Lys) variant in the GJB2 gene. In silico analysis supports that this missense variant has a deleterious effect on protein structure/function. This variant has been reported in multiple cases with hearing loss (Cryns et al., 2004; Frei et al., 2004), including homozygotes and individuals who were compound heterozygous for a second pathogenic variant. However, in the absence of a second pathogenic variant, the case remains inconclusive. Patient 96 harbored the c.5098G>C (p.Glu1700Gln) variant in the OTOF gene. This variant has been detected in at least 18 individuals with autosomal recessive non-syndromic hearing loss, including homozygotes and individuals who were compound heterozygous for a second pathogenic variant (Chiu et al., 2010; Zhu et al., 2021). However, in the absence of a second pathogenic variant, our case remains inconclusive. Patient 97 harbored the c.5816G>A (p.Arg1939Gln) variant in the OTOF gene. This variant is present in population databases (gnomAD 0.07%). This missense change has been observed in individuals with auditory neuropathy spectrum disorder and/or deafness (Bae et al., 2013; Iwasa et al., 2022). In at least one individual, the data are consistent with being in trans from a pathogenic variant. The variant was predicted as probably damaging by multiple computational algorithms in VarSome. However, in the absence of another causal variant, the case remains inconclusive.
4 Discussion
HL is one of the most etiologically heterogeneous disorders, encompassing over 400 genetic syndromes that include HL as a feature, more than 120 genes associated with NSHL, and a variety of non-genetic causes (Alford et al., 2014). With the advent of NGS technologies, it is now feasible to analyze hundreds of candidate HL genes simultaneously in a cost-effective manner. Nevertheless, the diagnostic yield varies across different patient cohorts and depends on the detection methods employed. Factors such as the degree of HL, the onset age of HL, the existence of family history, the ethnic origin, and the number of genes contained in the NGS panel may impact the rate of genetic diagnosis (García-García et al., 2020). Typically, the detection rate obtained usually increases in patients with a positive family history of HL or in cases where the HL was congenital and symmetric (Zhu et al., 2021). In the present study, ES was used to investigate the molecular etiology of HL in a Chinese cohort, resulting in genetic diagnoses for 78 of 171 probands, with an overall diagnostic yield of 45.6%. The diagnostic yield is comparable to that observed in other NGS-based panels on unselected heterogeneous HL patients, which ranged from 39% to 48% (Liu et al., 2022; Sloan-Heggen et al., 2016; Baux et al., 2017; Shearer et al., 2013). The study cohort consisted of patients with various types of sensorineural/mixed HL, including congenital, pre-lingual, and post-lingual HL, along with varying degrees of severity (mild, moderate, severe, and profound), and both stable and progressive HL and non-syndromic or syndromic HL. The heterogeneity within the patient cohort may affect the rate of genetic diagnosis. The positive diagnostic rate was higher in probands with a family history of HL (59.2%, 16/27), severe or profound HL (55.2%, 74/134), stable HL (54.4%, 68/125), symmetric HL (51%, 73/124), and pre-lingual HL (49.6%, 72/145), and it was lower in probands with mild or moderate HL (11.1%, 4/36), aminoglycoside exposure (19.0%, 4/21), a passed newborn hearing screening (20.0%, 6/30), post-lingual HL (23.1%, 6/26), and fluctuating HL (25.0%, 10/40). Moreover, a portion of the HL patients at our hospital opted for a stepwise approach and were pre-excluded for the GJB2, SLC26A4, and MT-RNR1 gene variations prior to ES. It also explains why the proportions of GJB2 and SLC26A4 gene variations are comparatively low in the study cohort (Figures 1, 2). In addition, WES and CES were used as diagnostic tools for HL in the present study, and their efficacy was evaluated. The diagnostic yields were 44.1% (52 out of 118 probands) for WES and 49.0% (26 out of 53 probands) for CES. The raw data volume was approximately 9.7 GB for each sample tested by WES and approximately 5.1 GB for each sample tested by CES. The average coverage depth was 148.3 X and 263.4 X for WES and CES, respectively, with >99.5% of the target regions covered by at least 20 reads. The cost was 824 USD and 668 USD for WES and CES, respectively. The turn-around time for both assays was similar, which was 3–4 weeks.

Figure 2. Detailed distribution of variants in MYO15A, GJB2, SLC26A4, and MYO7A genes. Note: Sector areas represent the frequencies or proportions of specific variant types detected in the gene, illustrating their relative distribution among the genetic alterations.
In the present study, several novel variants were detected, and their pathogenicity was estimated according to the ACMG guidelines. As shown in Table 4, 28 novel variants from 21 genes were detected. Among the 28 novel variants identified, there were 13 missense variants, five frameshift variants, five nonsense variants, three intronic variants affecting splicing, and two canonical splicing variants. Six variants predicted to generate direct stop codons and one variant predicted to affect splicing were classified as pathogenic or likely pathogenic, and the remaining 21 variants were classified as VUS. The detection and evaluation of novel variants are important for expanding the spectrum of variants associated with HL.
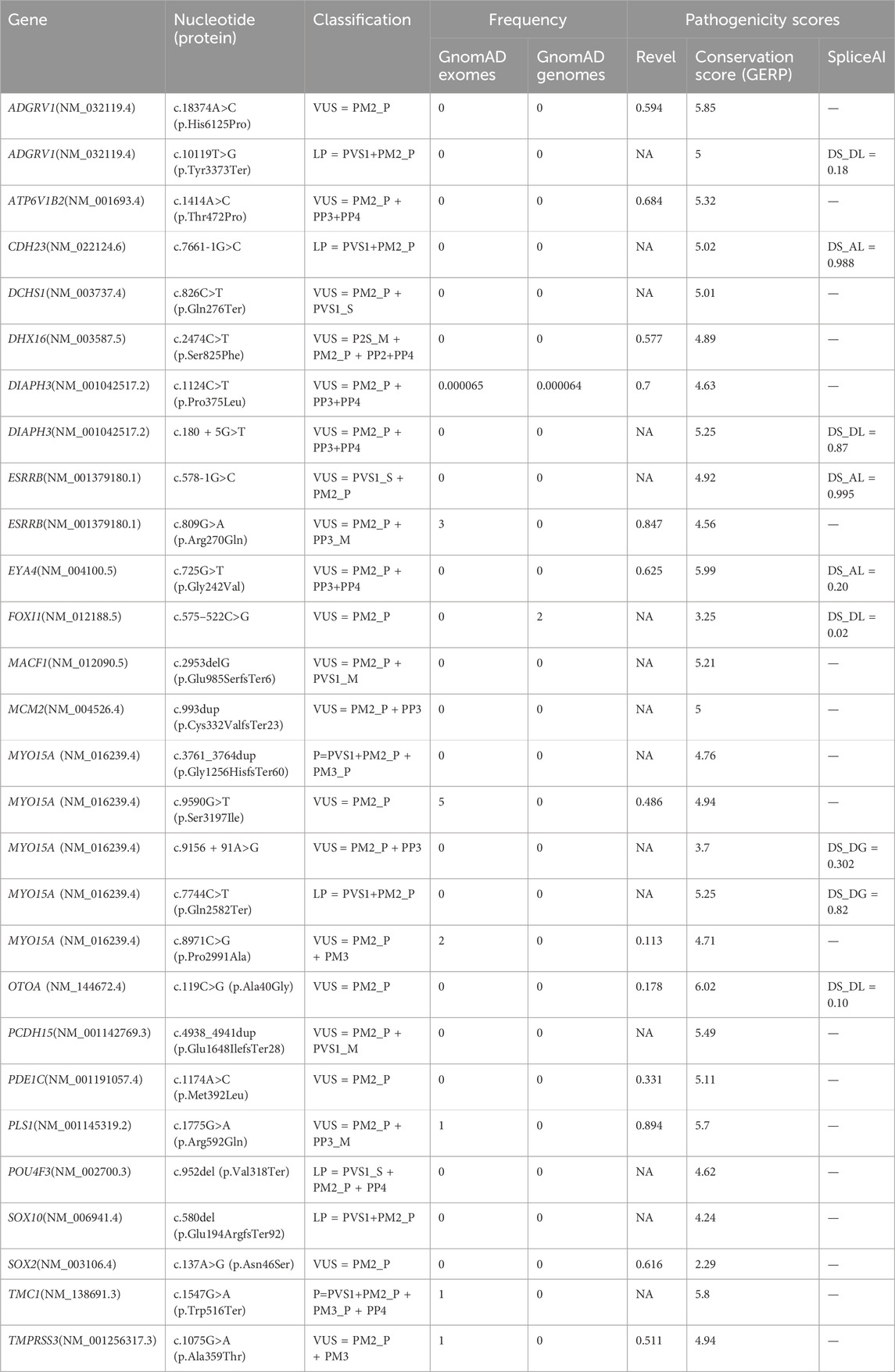
Table 4. Classification of the novel variants identified in this study.
Moreover, NGS panels enable the simultaneous analysis of hundreds of genes, and, in some cases, pathogenic variants in different genes can be identified in a single patient (García-García et al., 2020). In the study cohort, five cases harbored pathogenic variants in different genes or multiple pathogenic variants in the same gene. Patient 18 carried the c.676C>T (p.Arg226Ter) and c.880C>T (p.Arg294Trp) variants in the PEX13 gene in the compound heterozygous state, in addition to the homozygous c.109G>A (p.Val37Ile) variant in the GJB2 gene. Patient 23 carried the heterozygous c.119C>G (p.Ala40Gly) variant in the OTOA gene and 16p12.2 (chr16:21415697-21747711)×1 (including OTOA exon1-22), in addition to the homozygous c.109G>A (p.Val37Ile) variant in the GJB2 gene. Patient 49 harbored c.18374A>C (p.His6125Pro) and c.10119T>G (p.Tyr3373Ter) in the ADGRV1 gene in the compound heterozygous state, in addition to c.9690 + 1G>A and c.9590G>T (p.Ser3197Ile) in the MYO15A gene in the compound heterozygous state. Patient 39 harbored c.7744C>T (p.Gln2582Ter) and c.8971C>G (p.Pro2991Ala) in the MYO15A gene in the compound heterozygous state, in addition to c.993dup (p.Cys332ValfsTer23) in an AD-inherited MCM2 gene variant. The patient from case 4,377, who presented with HL and EVA, was found to carry three heterozygous LP variants in the SLC26A4 gene, namely, c.1547dupC (p.Ser517PhefsTer10), inherited from her father, and c.563T>C (p.Ile188Thr) and c.1746delG (p.Ala584ArgfsTer2), both inherited from her hearing mother. We speculate that c.1547dupC may contribute to the HL in this case, but we cannot determine whether c.563T>C and c.1746delG contribute respectively or collaboratively to the HL. These findings have important implications for reproductive genetic counseling, including risk assessment for the affected offspring and the potential application of pre-natal or pre-implantation genetic diagnosis.
Furthermore, the rapid accrual of knowledge in genomic medicine has prompted the reanalysis of existing data (Liu et al., 2019). As new disease genes are published, variants may be reclassified, and expanded phenotypic information becomes available (Ewans et al., 2018). This, combined with enhancements to bioinformatics pipelines and filtering strategies, contributes to an increased diagnostic yield, reinforcing the need for reanalysis (Ewans et al., 2018). In the present cohort, 93 probands (54.4%) remained undiagnosed after reanalysis and follow-up studies. One possible explanation for this is the clinical and genetic heterogeneity of HL. Additionally, causal variants may remain unidentified due to limitations in the analytical methods or inadequate knowledge in the literature regarding the genetics of the disease. Several factors may contribute to the improved diagnostic yield, including the strengthening of gene–disease associations, updates in the literature, the addition of detailed patient phenotype information, further sequencing of trios and other affected family members, improvements in sequencing data through re-sequencing, advancements in bioinformatics tools, and increased research collaborations facilitated by international case-sharing platforms (Shearer et al., 2013). These factors highlight the importance of reanalysis as the discovery of new gene– and variant–disease associations, the emergence of novel patient phenotypes, and the continued advancement of sequencing and bioinformatics technologies may collectively enhance the diagnostic yield of exome sequencing over time (Fung et al., 2020).
Nevertheless, ES has inherent technical constraints, including the inability to detect deep intronic variants, limited coverage of regulatory regions, and difficulty in identifying repeat expansions and complex structural variants. To overcome these limitations, multi-platform approaches should be integrated to enhance the diagnostic yield. Whole-genome sequencing (WGS) provides uniform coverage of coding and non-coding regions, enabling the detection of structural variants and regulatory mutations. Long-read sequencing resolves repetitive regions and phasing challenges. WGS-first pipelines are becoming clinically viable, although ES remains cost-effective for Mendelian disorders with clear phenotype–genotype correlations (Kang et al., 2025). Combining transcriptomics (RNA-seq) can reveal splicing impacts of non-coding variants, while machine learning models can improve non-coding variant interpretation.
5 Conclusion
Exome sequencing facilitates genetic diagnosis and improves the management of patients with HL in clinical practice. Identifying the etiology of HL may improve the management of patients with HL, refine genetic counseling, and facilitate the estimation of recurrence risk (Alford et al., 2014). The study highlights the importance of regularly reevaluating non-diagnostic exomes in light of updated gene discoveries, expanding variant databases, and improving bioinformatics pipelines to maximize diagnostic yield.
Data availability statement
The datasets generated and/or analyzed during the current study are not publicly available due to individual privacy but are available from the corresponding author (Aihua Yin, E-mail: eWluYWlodWEwMTMxQDE2My5jb20=) on reasonable request.
Ethics statement
These studies involving humans were approved by the Medical Ethics Committee of Guangdong Women and Children Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
CL: Writing – original draft, Formal analysis, Project administration, Data curation, Visualization, Validation, Methodology, Software, Investigation, Writing – review and editing, Supervision, Funding acquisition, Resources, Conceptualization. YH: Formal analysis, Visualization, Funding acquisition, Validation, Writing – original draft, Methodology, Data curation, Investigation, Conceptualization, Software. AF: Writing – review and editing, Investigation. YW: Writing – review and editing, Investigation. JW: Writing – review and editing, Investigation. YaZ: Writing – review and editing, Investigation. LD: Investigation, Writing – review and editing. HD: Investigation, Writing – review and editing. LY: Investigation, Writing – review and editing. FL: Investigation, Writing – review and editing. YI: Investigation, Writing – review and editing. YnL: Investigation, Writing – review and editing. XW: Investigation, Writing – review and editing. YuZ: Writing – review and editing, Investigation. LL: Investigation, Writing – review and editing. YX: Investigation, Writing – review and editing. YlL: Investigation, Writing – review and editing. XZ: Investigation, Writing – review and editing. LF: Writing – review and editing, Investigation. JJ: Investigation, Writing – review and editing. AY: Funding acquisition, Writing – review and editing, Project administration, Supervision. YY: Resources, Project administration, Writing – review and editing, Investigation, Supervision, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the sub-project of the National Key Research and Development Program (grant no. 2023YFC2705600), the Medical Science and Technology Research Project of Guangdong Province (grant no. A2023500, A2024303, and C2023030), the National Key Research and Development Program of China (2016YFC1000703), the Traditional Chinese Medicine Research Project of Guangdong Province (grant no. 20251042), the Guangzhou Science and Technology Planning Project (grant no. 202103000047), the Guangdong Women and Children Hospital Clinical Research Funding Project (grant no. 310103-1602), Guangzhou Science and Technology Program Project (grant no. 202201011788), and the National Key Research and Development Program of China (grant no. 2022YFC2703705). The funders did not play any role in the design of the study, collection, analysis, interpretation of data, or in writing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
HL, hearing loss; SHL, syndromic hearing loss; NSHL, non-syndromic hearing loss; NGS, next-generation sequencing; WGS, whole-genome sequencing; WES, whole-exome sequencing; CES, clinical exome sequencing; ES, exome sequencing; AR, autosomal recessive; AD, autosomal dominant; ACMG, American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; P, pathogenic; LP, likely pathogenic; VUS, variants of uncertain clinical significance; CNV, copy number variation; MAF, minor allele frequency.
References
Alford, R. L., Arnos, K. S., Fox, M., Lin, J. W., Palmer, C. G., Pandya, A., et al. (2014). American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 16 (4), 347–355. doi:10.1038/gim.2014.2
Bae, S. H., Baek, J. I., Lee, J. D., Song, M. H., Kwon, T. J., Oh, S. K., et al. (2013). Genetic analysis of auditory neuropathy spectrum disorder in the Korean population. Gene 522 (1), 65–69. doi:10.1016/j.gene.2013.02.057
Baux, D., Vaché, C., Blanchet, C., Willems, M., Baudoin, C., Moclyn, M., et al. (2017). Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 7 (1), 16783. doi:10.1038/s41598-017-16846-9
Chiu, Y. H., Wu, C. C., Lu, Y. C., Chen, P. J., Lee, W. Y., Liu, A. Y. Z., et al. (2010). Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol. Neurootol 15 (6), 364–374. doi:10.1159/000293992
Cryns, K., Orzan, E., Murgia, A., Huygen, P. L. M., Moreno, F., del Castillo, I., et al. (2004). A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J. Med. Genet. 41 (3), 147–154. doi:10.1136/jmg.2003.013896
Ewans, L. J., Schofield, D., Shrestha, R., Zhu, Y., Gayevskiy, V., Ying, K., et al. (2018). Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet. Med. 20 (12), 1564–1574. doi:10.1038/gim.2018.39
Frei, K., Lucas, T., Ramsebner, R., Schöfer, C., Baumgartner, W. D., Weipoltshammer, K., et al. (2004). A novel connexin 26 mutation associated with autosomal recessive sensorineural deafness. Audiol. Neurootol 9 (1), 47–50. doi:10.1159/000074186
Fu, Q., Xu, M., Chen, X., Sheng, X., Yuan, Z., Liu, Y., et al. (2017). CEP78 is mutated in a distinct type of Usher syndrome. J. Med. Genet. 54 (3), 190–195. doi:10.1136/jmedgenet-2016-104166
Fung, J. L. F., Yu, M. H. C., Huang, S., Chung, C. C. Y., Chan, M. C. Y., Pajusalu, S., et al. (2020). A three-year follow-up study evaluating clinical utility of exome sequencing and diagnostic potential of reanalysis. npj Genom Med. 5 (1), 37. doi:10.1038/s41525-020-00144-x
García-García, G., Berzal-Serrano, A., García-Díaz, P., Villanova-Aparisi, R., Juárez-Rodríguez, S., de Paula-Vernetta, C., et al. (2020). Improving the management of patients with hearing loss by the implementation of an NGS panel in clinical practice. Genes (Basel) 11 (12), 1467. doi:10.3390/genes11121467
Geoffroy, V., Herenger, Y., Kress, A., Stoetzel, C., Piton, A., Dollfus, H., et al. (2018). AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34 (20), 3572–3574. doi:10.1093/bioinformatics/bty304
Iwasa, Y. I., Nishio, S. Y., Yoshimura, H., Sugaya, A., Kataoka, Y., Maeda, Y., et al. (2022). Detailed clinical features and genotype-phenotype correlation in an OTOF-related hearing loss cohort in Japan. Hum. Genet. 141 (3–4), 865–875. doi:10.1007/s00439-021-02351-7
Kang, H., Li, J., Duan, H., Su, L., Xia, Y., Li, Z., et al. (2025). Clinical application of whole exome sequencing (WES) in the genetic diagnosis of 768 Chinese patients with bilateral hearing loss. Eur. J. Hum. Genet. doi:10.1038/s41431-025-01896-9
Kral, A., and O’Donoghue, G. M. (2010). Profound deafness in childhood. N. Engl. J. Med. 363 (15), 1438–1450. doi:10.1056/NEJMra0911225
Lieu, J. E. C., Kenna, M., Anne, S., and Davidson, L. (2020). Hearing loss in children: a review. JAMA 324 (21), 2195–2205. doi:10.1001/jama.2020.17647
Liu, P., Meng, L., Normand, E. A., Xia, F., Song, X., Ghazi, A., et al. (2019). Reanalysis of clinical exome sequencing data. N. Engl. J. Med. 380 (25), 2478–2480. doi:10.1056/NEJMc1812033
Liu, C., Huang, Y., Zhang, Y., Ding, H., Yu, L., Wang, A., et al. (2022). Next-generation sequencing facilitates genetic diagnosis and improves the management of patients with hearing loss in clinical practice. Int. J. Pediatr. Otorhinolaryngol. 161, 111258. doi:10.1016/j.ijporl.2022.111258
Michels, T. C., Duffy, M. T., and Rogers, D. J. (2019). Hearing loss in adults: differential diagnosis and treatment. Am. Fam. Physician 100 (2), 98–108.
Nambot, S., Thevenon, J., Kuentz, P., Duffourd, Y., Tisserant, E., Bruel, A. L., et al. (2018). Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet. Med. 20 (6), 645–654. doi:10.1038/gim.2017.162
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Schimmenti, L. A., Martinez, A., Fox, M., Crandall, B., Shapiro, N., Telatar, M., et al. (2004). Genetic testing as part of the early hearing detection and intervention (EHDI) process. Genet. Med. 6 (6), 521–525. doi:10.1097/01.gim.0000144187.21727.28
Shearer, A. E., Black-Ziegelbein, E. A., Hildebrand, M. S., Eppsteiner, R. W., Ravi, H., Joshi, S., et al. (2013). Advancing genetic testing for deafness with genomic technology. J. Med. Genet. 50 (9), 627–634. doi:10.1136/jmedgenet-2013-101749
Sloan-Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., et al. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135 (4), 441–450. doi:10.1007/s00439-016-1648-8
Talevich, E., Shain, A. H., Botton, T., and Bastian, B. C. (2016). CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12 (4), e1004873. doi:10.1371/journal.pcbi.1004873
Tavtigian, S. V., Harrison, S. M., Boucher, K. M., and Biesecker, L. G. (2020). Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum. Mutat. 41 (10), 1734–1737. doi:10.1002/humu.24088
Wilson, B. S., Tucci, D. L., Merson, M. H., and O’Donoghue, G. M. (2017). Global hearing health care: new findings and perspectives. Lancet 390 (10111), 2503–2515. doi:10.1016/S0140-6736(17)31073-5
World Health Organization (2018). Addressing the rising prevalence of hearing loss. Geneva: World Health Organization. Available online at: https://iris.who.int/handle/10665/260336.
Yin, A., Liu, C., Zhang, Y., Wu, J., Mai, M., Ding, H., et al. (2014). Genetic counseling and prenatal diagnosis for hereditary hearing loss in high-risk families. Int. J. Pediatr. Otorhinolaryngol. 78 (8), 1356–1359. doi:10.1016/j.ijporl.2014.05.030
Keywords: hearing loss, exome sequencing, molecular analysis, clinical evaluation, reanalysis
Citation: Liu C, Huang Y, Fu A, Wang Y, Wu J, Zhang Y, Du L, Ding H, Yu L, Li F, Qi Y, Liu Y, Wang X, Zeng Y, Liu L, Xiong Y, Liu Y, Zhao X, Fang L, Jian J, Yin A and You Y (2025) Clinical utility of exome sequencing in hearing loss: a retrospective cohort study. Front. Genet. 16:1643537. doi: 10.3389/fgene.2025.1643537
Received: 09 June 2025; Accepted: 22 August 2025;
Published: 10 September 2025.
Edited by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Yuzuru Ninoyu, Kyoto Prefectural University of Medicine, JapanAlessandro Castiglione, Örebro University Hospital, Sweden
Copyright © 2025 Liu, Huang, Fu, Wang, Wu, Zhang, Du, Ding, Yu, Li, Qi, Liu, Wang, Zeng, Liu, Xiong, Liu, Zhao, Fang, Jian, Yin and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aihua Yin, eWluYWl3YUB2aXAuMTI2LmNvbQ==; Yanqin You, eW91eWFucWluNzZAMTYzLmNvbQ==
†These authors have contributed equally to this work