 Debasish Swapnesh Kumar Nayak1,2,3
Debasish Swapnesh Kumar Nayak1,2,3 Abhilash Pati1*
Abhilash Pati1* Amrutanshu Panigrahi1
Amrutanshu Panigrahi1 Mudassir Khan4,5*
Mudassir Khan4,5* Bayan Alabdullah6
Bayan Alabdullah6 Santanu Kumar Sahoo7
Santanu Kumar Sahoo7 Bibhuprasad Sahu8Abrar Almjally9
Bibhuprasad Sahu8Abrar Almjally9 Saurav Mallik10,11*
Saurav Mallik10,11* Tripti Swarnkar12
Tripti Swarnkar12- 1Department of Computer Science & Engineering, Siksha “O” Anusandhan (Deemed to be University), Bhubaneswar, Odisha, India
- 2Department of Computer Science and Engineering, Centurion University of Technology and Management, Bhubaneswar, Odisha, India
- 3Department of Computer Science and Engineering, Indian Institute of Technology, Bhilai, Chhattisgarh, India
- 4Department of Computer Science, College of Computer Science, Applied College Tanumah, King Khalid University, Abha, Saudi Arabia
- 5Jadara University Research Center, Jadara University, Irbid, Jordan
- 6Department of Information Systems, College of Computer and Information Sciences, Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia
- 7Department of Electronics and Communication Engineering, Siksha “O” Anusandhan (Deemed to be University), Bhubaneswar, Odisha, India
- 8Symbiosis Institute of Technology, Hyderabad Campus, Symbiosis International University, Pune, India
- 9College of Computer and Information Sciences, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh, Saudi Arabia
- 10Department of Environmental Health, Harvard T H Chan School of Public Health, Boston, MA, United States
- 11Department of Pharmacology & Toxicology, University of Arizona, Tucson, AZ, United States
- 12Department of Computer Application, National Institute of Technology, Raipur, India
Background: Infectious diseases pose a global health threat, with antimicrobial resistance (AMR) exacerbating the issue. Considering Escherichia coli (E. coli) is frequently linked to urinary tract infections, researching antibiotic resistance genes in this context is essential for identifying and combating the growing problem of drug resistance.
Objective: Machine learning (ML), particularly deep learning (DL), has proven effective in rapidly detecting strains for infection prevention and reducing mortality rates. We proposed aiGeneR 3.0, a simplified and effective DL model employing a long-short-term memory mechanism for identifying multi-drug resistant and resistant strains in E. coli. The aiGeneR 3.0 paradigm for identifying and classifying antibiotic resistance is a tandem link of quality control incorporated with DL models. Cross-validation was adopted to measure the ROC-AUC, F1-score, accuracy, precision, sensitivity, specificity, and overall classification performance of aiGeneR 3.0. We hypothesized that the aiGeneR 3.0 would be more effective than other baseline DL models for antibiotic resistance detection with an effective computational cost. We assess how well our model can be memorized and generalized.
Results: Our aiGeneR 3.0 can handle imbalances and small datasets, offering higher classification accuracy (93%) with a simple model architecture. The multi-drug resistance prediction ability of aiGeneR 3.0 has a prediction accuracy of 98%. aiGeneR 3.0 uses deep networks (LSTM) with next-generation sequencing (NGS) data, making it suitable for novel antibiotics and growing resistance identification in the future.
Conclusion: This work uniquely integrates SNP-level insights with DL, offering potential clinical utility in guiding antibiotic stewardship. It also enables a robust, generalized, and memorized model for future use in AMR analysis.
1 Introduction
One of the biggest concerns for global public healthcare is the issue of diseases brought on by bacteria that are resistant to antibiotics, often known as antimicrobial resistance (AMR). According to estimates from the World Health Organization (WHO), there were over 700,000 fatalities from drug-resistant illnesses in 2019, and that number might increase to 10 million deaths by 2050 (Sharma et al., 2024; Chandra et al., 2021). Identifying antibiotic resistance genes (ARGs) is important for discovering the patterns of AMR and plays a key role in personalized treatment and drug discovery.
Urinary tract infections (UTIs) are among the many infectious diseases that pose a serious threat to world health (Tan and Chlebicki, 2016). Escherichia coli (E. coli) bacteria are the main cause of UTIs, which affect millions of people each year (Vasudevan, 2014). If left untreated, many infections that affect the urinary system carry the potential to cause consequences like kidney damage. The problem is heightened by the advent of antibiotic resistance in E. Coli strains (Totsika et al., 2012), which restricts available treatments and calls into question accepted ideas of antimicrobial stewardship (Niranjan and Malini, 2014). E. coli is the primary cause of UTIs and provides a statistical analysis of other bacteria that can cause UTIs, as shown in a short research conducted in the northern region of India. E. coli (76.60%) was the most common gram-negative bacterium among the 47 positive isolates out of a total of 83 positive samples (Das et al., 2018). As shown in Figure 1, E. coli is the main cause of UTI in more than 53% of the cases, which is significant and needs to be addressed in the AMR pattern, antibiotic-resistant strains, and ARGs in E. coli for further effective drug development.
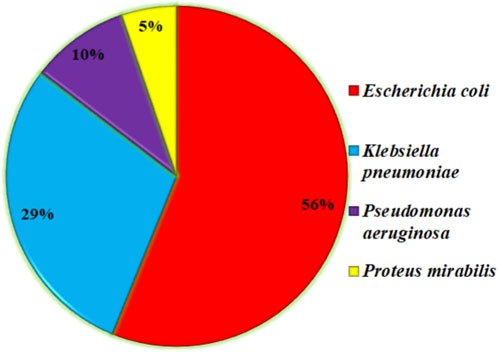
Figure 1. Most UTI cases are with selected bacteria.
The robust identification of antibiotic resistance determinants and their curation in specialized databases has been made possible by the growing availability and affordability of whole-genome sequencing data from clinical strains. Computational techniques can then search these resources for known causative genes, given the sequence from a new strain (McArthur et al., 2013; Zankari et al., 2012; Stoesser et al., 2013). By detecting mutations, examining entire genomes, and pinpointing particular resistance genes, the genetic study of E. coli shows antibiotic resistance patterns. To effectively tackle the global challenge of antibiotic resistance, this technique aids in understanding the genetic basis of resistance, tracking its spread, and predicting emerging patterns. This information guides focused treatments for antibiotic stewardship (Truswell et al., 2023; Wilson et al., 2016; Malekian et al., 2022). Antibiotic resistance, particularly in bacteria like E. coli, makes urinary tract infections (UTIs) a serious concern to human health. UTIs are among the most prevalent bacterial illnesses worldwide, with millions of cases occurring yearly. UTIs can cause serious side effects such as kidney infections, sepsis, and long-term harm to the urinary system if they are not addressed. The danger is exacerbated by introducing strains resistant to antibiotics, particularly in E. coli (Reza Asadi et al., 2019; Jafri et al., 2014; Bryce et al., 2016).
Novel approaches are needed to address the growing epidemic of antibiotic resistance in urinary tract infections (UTIs). Through genetic insights, DNA data advances several sectors, including disease diagnosis, customized medicine, AMR analysis, and microbial diversity study (Satam et al., 2023). Due to its capacity to handle high-dimensional data, identify intricate correlations, and integrate various data sources, deep learning (DL) performs very well when evaluating DNA sequencing data for the identification of antibiotic resistance (Lueftinger et al., 2021; Shi et al., 2019). DL is a revolutionary method that reduces the need for wasteful antibiotic treatment by providing precision medicine through the identification of distinct resistance profiles (Bryce et al., 2016; Taylor et al., 2018). Real-time decision assistance is empowered by DL, allowing for quick and knowledgeable antibiotic selection decisions. Additionally, it makes it easier to identify new resistance trends early on, which supports preventative measures (Ren et al., 2022a; Ren et al., 2022b). This work holds the potential to completely transform the way that UTIs are managed and the identification of resistance patterns in E. coli utilizing the next-generation WGS data, providing efficient solutions to the ever-changing problem of antibiotic resistance. We proposed our aiGeneR 3.0 model, which can identify the multidrug resistance genes in E. coli. In our work, we deal with a highly imbalanced and small dataset to assess the efficacy of our aiGeneR 3.0 model. We also compare the performance of aiGeneR 3.0 with well-accepted state-of-the-art ML and DL models. The generalization of our model boosts the adaptability and robustness. The simplified architecture and less computational time are the major advantages of our aiGeneR 3.0 model. We hypothesized that the aiGeneR 3.0 can reduce the cost and time for multi-drug resistance identification utilizing the WGS data. The dataset (NGS single-nucleotide polymorphism (SNP) WGS) utilized in this work is small and imbalanced; still, our aiGeneR 3.0 performs exceptionally well; the ROC value achieved during the deployment phase has already proven this. The detailed architecture of our study is shown in Figure 2.
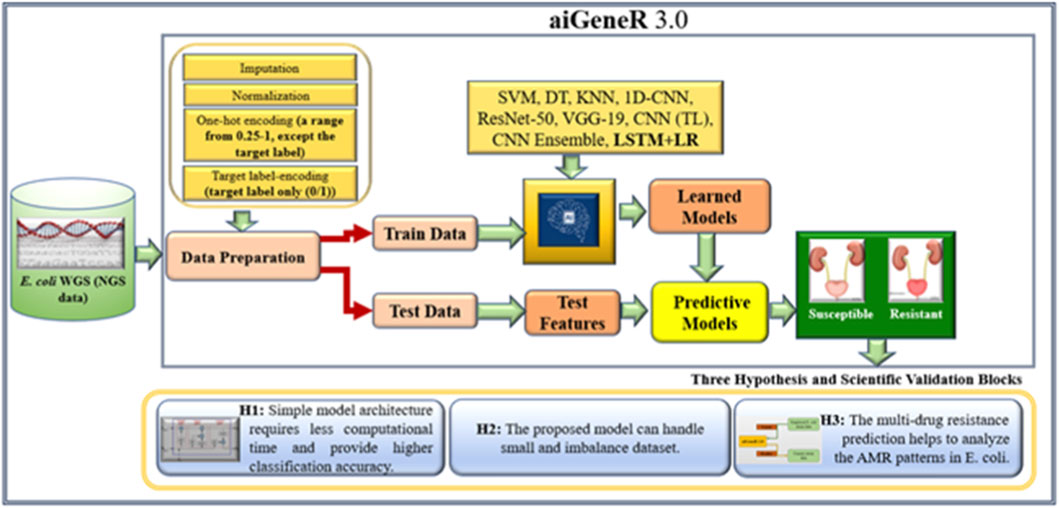
Figure 2. The overall architecture of our study.
The following describes the paper’s structure and major contributions. The relevant work for classifying and identifying E. coli antibiotic resistance is included in Section 2 to set up our research pipeline. We go over the aiGeneR 3.0 content and overall design in Section 3. The AI models and the experimental technique are presented in Section 4. Section 5 has the experimental results presentation. The validation and discussion of our aiGeneR 3.0 outcome are conducted in Section 6 and Section 7, respectively. We benchmarked our aiGeneR 3.0 in Section 8, and Section 9 held the conclusion.
2 Literature surveys
Researchers Moradigaravand et al. (2018) used gradient-boosted decision trees to achieve a 91% success rate in predicting antibiotic resistance in 1,681 Escherichia coli strains. Researchers found that using population structure and gene content greatly improved prediction accuracy. Based on these findings, machine learning (ML) shows promise as a clinical tool for identifying antibiotic resistance. Introduced by Arango-Argoty et al. (2018), the DeepARG-SS model outperformed conventional approaches with a recall of 91% and an accuracy of 97% over 30 antibiotic categories. Applying the DeepARG-LS model to the MEGARes database confirmed its great recall and accuracy. When used in conjunction with the DeepARG-DB database, these models allow for more precise gene identification by producing predictions of antibiotic resistance genes. The difficulties and limits of using ML to forecast antibiotic resistance were addressed by Boolchandani et al. (2019). In order to improve the accuracy of predictions, the study highlighted the necessity for extensive databases that connect resistance genes to test results. The significance of continuously improving computational methods to combat antibiotic resistance was highlighted by recognizing Resfams, Resfinder, and CARD as effective techniques for finding resistance genes. Among the multi-label classification models used by Ren et al. (2022a) to forecast E. coli multi-drug resistance, the ECC model proved to be the most accurate. In order to have a whole picture of resistance, the study stressed the significance of non-chromosomal genetic variables. Researchers Gunasekaran et al. (2021) used DL methods to classify DNA sequences, successfully determining the origins of viruses and DNA mutations with a high degree of accuracy. This research proved that DL could be useful for a variety of genetic analysis, drug discovery, and viral identification tasks. The accuracy of antimicrobial resistance predictions for underrepresented groups was greatly enhanced by the deep transfer learning model put forth by Ren et al. (2022b) while dealing with tiny, imbalanced datasets. Rapid diagnosis and focused therapies could both benefit from this strategy.
Over the last decade, various tools, quality control pipelines, and AI models have been gaining attention in AMR analysis. The AMR mechanism is too complex and requires trained manpower to access the laboratory test for the identification of resistance patterns, resistance strains, and multi-drug resistant percentages. In addition to this, the procedure of resistant strain identification is associated with massive cost and time. However, AI models have been found to perform well compared to traditional approaches for resistant strain identification. It was also found that existing AI studies for resistant strain identification lack a comparative analysis that includes ensemble and simplified model architectures. In addition to this, the computational cost associated with the resistant strain identification is still an open issue. This study aims to bridge this gap and provide evidence for the superiority of ensemble-based DL, transfer learning, and solo simplified architecture-based DL models regarding prediction accuracy and computational cost. Additionally, it is observed from the literature that researchers used transfer learning (TL) on a small dataset to identify the resistant strains. We aim to achieve a more effective outcome with less model complexity and computational time. Interventional studies involving animals or humans, as well as other studies that require ethical approval, must list the authority that provided approval and the corresponding ethical approval code.
In this section, where applicable, authors are required to disclose details of how generative artificial intelligence (GenAI) has been used in this paper (e.g., to generate text, data, or graphics, or to assist in study design, data collection, analysis, or interpretation). The use of GenAI for superficial text editing (e.g., grammar, spelling, punctuation, and formatting) does not need to be declared.
3 Materials and methods
The methods, resources, and materials used in this study to accomplish the study’s goals are described in this section. This section seeks to present a clear and thorough explanation of the experimental design, data collection, and data analysis methods.
3.1 Dataset
The E. coli WGS dataset utilized in this study is openly available and was collected from GitHub (2025), and Moradigaravand et al. (2018). Both these datasets have the susceptible and resistant information of the WGS of the E. coli K-12 strain. Due to the common association between these mutations and increased antibiotic resistance in both environmental and clinical contexts, the double-mutated E. coli genome dataset was chosen for its practical and clinical importance. The genetic variety of resistant strains is reflected in this dataset, which captures changes linked to resistance to several classes of antibiotics. This provides valuable insights into the complicated processes of resistance. Previous studies have mostly concentrated on single mutations or resistance genes specific to individual isolates; this work fills a significant void by shifting the focus to double mutations.
3.2 Dataset collection
We employed two datasets of E. coli in this study, which included WGS, SNP, and resistance-susceptible data for four antibiotics: gentamicin (GEN), cefotaxime (CTX), ciprofloxacin (CIP), and ceftazidime (CTZ), as shown in Table 1. The first dataset has 809 E. Coli strains, which are generated by Ren et al. (2022b). Clinical samples from both humans and animals were used to get the isolates. Using the VITEK® 2 system (bioMérieux, Nurtingen, Germany), antimicrobial susceptibility testing was carried out, and results were evaluated by EUCAST criteria. The proportion of isolates resistant to CTX, GEN, CTZ, and CIP is 23%, 44%, 34%, and 45% in that sequence.
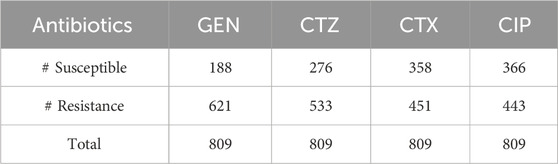
Table 1. Strain distribution to all the studied antibiotics.
It is observed from Figure 3 that the dataset utilized in our work has a high imbalance ratio of resistance-susceptible strains for GEN and CTZ antibiotics, with a slight improvement in the case of inconsistency for CTX and CIP antibiotics. There is a significant imbalance ratio in susceptible (S): resistant (R) of 1:3 and 1:2 in the case of GEN and CTZ antibiotics, respectively. While considering all four antibiotics, the ratio of S: R is 1:2 (1,188:2048).
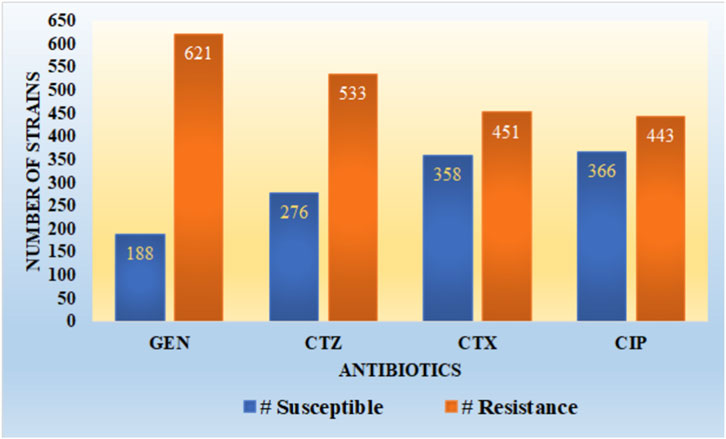
Figure 3. Susceptible and resistant strains for all four antibiotics.
3.3 Quality control
Data quality is the key to various AI model performances (Nayak et al., 2022; Nayak et al., 2023; Swain et al., 2023). Ren, Y. et al. developed the dataset (GitHub, 2025) to preprocess the raw WGS data; it uses BWA-MEM, and clean reads were mapped to the E. coli reference genome (E. coli K-12 strain, MG1655) after low-quality reads were filtered using fastp (v0.23.2) (Chen et al., 2018). By extracting reference and variant alleles and combining isolates according to reference allele positions, single-nucleotide polymorphisms (SNPs) were found using bcftools (v1.14) (Danecek et al., 2011; Li and Durbin, 2009). Preserving alleles that were found to be variations in more than half of the samples and creating an SNP matrix. One-hot encoding transformed the matrix into a binary format for further ML analysis.
3.4 Data preparation
This phase is the most crucial and contributes the most toward the model’s performance (Nayak et al., 2024; Mohanty et al., 2023). We utilized the dataset developed by GitHub (2025) for our study. Hence, we restructured the dataset to meet our study objective. The one-hot encoding in the original data ranges from 1-4, while in our study, we modified this to 0.25-1. In addition, we aim to study the effect of this one-hot encoding on computational costs.
3.5 Proposed model aiGeneR 3.0
To identify E. coli strains that have gained resistance, we created the state-of-the-art aiGeneR 3.0 model; this model is based on DL and ML. The approach we have created is multi-staged and uses modern techniques to boost accuracy and robustness. Beginning with processed Next-Generation Sequencing (NGS) WGS data (GitHub, 2025), which offers a solid foundation for comprehensive inquiry, we use it as our primary source. To prepare the dataset, we use the quality control (QC) pipeline that we developed before (Nayak et al., 2022). In the final phase, highly-trained deep neural networks (LSTM) and Linear regression (LR) are used to reliably identify susceptible and resistant bacteria and to forecast the likelihood of multi-drug resistance in any given strain that shows resistance to any of the four antibiotics under investigation. The design and execution of aiGeneR 3.0 are depicted in Figure 4. To describe the association between gene regulation variables and resistance extent, we used Linear Regression with least-squares optimization to reduce prediction errors and identify resistance-associated markers. Linear Regression provided a baseline prediction framework for deep learning model comparison, making the aiGeneR 3.0 pipeline robust in multidrug resistance categorization. Ultimately, a comprehensive evaluation of its efficacy using a predetermined set of assessment measures ensures the model’s reliability. Biological confirmation also adds credence to its real-world utility. The aiGeneR 3.0 model is an all-inclusive and potent tool that could change the game when finding E. coli antibiotic resistance.
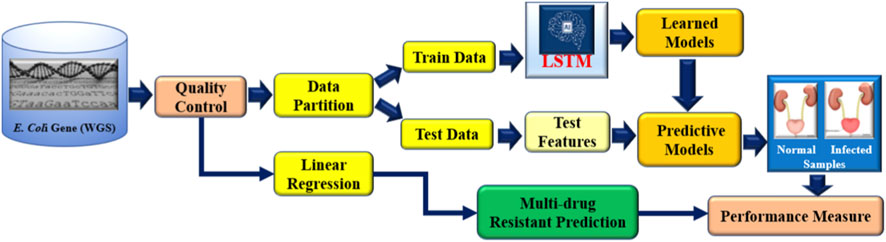
Figure 4. Overall architecture of our proposed aiGeneR 3.0 model.
3.6 Algorithm: the proposed model aiGeneR 3.0
Step 1 Read the data
Gather E. coli whole-genome sequencing data using NGS.
Use fastp for sequencing reads and quality assurance (Chen et al., 2018).
Align the filtered reads using BWA-mem.
Adopt Bfctools for calling variants (Danecek et al., 2011).
Sort and filter the aligned reads using Samtools (Li and Durbin, 2009)
Let ED be the processed dataset containing SNPs.
Step 2 Preparing the data
Align up (A) the data and eliminate duplicates.
To remove duplicates,
Step 3 Data Engineering
Use a one-hot encoding method (OHE) as
Decide on normalized values between 0.25 and 1 as follows: Equation,
Step 4 Split the train and test
Split the data into sets for training and testing.
Step 5 aiGeneR 3.0 application (train data)
Customize the model by
Utilizing the training set
Acquire the predictive model.
Find out what percentages of the various types are resistant to antibiotics with the Equation
Step 6 Multi-drug resistant prediction and identification of resistant strains (test data)
Determine which strains are resistant in the test data by following the Equation.
Estimate the resistance of strains to multiple drugs as per the following Equation,
Achieve outcomes with
Step 7 Assessment of the Model
Analyze aiGeneR 3.0’s effectiveness.
3.7 Architecture and parameter
The primary step in using aiGeneR 3.0 is careful data preprocessing. The genomic sequences of different strains of bacteria are encoded into numerical formats that are suitable for input into neural networks. Enabling further calculation usually involves converting categorical genetic data into a numerical format using methods like one-hot encoding (Dahouda and Joe, 2021). The proposed architecture consists of a total of eight layers, and four types of layers are utilized, out of which three are dense, two are dropout, and one each for regularization, flattening, and softmax make up the model’s architecture. The first, second, and third dense layers contain 64, 64, and 32 neurons, respectively. Comparably, our work employs many values for the dropout and regularization layers. The different regularization values experimented with in our work are 0.01, 0.001, and 0.0001, and the dropout rates are 0.25, 0.5, 0.7, and 0.9. Our proposed local architecture of the LSTM model with other added layers utilizing the random search is shown in Figure 5.
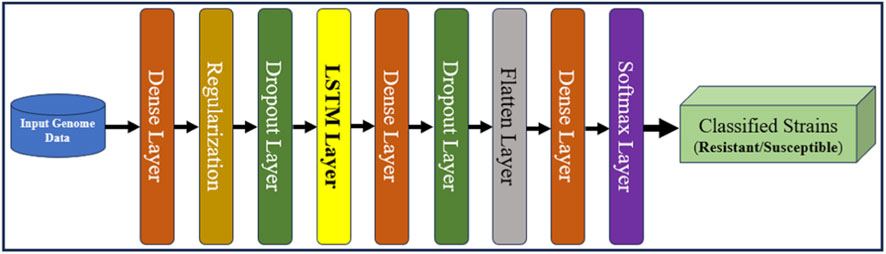
Figure 5. Local architecture of the LSTM deployed in our study.
The main objective of this study is to examine and compare the effectiveness of our proposed aiGeneR 3.0 model with different parameters to achieve the best classification accuracy for identifying the resistant strains utilizing the WGS E. coli NGS data. Thus, we adopt several changes during the implementation phase to the parameters of aiGeneR 3.0. We discuss some key phases of our experiments in the following sub-sections and the updation in several parameters of our implemented model, among which a few are shown in Table 2.
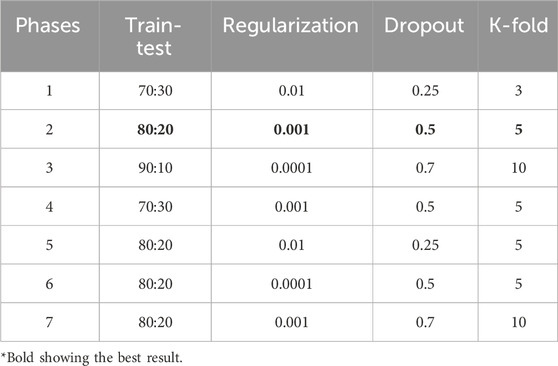
Table 2. All the phases of model implementations have different parameter configurations.
4 Experimental setup and implementation
The proposed aiGeneR 3.0 is a complete package of DL and ML models for identifying resistant strains and predicting multidrug resistance in strains. The architecture of aiGeneR 3.0 is simple and less complex than that of previously proposed DL models for resistant strain identification. In our experimental setup, we implemented several versions of aiGeneR 3.0 with different model parameters and finally proposed the architecture that consumes less computational time and produces the most significant results. In this section, we discuss a few of the several implementation versions of aiGeneR 3.0 with different hyperparameters.
4.1 Experiment I: high learning rate with smaller training data
During the initial development phase, we are refining the aiGeneR 3.0 model, which employs an LSTM architecture, for our analysis. A softmax layer was incorporated to facilitate classification tasks. A learning rate of 0.01 and a dropout rate of 0.25 were utilized to optimize the training process and mitigate overfitting. To conduct a comprehensive assessment of the model’s performance, we partitioned the dataset into two distinct subsets: the training set and the testing set. The ratio of the train-test split was 70:30. Furthermore, a K-fold cross-validation technique was employed with K = 3 to assess the model’s ability to generalize. Through iterative training and evaluation on various subsets of the dataset, we successfully enhanced the accuracy and dependability of the model in identifying resistant strains in the data.
4.2 Experiment II: moderate learning rate with increasing training data
In this implementation phase, we continued our research by iteratively improving the aiGeneR 3.0 model by changing several critical hyperparameters. We adjusted the learning rate to 0.001 to address overfitting and raised the dropout rate to 0.5. These changes should promote more regularization. To keep the assessment process consistent, we partitioned the dataset at an 80:20 train-test split ratio. We also used a K-fold cross-validation method with K = 5 to strengthen our model evaluation and thoroughly examine its generalizability capabilities, which improved the validation procedure. The model’s training dynamics were fine-tuned using these improvements so that it could better use features from the NGS data to identify resistant bacteria.
4.3 Experiment III: low learning rate with maximum training data
During this experiment phase, we kept tweaking the hyperparameters of the aiGeneR 3.0 model to make it even better. Now, we are trying to find the sweet spot by gradually adjusting the model weights during training with a learning rate of 0.0001. We raised the dropout rate to 0.7 to improve model regularization and alleviate overfitting worries; this should lead to more diverse and resilient learned representations. To keep things uniform throughout the assessment, we kept the train-test split ratio at 90:10. To further evaluate the model’s efficacy across different data subsets, we also used a more stringent K-fold cross-validation method with K = 10. This broadened the scope of our validation strategy.
We consistently obtained the best performance metrics with an 8:2 train-test ratio throughout all stages of our model, aiGeneR 3.0, as shown in Table 2. This ratio consistently produced the best outcomes, even with changing parameter combinations during the different phases. After training on 80% of the data and testing on the remaining 20%, our model showed exceptional accuracy, precision, sensitivity, and specificity. This strategy ensured that generalization and model complexity were balanced, enabling reliable performance on several splits of the datasets. Furthermore, at each step, regularization strategies, dropout rates, and K-fold cross-validation were methodically investigated to improve the performance of our models. Notably, the 8:2 train-test ratio was a stable base for attaining optimal outcomes across all of our implementations, even though changes to the parameters affected the model’s behavior.
In addition to the above experiments, we also implemented our aiGeneR 3.0 in several other phases, with the model parameters fine-tuned. We also take the different train-test splits to the above experiments and add various other possible dropout rates. However, we observe different model matrices with each of these implementation phases of our aiGeneR 3.0 and consider the best performance, which is described in sections 5 (results) and 6 (discussions).
5 Performance evaluation
This section presents a thorough performance evaluation of aiGeneR 3.0 and discusses the various evaluation processes adopted in our study. Our study employs a distinct blend of methodologies: power analysis, empirical analysis, and evaluation of model generalization. Empirical analysis assesses the practical value of the model in real-world situations, while power analysis evaluates its ability to detect meaningful effects. The analysis of model generalization focuses on its ability to acquire knowledge from training data and adjust to various unseen datasets. This comprehensive evaluation technique will unveil the intricate complexities of aiGeneR 3.0, providing insights into its effectiveness and robustness.
5.1 Power analysis
Power analysis is a statistical method employed to ascertain the minimal sample size necessary for a study to attain a specified degree of statistical power (Nayak et al., 2024; Jamthikar et al., 2020). Power analysis is essential in the realm of deep learning models as it allows for the estimation of the required sample size to effectively detect significant impacts or disparities in the model’s performance while maintaining a desired level of confidence.
We conducted a power analysis to determine the minimum sample size needed to calculate a population proportion with precision and accuracy. The experiments were conducted utilizing the methodology described in (Jamthikar et al., 2019; Skandha et al., 2020). The formula for sample size calculation, represented by the symbol Sn, is shown in Equation 1:
In this context, MoE represents the margin of error,
5.2 Empirical analysis
The confusion matrix, a real-to-anticipated-class matrix with multiple evaluation standards, is the primary target of performance parameters. TP and FP stand for true positives and false positives, respectively, in the confusion matrix. Similarly, TN and FN represent true negatives and false negatives, respectively. There are four types of predictions: TP, which accurately predicts that samples with resistance will be resistant; TN, which accurately predicts that samples without resistance will be susceptible; FP, which inaccurately predicts that susceptible samples will be resistant; and FN, which inaccurately predicts that resistant samples will be susceptible.
Measures such as accuracy (Acc), precision (Pre), specificity (Spe), sensitivity (Sen), F1-score (F1), Matthews Correlation Coefficient (MCC), and area under the curve (AUC) are some of the classification performance measures that were studied in this study. The total number of input samples divided by the number of valid predictions is the “processor, ranging from 1 to 4 (dataset A), while the second dataset consists of one-hot encoding. We call the recall the percentage of positive observations that were projected to be positive compared to the total number of positive observations. F1 is the weighted mean of precision and recall. All the model metrics are calculated based on the following equations (Equations 2–7).
6 Results
The Anaconda environment and Jupyter Notebook are used to carry out the model architecture design and parameter configuration. The learning models are implemented in Python (version 3.7) (Python, 2025). Here, we present the findings from the exploratory data analysis, together with a discussion of the results obtained using the suggested methodology. Our study optimized K-Nearest Neighbors (KNN), Decision Tree (DT), Support Vector Machine (SVM), VGG-19, 1-Dimensional Convolutional Neural Network (1-D CNN), and ResNet-50 to ensure resilient performance. Grid search found the best k for KNN, balancing classification accuracy and computing economy. DT used a Gini impurity-based criterion with a maximum depth to avoid overfitting. SVM utilized an RBF kernel with optimized hyperparameters C and γ by cross-validation. Transfer learning adjusted VGG-19 and ResNet-50 architectures were adapted to gene expression data, where both models were trained from scratch with customized input layers and trained using the Adam optimizer. Convolutional, pooling, and dense layers were added to the 1-D CNN architecture with sequential data pattern kernel sizes and activation functions. All models were hyperparameter-tuned and evaluated for optimal performance.
We used a grid search to optimize the learning rate, batch size, hidden layers, and neuron counts hyperparameters for aiGeneR 3.0; we then tested each combination using 5-fold cross-validation to make sure it generalized well and did not overfit. The input data was meticulously preprocessed to remove duplicates, remove samples with too many missing values, and impute missing entries using nearest-neighbor averaging. The magnitude-driven biases in deep learning models were mitigated by scaling numerical features to 0.25–1. To classify resistance, we used a 0.5 threshold to turn projected probabilities into binary calls, and we fine-tuned for unbalanced medicines using ROC to maximize F1-score and minority-class detection. Stable training, repeatable performance, and accurate resistance strain prediction were all achieved by means of this integrated technique.
The proposed aiGeneR 3.0 architecture was constructed using two different machines. The main machine, also known as machine-1, is a workstation running Ubuntu 20.04 that has an Intel Core i7 CPU, 32 GB of RAM, and 1 TB of solid-state drive storage, among other characteristics. The second machine, called Machine-2, is equipped with an Intel Core i5 processor, ranging from 1 to 4 (dataset A), while the second dataset consists of one-hot encoding, utilizing two different datasets. The first dataset consists of the one-hot encoding, ranging from 1 to 4 (dataset A), while the second dataset consists of one-hot encoding, ranging from 0.25 to 1 (dataset B). The comparison of the two systems’ computing time performance using the implemented model is presented in Table 3.
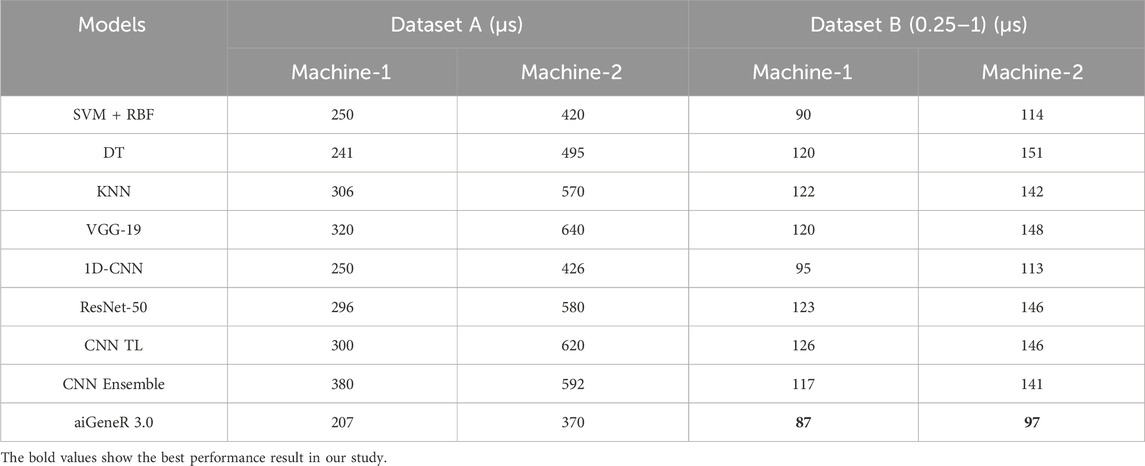
Table 3. Computational time taken by all the studied models.
From the above table, we observed notable variations in the computation times of different deployed models when they were assessed during both the training and testing stages, including aiGeneR 3.0. Notably, our suggested aiGeneR 3.0 model leads other studied models in terms of efficiency for the two datasets, consuming just 207 µs for machine-1 and 87 µs for machine-2. Due to its better hardware, machine-1 constantly shows faster computational times than machine-2; yet, aiGeneR 3.0 is the most effective model, with quick processing times that boost output and facilitate quick decision-making. On the other hand, other models like SVM + RBF, DT, KNN, VGG-19, 1D-CNN, ResNet-50, CNN TL, and Ensemble approaches have significantly longer training and testing times on both the datasets studied. All things considered, aiGeneR 3.0’s effectiveness highlights how quickly it can train and assess models, which shows its potential for quick learning capacity. We evaluate our aiGeneR 3.0 with a previously developed TL model. Ren et al. (2022b) and found that it consumes a remarkably less computational time of 31% and 45% in machine-1 for dataset A, and similarly takes 40% and 38% less in machine-2 for dataset B, as seen in Table 3. In addition to this, it can be seen from the table that the one-hot encoding approach adopted in our study (dataset B) shows a remarkably lower computational time compared to dataset A (Ren et al., 2022b) for producing the classification result for all the studied models.
This section quantifies and thoroughly examines the accuracy of the proposed framework, aiGeneR 3.0. Regarding its simple bending model architecture and predictive abilities, aiGeneR 3.0 performs admirably in various tasks, including prediction and classification. The pipeline of aiGeneR 3.0 is the adaptation of the LR and LSTM algorithms. Through an in-depth evaluation of its accuracy, we aim to gain insight into how well aiGeneR 3.0 works when it comes to resistance strain classification with limited computational capabilities and unbalanced data. This section discusses the outcome of our work in the following manner,
A. The ability of our aiGeneR 3.0 model to identify resistant strains by utilizing a single antibiotic.
B. The performance outcome of aiGeneR 3.0 on all four antibiotics taken together to identify the resistant strains.
C. Comparison of all the studied AI models
D. The aiGeneR 3.0 and multi-drug resistance prediction
A: We assess the performance of all our studied models on four different antibiotics. The antibiotics considered for our work are CTX, GEN, CTZ, and CIP. We observed better model metrics while we deployed our proposed model on the CIP dataset, as this dataset is quite balanced compared to other datasets. The detailed model metrics for the CIP dataset of implementations are shown in Table 4 below.
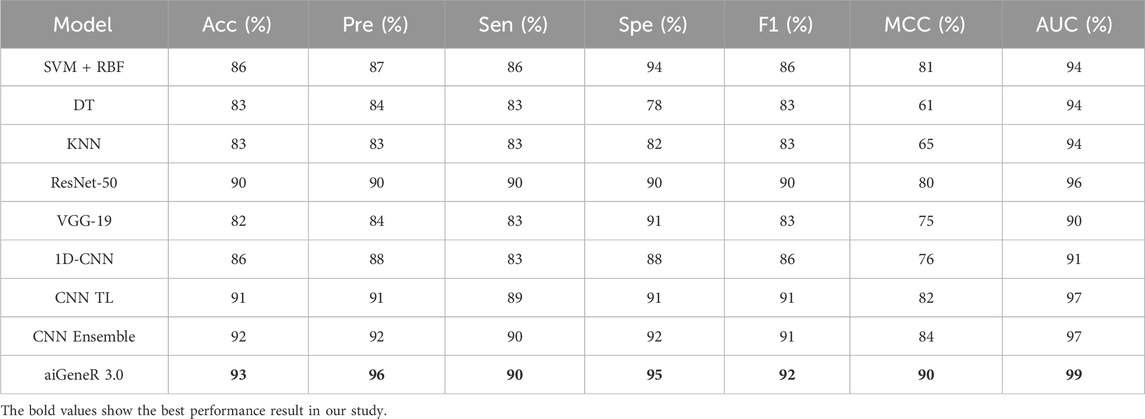
Table 4. Performance metrics of all the studied models on the CIP dataset.
The identification of resistant strains by our proposed aiGeneR 3.0, utilizing the CIP dataset, has an accuracy of 93%, which is higher than that of all the studied models. In addition to this, our proposed approach achieves higher sensitivity and specificity of 90% and 95%, respectively, as shown in Figure 6.
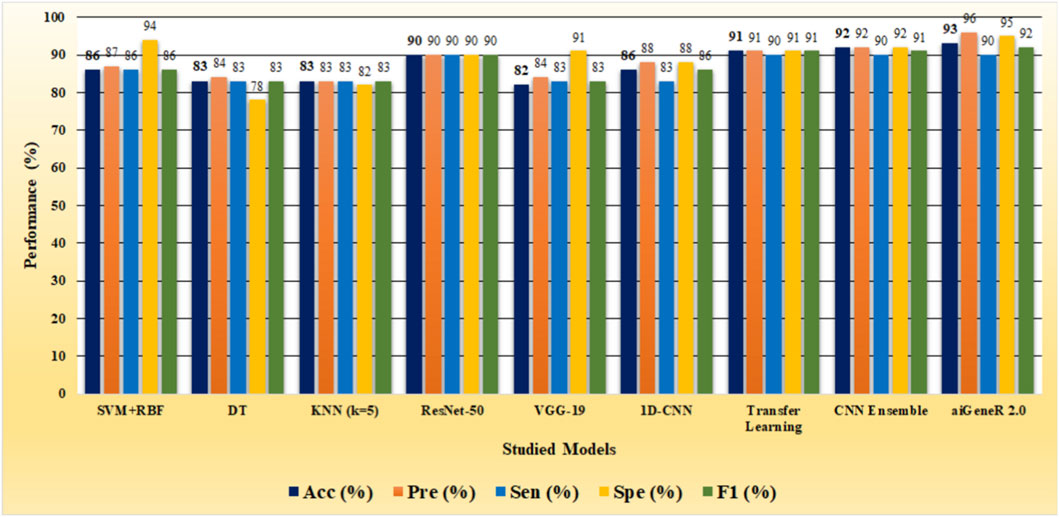
Figure 6. Performance metrics of all the deployed models on the CIP dataset.
We also evaluate our proposed model on the CTX, CTZ, and GEN antibiotics datasets. aiGeneR 3.0 achieves the highest classification accuracy of 82%, 88%, and 80% for CTX, CTZ, and GEN data, respectively. It is observed that the GEN dataset is highly imbalanced and contains a susceptible-to-resistant ratio of 4:1, and notably, our aiGeneR 3.0 reaches the highest classification accuracy of 80% among all the studied models. In addition to this, aiGeneR 3.0 sustains good specificity and sensitivity values for all three antibiotics, which shows its potential to classify the resistant strains with a very minimal false negative rate. The model metrics for CTX, GEN, and CTZ are summarized in Table 5 below. However, among all the studied models, CNN ensemble, CNN TL, and 1D CNN perform better compared to other models in terms of classification accuracy.
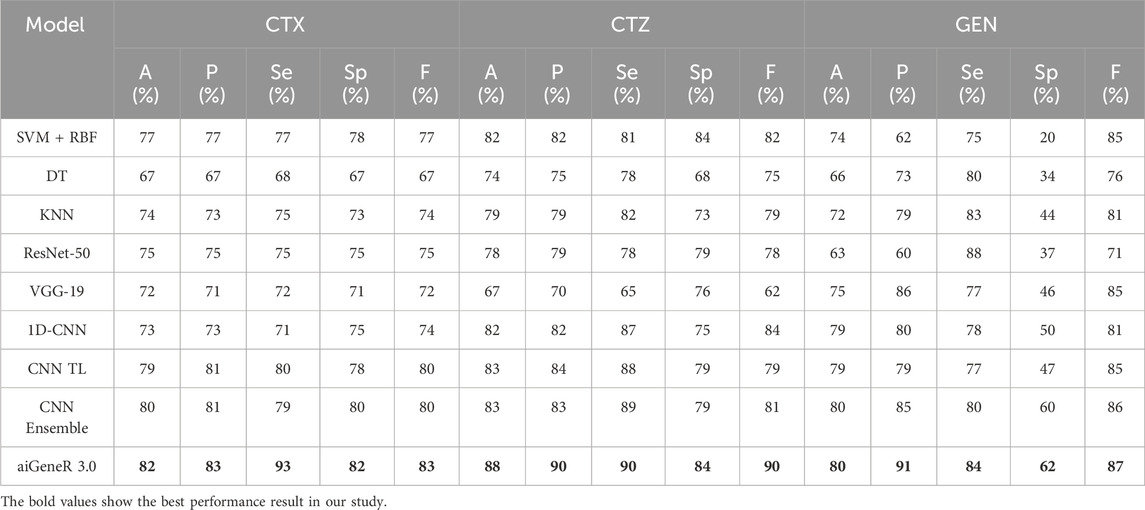
Table 5. Performance metrics of all the studied models on the CTX, CTZ, and GEN datasets.
The performance of aiGeneR 3.0, while we are utilizing the CTX, CTZ, and GEN antibiotics, excels in terms of classification accuracy, sensitivity, and specificity. In addition to this, we obtained a notable sensitivity and specificity while deploying our aiGeneR 3.0 on these three datasets. This result showcases the potential of aiGeneR 3.0 to identify the resistant strains in E. coli and can further be tested with other bacterial agents causing antibiotic resistance. The performance of the top-4 models on CTX, CTZ, and GEN datasets based on the accuracy (A), precision (P), sensitivity (Se), specificity (Sp), and F score (F) is visualized in Figure 7.
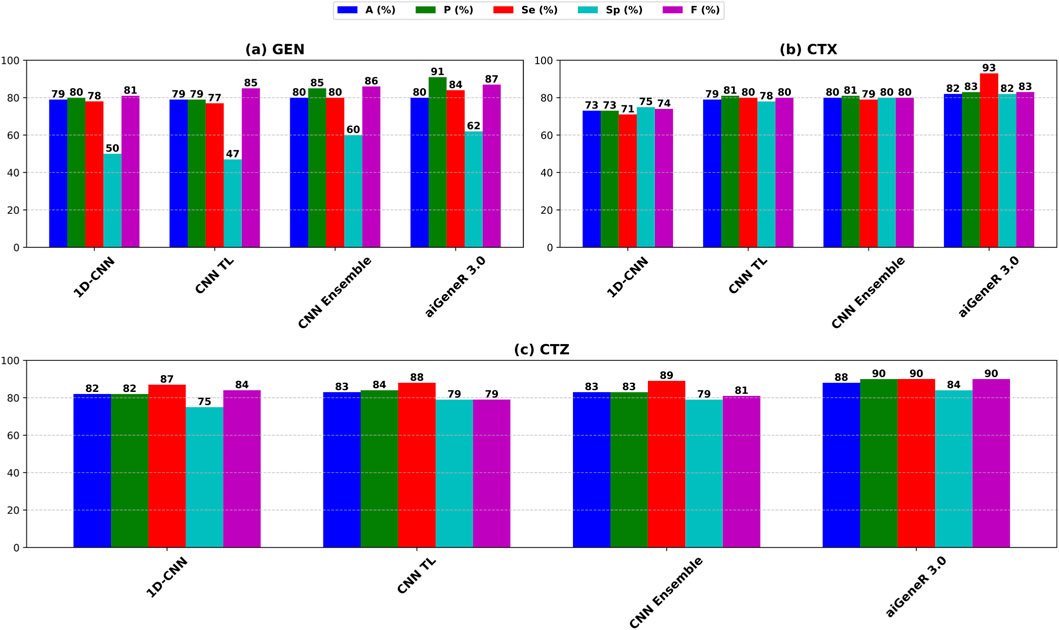
Figure 7. Performance metrics of the top-4 studied models on (a) CTX, (b) CTZ, and (c) GEN datasets.
B: We evaluate the efficacy of our proposed aiGeneR 3.0 to predict the resistance strains by taking all four antibiotics. This pipeline is designed by taking all the strains of the dataset along with all four antibiotics. We refined the dataset by keeping the original susceptible strains and updating the strain resistance to more than two antibiotics as multidrug resistance.
Based on our evaluations with a dataset that included all antibiotics, a learning rate of 0.01, a dropout rate of 0.5, and k-fold cross-validation with k = 5, we found that the aiGeneR 3.0 model performed significantly better than other models. The model metrics for all the studied models are shown in Table 6 below and can be visualized in Figure 8. With an impressive 92% accuracy, 92% precision, 91% sensitivity, and 95% specificity, the model accurately identified resistant strains while reducing false positives and negatives. Additionally, aiGeneR 3.0 demonstrated excellent discriminative ability in differentiating between susceptible and resistant strains with an impressive AUC value of 0.99. These results highlight the efficacy of our model architecture and training methodology, confirming that it is suitable for precise antibiotic resistance prediction and indicating that it may prove to be a helpful tool for improving therapeutic strategies in clinical settings. In addition to this, the classification accuracy of our proposed aiGeneR 3.0 model is 3% higher than that of the previously studied CNN TL model (Ren et al., 2022b). On the highly unbalanced all-antibiotic dataset, the highest Matthews Correlation Coefficient (MCC) obtained by aiGeneR 3.0 is 87%, while the lowest MCC acquired by DT is 28%. Because the CIP dataset is more balanced than the all-antibiotic datasets, the MCC on this dataset is excellent across all models that have been assessed.
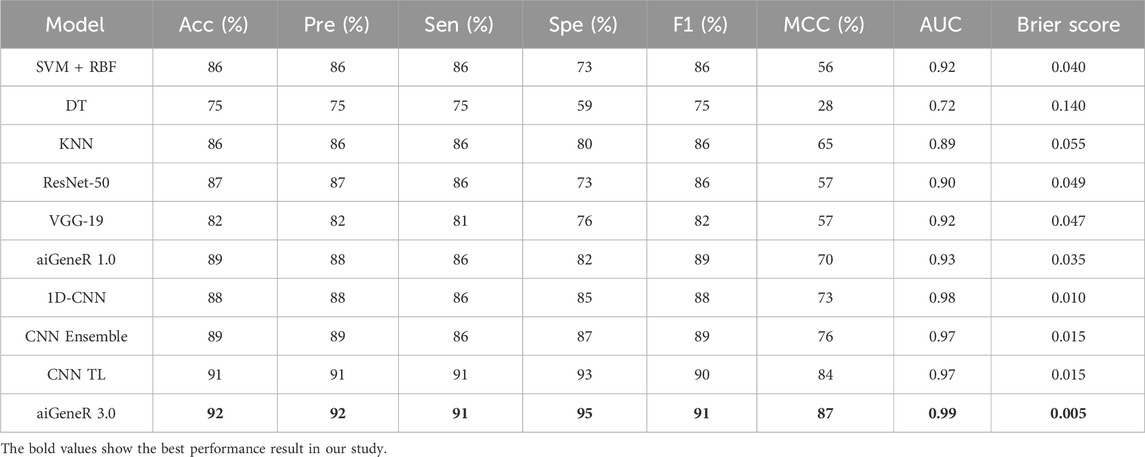
Table 6. Model metrics of all the studied models utilizing the dataset with cases having resistance/susceptibility to all four antibiotics.
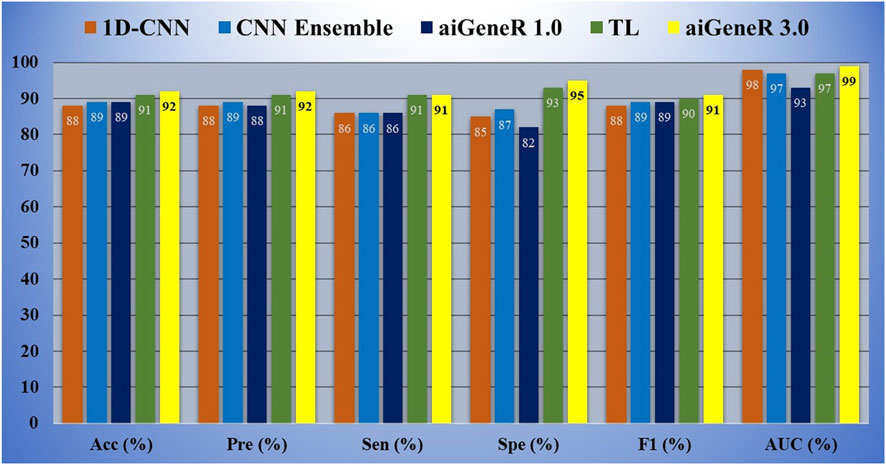
Figure 8. Performance of the top 5 models on all the antibiotics data.
C: Comparison of studied models. The studied NGS E. coli WGS includes 810 strains and 14,972 SNPs. Our study made use of all 14,972 SNPs with data standardization. With a ratio of 8:2, 648 samples were used for training, and 162 samples were used for testing. The complexity and processing demand of each strategy were evaluated as we explored different models for resistant strain identification using NGS E. coli WGS. DT has the potential to overfit as the depth increases, while SVM with RBF kernels is computationally demanding and produces higher classification accuracy compared to DT and KNN, as shown in Table 4. When it comes to prediction, KNN requires more memory and has more computational complexity (Kuang and Zhao, 2009)Thus, we observed a higher computational time for KNN in Table 3. The CNNs like ResNet-50 and VGG-19 deployed in our study have complex architecture and consume more memory and computational cost, as shown in Table 3. The level of complexity in aiGeneR 1.0 is moderate (Nayak et al., 2024). Despite its simplicity, the 1D-CNN still requires a lot of resources. CNN Ensemble adds complexity by combining different models (Zhang et al., 2020) and consumes the highest computational time compared to all the studied AI models, as shown in Table 3. Despite keeping complexity high, CNN TL shortens training time. The proposed aiGeneR 3.0 strikes the perfect balance between processing time and significant classification accuracy, especially due to its streamlined LSTM architecture.
The overall performance of all the models is assessed in terms of classification accuracy, precision, sensitivity, and specificity, as discussed in the performance evaluation section. In addition to this, we consider computational time to be one of the major performance parameters used to evaluate all the studied models. We observe that our proposed model, aiGeneR 3.0, achieves higher performance metrics compared to all other studied models. There is a slight increasing trend in the classification accuracy of aiGeneR 3.0 compared to the previously deployed CNN TL model. Ren et al. (2022b) with a remarkable AUC of 0.99. The most powerful aspect of our aiGeneR 3.0 model is the computational cost; it consumes very little computational power compared to all other studied models. The overall architecture and one-hot encoding techniques adopted by aiGeneR 3.0 make it robust and computationally cost-effective. The multi-drug resistant prediction is one of the significant contributions of aiGeneR 3.0 compared to previous works (Ren et al., 2022a), and it will be discussed in the next section. Additionally, the computational time taken by our aiGeneR 3.0 is much lower compared to all other studied models. The average learning time taken by aiGeneR 3.0 is 86µs less taken from all the studied models together and 83µs less compared to the previously studied TL model.
6.1 The aiGeneR 3.0 and multi-drug resistant prediction
Our proposed aiGeneR 3.0 model’s experimental results show potential in predicting multi-drug resistant (MDR) in E. coli strains. We considered the strain that resists more than two antibiotics to be in the multidrug-resistant category. We used the prediction model of logistic regression (Vermeiren et al., 2007) to estimate the percentage of bacteria resistant to four frequently given antibiotics: CIP, CTX, CTZ, and GEN. The percentage of resistance to each antibiotic is determined by counting the number of antibiotics that the strain is resistant to; the range is 0.25 for resistance to one antibiotic and 1 for resistance to all four antibiotics. The performance of our deployed resistant prediction model achieves a prediction accuracy of 98%, and the other model metrics for all the studied models are shown in Table 7.
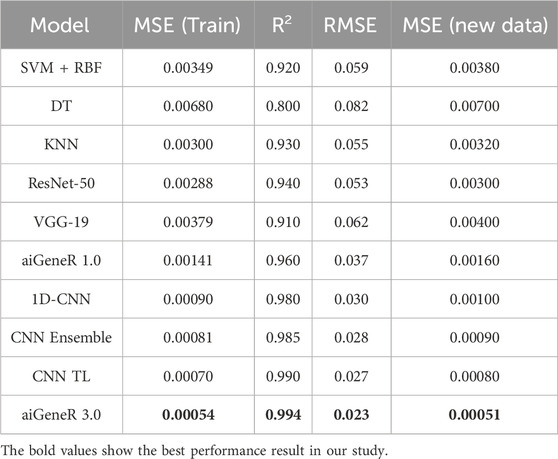
Table 7. Predictive model metrics for MDR (all studied models).
The experimental result for MDR prediction witnessed a 98% accuracy rate; our model demonstrated exceptional predictive performance and resilience in detecting variations in MDR. The model’s lowest mean squared error (MSE) during training (0.00054) was found during the model performance evaluation, demonstrating how closely the predicted resistance percentages matched the actual values. Moreover, the high R-squared value of 0.9940 indicates that our model may explain a considerable amount of variability in the resistance percentages across strains. The model’s accuracy for predicting levels of resistance is further demonstrated by the root mean squared error (RMSE) of 0.02327.
Our model’s active predictive capacity was tested using fresh data, and its MSE of 0.00051 confirmed its generalizability and dependability in practical settings. Together, these findings highlight the precision and effectiveness of our suggested aiGeneR 3.0 model in identifying multi-drug resistance in E. coli strains, providing crucial data for directing antibiotic treatment procedures and battling antibiotic resistance. As we obtained the best version of our proposed aiGeneR 3.0 with a moderate learning rate, with an increase in training data (80%), we intended to keep this for multi-drug identification. In addition to this, the model parameters like learning rate, CV, train-test split, and dropout rate of aiGeneR 3.0 are 0.001, 5, 8:2, and 0.5, respectively.
7 Validation
Accurate evaluation of model performance is a crucial component in building reliable and efficient prediction models. The ability to assess how well a model performs is an important indicator of its suitability for solving practical issues in many fields, including ML and scientific inquiry (Bellazzi and Zupan, 2008). In this section, we take a close look at our proposed models and evaluate them thoroughly, taking into account many criteria so that users can understand their strengths and weaknesses. We examine numerous critical aspects to evaluate the model’s performance in different contexts. Each section delves into a different facet of the model’s performance and thoroughly analyzes its efficacy.
7.1 Effect of Training size
The comparison of classification accuracy on test data and all conceivable train-test splits on the used dataset is shown in Table 8. According to the PE (section 4), the objective is to monitor the effect of data size on the model’s performance. As the proportion of training data increases, the accuracy of the aiGeneR 3.0 classification model rapidly increases. It is observed that the model achieves its highest accuracy of 92% when the train-to-test split ratio is 80:20, as shown in Table 8.
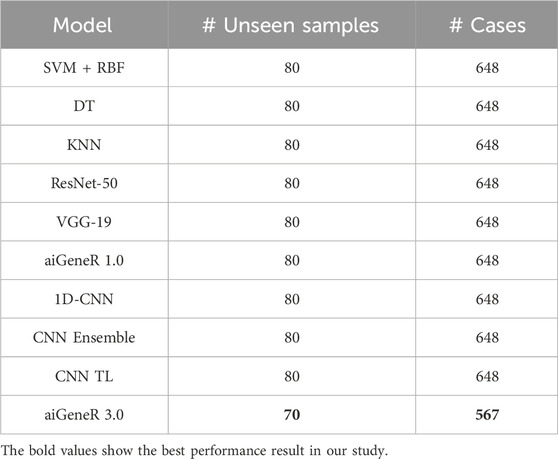
Table 8. Minimum unseen cases and samples are required for the generalization of individual models.
During our experiments, we observed that the studied AI models require more training data for generalization compared to our proposed aiGeneR 3.0 model. If we set the performance threshold as classification accuracy, then our aiGeneR 3.0 takes only 70% (567 unseen cases) of the data to achieve this trademark. Similarly, all of the implemented ML and DL models take 80% of the unseen data to obtain the generalization standard. The generalization of our aiGeneR 3.0 requires 10% less unseen data to obtain the best classification accuracy, proving that our model can be generalized by utilizing fewer strains than all other studied models.
7.2 Confusion matrix
The matrix shown in Figure 9 has significant diagonal dominance, indicating that the model predicted the proper class with few misclassifications. Most GEN-resistant strains (142 of 154) were correctly classified, with only a few CTZ and CTX misassignments. The model also predicted CIP, a smaller class, with great accuracy (148 out of 156 properly classified), demonstrating its class imbalance resilience. The confusion matrix yielded class-wise measurements. Each class has good precision, sensitivity (recall), and specificity, indicating that the model minimized false positives and negatives. Minority class CIP had good sensitivity and specificity, showing that the model did not underperform on underrepresented categories, a major antimicrobial resistance prediction difficulty.
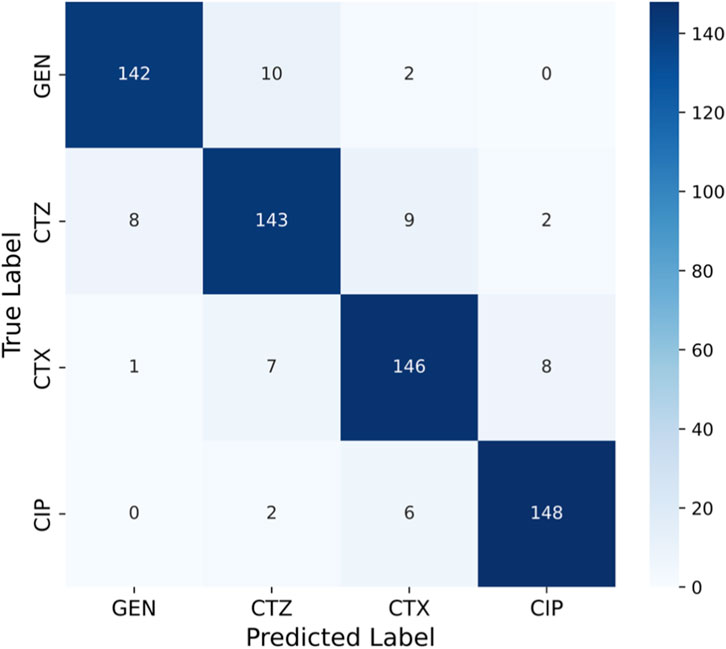
Figure 9. Confusion matrix of aiGeneR 3.0 on all four antibiotics.
The model for GEN has lower specificity (76%) than sensitivity (93%), suggesting reliable identification of susceptible strains but a little probability of under-detection of resistant isolates. CTZ had 91% sensitivity and 85% specificity, recognizing resistant bacteria with minimal false-positive rates. CTX and CIP had a stable finding, with sensitivity and specificity exceeding 92%, indicating robust class classification. CIP’s sensitivity (93%) and specificity (94%) were the best, detecting resistant bacteria and identifying vulnerable ones. These results show that aiGeneR 3.0 consistently supports better specificity while maintaining excellent sensitivity, ensuring reliable detection of minority resistant strains without inflating misleading resistance predictions. The confusion matrix confirms that aiGeneR 3.0 demonstrated balanced predictive performance among antibiotics, with ∼92% accuracy, 92% precision, 91% sensitivity, 95% specificity, 91% F1-score, and 87% MCC.
7.3 Receiver operating curves
The Receiver Operating Characteristic (ROC) curve is an essential metric for evaluating the effectiveness of a classification model. In this study, we conduct a performance analysis of our suggested aiGeneR 3.0 model in comparison to other studied models, with a significance level of p = 0.001. K-5 cross-validation is employed to determine the variation in the accuracy of each model as the quantity of training data changes.
The ROC performance of all the studied models is shown in Figure 10. The proposed model, aiGeneR 3.0, has achieved a significant milestone by achieving a strong Area Under the Curve (AUC) value of 98.48%. Nevertheless, when compared to other classification models, the ROC value of the 1-D CNN is the lowest (93.47%). Compared to the previously developed CNN TL model and despite the hurdles posed by the imbalance and small dataset, our proposed aiGeneR 3.0 achieves the highest AUC value in the identification of the resistant strains.
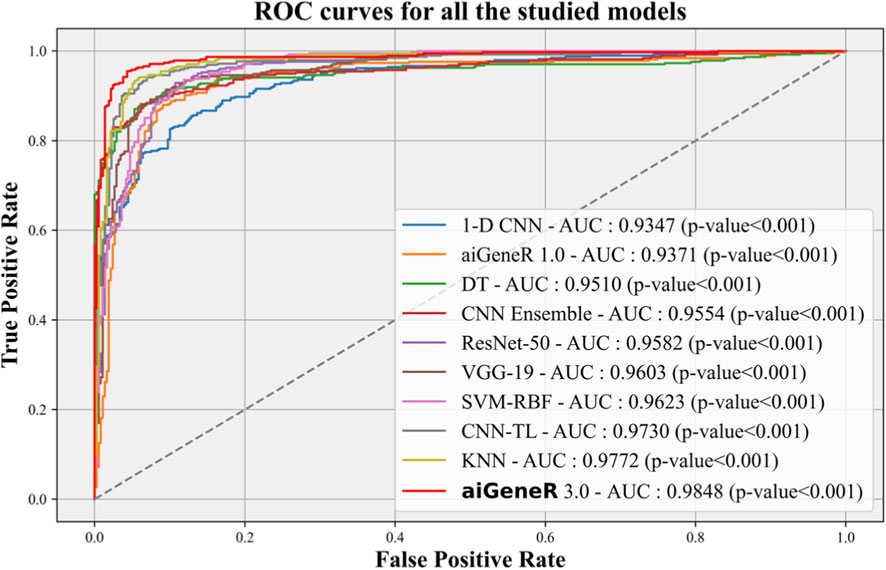
Figure 10. ROC-AUC of all the studied models with p-value <0.001.
7.4 Model generalization
In the validation phase of our aiGeneR 3.0 model, we utilize an openly available and highly imbalanced dataset (Moradigaravand et al., 2018). The detailed characteristics of the dataset are summarized in Table 9. There is a high imbalance in the susceptible-to-resistant ratio in all four antibiotics taken for validation of our aiGeneR 3.0 model. It can be seen from the table that the ratio is very high in the case of CTZ and GEN (≈1:7) there is a slight increase in the ratio for CTX and CIP (≈1:4). We perform the model validation in two different phases first, we have considered four different datasets based on four individual antibiotics and secondly, prepare the dataset by combining all the four antibiotics into one dataset.
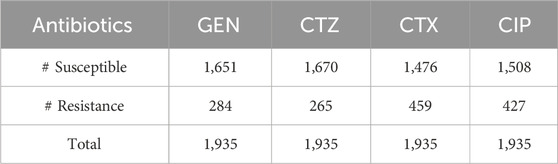
Table 9. Characteristics of the validation dataset.
We tested the efficacy of our proposed model on the four individual antibiotics considered for our experiments in the publicly available data, and aiGeneR 3.0 holds the consistency and remains the best performer in terms of classification accuracy, specificity, and sensitivity. In Table 10, we summarize the performance of aiGeneR 3.0 on individual datasets.
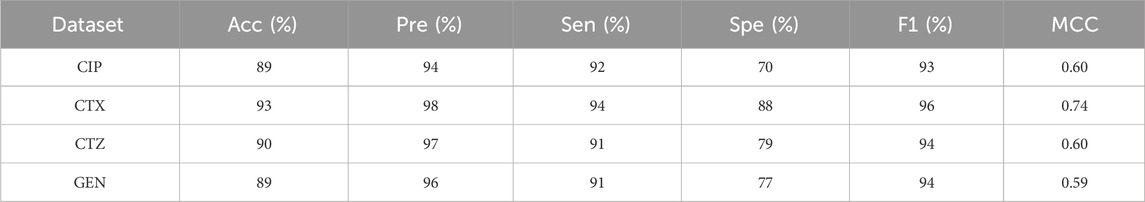
Table 10. Validation model metrics of aiGeneR 3.0.
It is observed from the above table that, during validation of aiGeneR 3.0 with individual antibiotics data, we obtained a higher classification accuracy in the case of CTX, and this is due to the higher strain ratio compared to the other three antibiotics datasets. aiGeneR 3.0 achieves the second-highest classification accuracy in the case of CTZ (90%), followed by CIP and GEN (89%).
Similarly, while we tested aiGeneR 3.0 along with other studied models on the dataset that combines all four antibiotics, we observed that aiGeneR 3.0 achieves the highest classification accuracy (90%), as shown in Table 11. The sensitivity and specificity of aiGeneR 3.0 are 97% and 76%, respectively, which is the highest among all the studied models, and this is due to the one-hot encoding we adopt in our study. In addition to this, the SVM, aiGeneR 1.0, CNN TL, CNN ensemble, and ResNet-50 achieve a remarkable sensitivity of more than 90%, whereas CNN TL and ResNet-50 achieve a specificity just higher than 70%. The studied model metrics of the validation phase are shown in Figure 11. However, in the validation phase, we observed that the ResNet-50, CNN ensembled, and TL performed better than aiGeneR 1.0. This is due to the effectiveness and automated feature extraction techniques with these models compared to our previously developed aiGeneR 1.0, which is based on traditional feature selection techniques. This validation outcome may provide insight into the use of automated and effective feature selection techniques, especially DL, for future resistant strain identification.
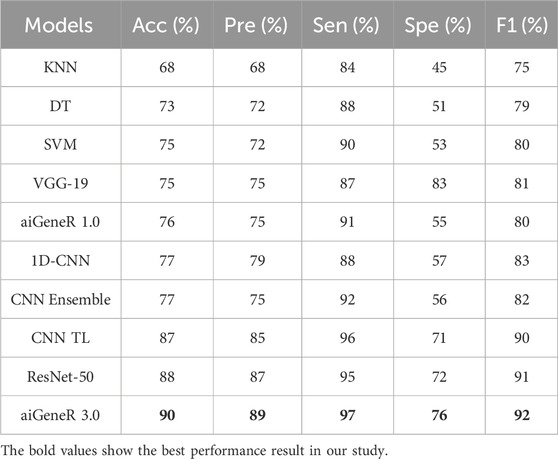
Table 11. Model metrics of all the studied models on the validation dataset (all four antibiotics).
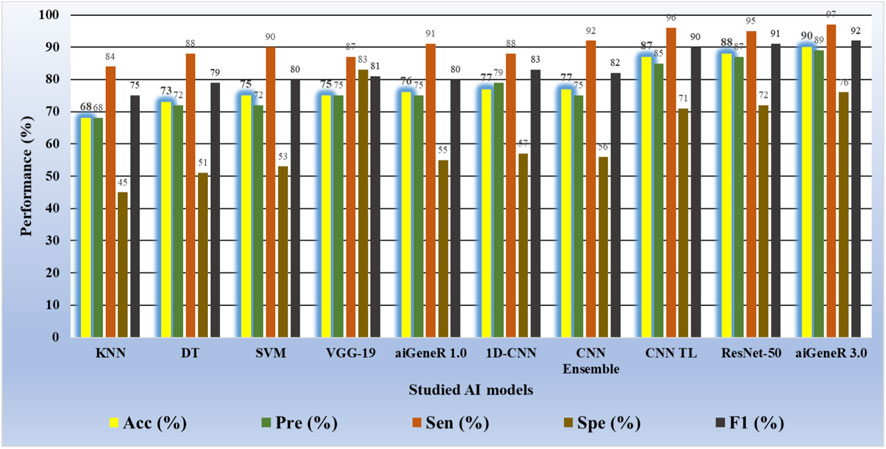
Figure 11. Model metrics of all the studied models during the model validation phase.
The performance of the designed model on every conceivable train-test split and the comparison of classification accuracy on test data were also explored in this study. The learning model is impacted by the amount of training data, which also helps the model generalize effectively to new data. Using a dataset with various train-test splits, we assess our suggested model, aiGeneR 3.0, and the four other classifiers employed in this investigation. It has been noted that while other models require a more significant number of cases for generalization, aiGeneR 3.0 requires a small number of cases. This section thoroughly explains how data size affects our suggested model. Figure 12 displays the comparison of classification accuracy on test data as well as all conceivable train-test splits on the utilized dataset.
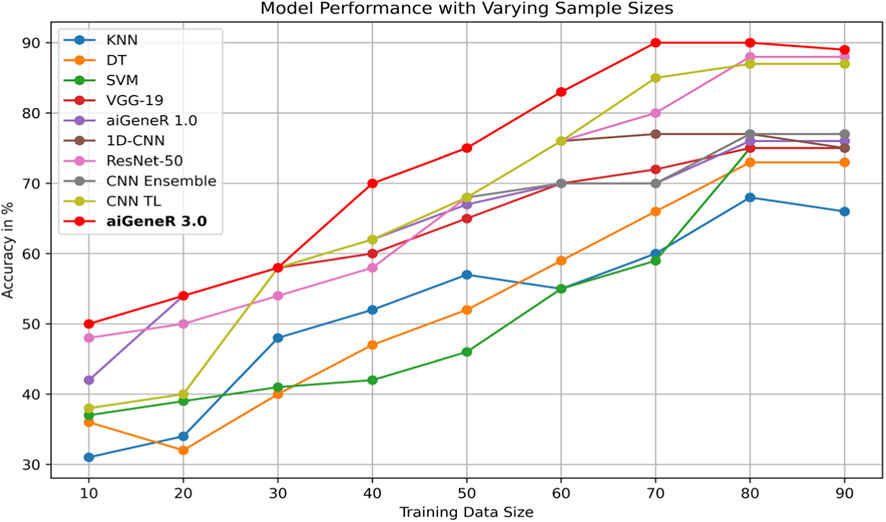
Figure 12. Performance of all the studied models with different train-test splits.
It can be observed from the figure that the ML models require more training samples to obtain generalization in classification accuracy than the DL models. While we compare the top three ML (SVM + DT + KNN) with the top three DL (CNN TL + aiGeneR 1.0 + aiGeneR 3.0) models, there is a significant difference in the train-test split for learning models to achieve their best results. Compared to the top three DL models, the top three ML models take 65.9% more data to be generalized. The other DL models studied in this work take a range of 55%–75% unseen cases to obtain their generalization. In addition to this, our proposed aiGeneR 3.0 requires 70% (567 cases) of data to generalize and obtain a stable classification accuracy. This performance outcome of aiGeneR 3.0 showcases the model’s generalization ability with a very small number of unseen data, which leads to its chances of better performance with real-time data.
8 Discussion
The results show that the aiGeneR 3.0 model effectively detects resistance strains without using any known resistance strains during model training. However, there are some limitations to be aware of due to variations in dataset sizes and methods. We implemented our suggested aiGeneR 3.0 model using a basic model architecture and then applied it to a publicly available, imbalanced, and noisy dataset. When given balanced antibiotic data, learning models perform much better in terms of accuracy, and we fine-tuned aiGeneR 3.0 to consistently classify each drug. In comparison to other conventional ML models used in our study, aiGeneR 3.0’s computational time is much lower.
We observed that, because typical one-hot encoding introduces a relative scale with numbers like 1, 2, 3, and 4, higher numerical values may inadvertently dominate or introduce bias during the learning process in certain deep learning models. Using bigger numerical representations may result in learning disparities in certain deep learning models, especially those that are sensitive to input magnitudes (models that ineffectively normalize weights), even if one-hot encoding is categorical and theoretically scale-invariant. By limiting the range to 0.25–1, biases resulting from magnitude are less likely to occur, and a more consistent expression is assured. We found that scaling to a smaller range improved training convergence and made the gradient updates of our models more reliable.
The deployed aiGeneR 3.0 model has a straightforward design that can deliver good classification accuracy. Among the most advanced ML and DL models we tested, aiGeneR 3.0 yielded the best classification accuracy. The following is a list of some of the major study findings we came across while doing this work: a simple and effective model architecture can achieve better classification accuracy, minimal computational cost, antimicrobial resistance (AMR) analysis, and antibiotic resistance strain identification.
• The proposed aiGeneR 3.0 has a simple deep network architecture and has the potential of a good learning model by providing relatively higher classification accuracy to identify the resistant strains.
• The aiGeneR 3.0 requires less computational time compared to all the studied models in this work.
• The multi-drug prediction ability with significant minute errors is a major contribution of our proposed aiGeneR 3.0 model.
• The aiGeneR 3.0 can effectively identify the resistant strains with a classification accuracy of 92% which is the highest among all the studied models.
• Model generalization of aiGeneR 3.0 persists in its classification potential and proves the ability of our proposed model to handle imbalanced and unseen data.
8.1 Claim
Our cutting-edge study reveals that aiGeneR 3.0 is an excellent resource for identifying strains of antibiotic resistance; it can handle imbalanced and constrained datasets with ease. Through the utilization of sophisticated DL algorithms, aiGeneR 3.0 achieves better classification accuracy, as shown in Table 12.
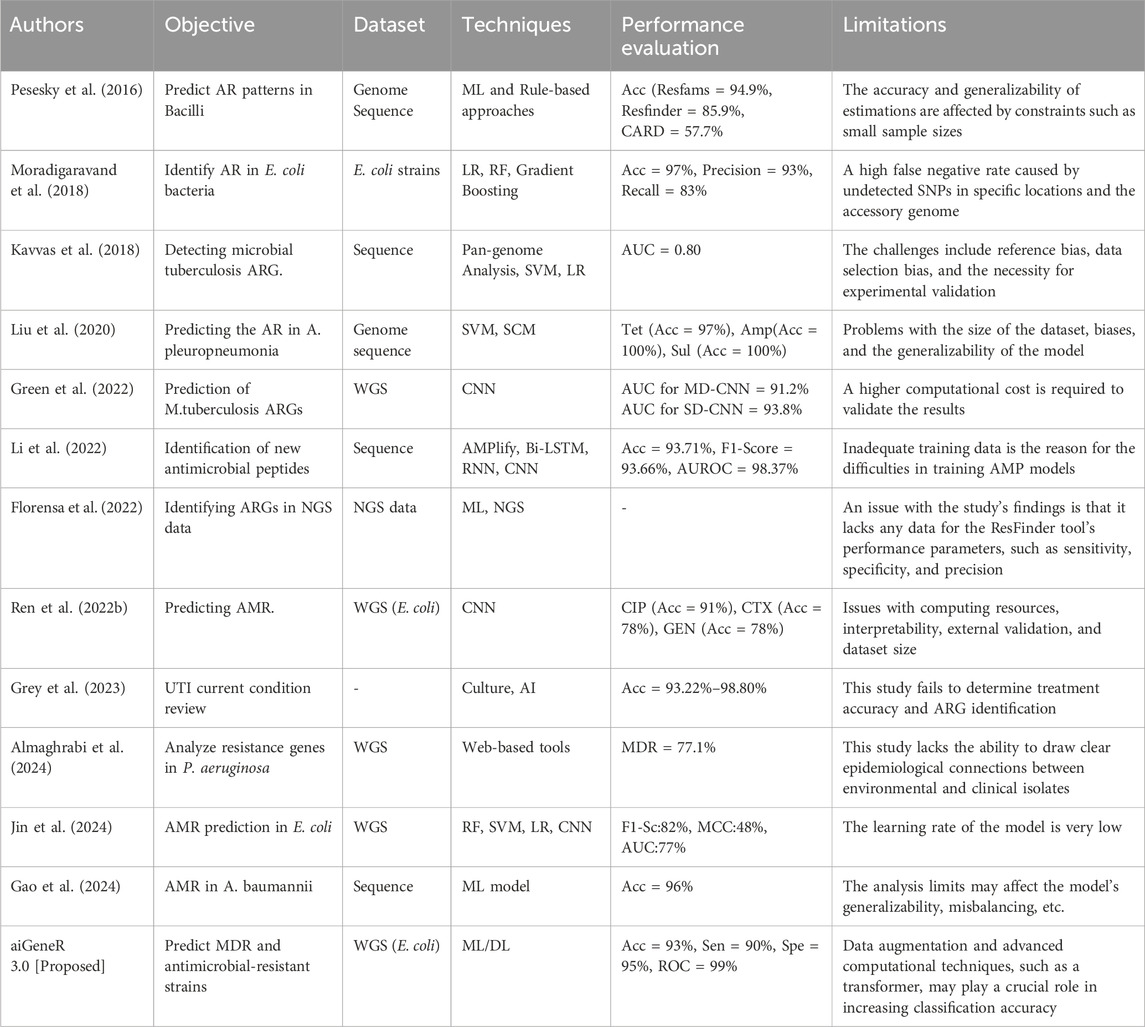
Table 12. Benchmarking parameters of the studied state-of-the-art ML and DL techniques for AMR analysis.
By minimizing variance and keeping feature scaling consistent, this method stabilizes the training process, which in turn produces smoother gradients and avoids problems like bursting gradients (dos Santos and Papa, 2022). We found that our studied DL models performed much better when we used a one-hot encoding range of 0.25-1 rather than 1–4. With a 2% increase in specificity and a 1% improvement in precision, our empirical data demonstrated better accuracy and generalizability on both the validation and test sets. In addition, our model was able to generalize well to a different dataset (Moradigaravand et al., 2018), which further proves how effective and resilient this proposed normalized feature range is for the learning of the deployed DL models.
The validation confirms the edge of aiGeneR 3.0, demonstrating its capacity to surpass rivals with small input data. Its processing cost is minimized, and its simple design gives it the ability to run on typical personal computers and laptops, ensuring better classification accuracy. Furthermore, a comprehensive power analysis reveals aiGeneR 3.0’s capacity to surpass the desired number of training cases, underscoring its potential for further refinement and expansion. Additionally, the significant AUC value of 98.48% shows the potential of our aiGeneR 3.0 toward its adaptability and learning capacity with imbalanced datasets. Overall, our research shows that aiGeneR 3.0 is an innovative breakthrough that will change how we diagnose diseases and, more generally, not just when identifying strains of antibiotic resistance.
8.2 Special notes
We designed the cutting-edge aiGeneR 3.0 model, a DL-based AMR analysis tool, to use double-mutated gene data to predict multi-drug resistance and detect antibiotic resistance strains without prior knowledge of known ARGs. The powerful DL model, LSTM, and LR combination in aiGeneR 3.0 advances AMR analysis, particularly multi-drug prediction. The primary notable accomplishments of our aiGeneR 3.0 framework are as follows:
• We proposed aiGeneR 3.0, an AI model with a simple and robust architecture that can handle imbalances and small genomics data.
• The aiGeneR 3.0 can predict the resistant strain from a double-mutated gene sequence with higher classification accuracy compared to previous studies.
• aiGeneR 3.0 offers an ultimate ability to predict the multi-drug resistance in strains with 98% prediction accuracy.
• The generalization and scientific validation of aiGeneR 3.0 prove its potential to handle small and imbalanced (curse of dimensionality) gene data.
• The benchmarking of aiGeneR 3.0 with other state-of-the-art- AI models enhance its adaptability for real-time implementation.
This proposed aiGeneR 3.0 model has the ability to identify E. coli bacteria that are resistant to antibiotics, which could be useful for antimicrobial stewardship initiatives. The approach has the potential to lower the usage of inefficient medicines and limit the use of broad-spectrum antibiotics by offering early insights about resistance profiles, which could support antibiotic selection that is tailored to individual patients. Subject to medical validation, the fast prediction capacity suggests real-time clinical decision support. Hospital monitoring systems can benefit from aiGeneR 3.0, which could have applications beyond individual patient care and bolster initiatives to combat new resistance tendencies. However, before these applications are widely used in ordinary practice, more multicenter clinical trials and prospective validation must be conducted.
8.3 Limitations
This highly motivated study focuses on identifying resistant strains utilizing imbalances and small datasets. Data augmentation can be used to further this study and potentially improve model performance. However, because it is medically incorrect, experts do not advise using this approach (the augmentation of medical data). Better model metrics might be obtained if the model were trained using synthetic data. Further study could address a few biases in our model, such as (i) smaller studies are found related to our work, (ii) SNP filtering threshold applied during preprocessing, which may have influenced the set of variants included for model training (iii) the use of data augmentation, (iv) comparisons with other ML and recently trending DL models like deep network with an attention mechanism. (v) a summary of the benchmarking studies and (vi) no remarks regarding the clinical validation (Paul et al., 2022; Eskofier et al., 2016; Hu et al., 2021).
In addition to the above, AiGeneR 3.0 has a few limitations despite its better predictive accuracy. First, despite balancing and preprocessing, the datasets had class imbalances that potentially bias predictions toward the majority class. The model may learn dataset-specific artifacts instead of generalizable biological patterns when training on short or noisy datasets, increasing the risk of overfitting.
8.4 Extension
This work focuses on applying DL and AI models to resistant strain identification and classification. The proposed aiGeneR 3.0 is now considered a benchmark in the field of AMR analysis due to its great improvement in detecting resistant strains. The aiGeneR 3.0 model performs highly compared to earlier research (Ren Y. et al., 2022) on resistant strain identification. Furthermore, cross-validation and unseen implementations show the system’s endurance, domain flexibility, and capacity to function well in domains other than the one on which it was trained. In extension, the application of other DL models, especially transformer architecture with attention mechanism, and Dl model hyper-parameter optimization may be adopted to validate their efficacy in identifying multi-drug resistance in double-mutated genome sequences.
Despite aiGeneR 3.0’s capabilities, small or imbalanced datasets risk overfitting, when the model learns training data-specific patterns or noise rather than generalizable correlations. Even high accuracy on the training set may not guarantee accurate predictions on unseen data. Imbalanced class distributions bias the model toward majority classes, making unusual antibiotic-resistant organisms harder to detect. Additional variability can worsen model performance in real-world deployment. Different laboratory techniques, sample preparation methods, sequencing platforms, and batch effects introduce systematic variances that may not be in the training data. Sequencing errors or missing data can skew input features, and variable sample distributions across populations may produce patterns the model has not learnt, raising misclassification risk. Parameters and techniques like Cross-validation, data augmentation, and regularization (dropout, weight decay, and early stopping) need to be tested in a wider range. Finally, ongoing retraining with new datasets adapts the model to changing data distributions, ensuring robustness and reliability in clinical or laboratory contexts. Future research should use explainable AI methods like feature attribution or pathway-level analysis to identify resistance-predicting genes or biological markers. For clinical implementation, the model needs to be verified across larger, multi-center cohorts to account for sequencing procedures, sample handling, and patient demographics. To maintain accuracy and dependability in clinical operations, rigorous benchmarking, seamless software interfaces, real-time processing, and constant retraining with new resistance data are needed.
9 Benchmarking
At its core, our study centered on identifying resistant strains using a DL model that combined advanced techniques with a simple design. Along with this, we also want to make sure that our pipeline does not lose consistency when applied to small or imbalanced datasets. Research shows that few studies have used DL models to identify resistance strains in double-mutated WGS. This set of AI models was constructed by merging several deep networks. Hence, it is essential to evaluate our method for previous AI models. In light of this, we choose to tackle the benchmarking efforts head-on by comparing our proposed models to earlier DL/ML models used in AMR for resistant strain classification and other disease investigations.
Our suggested aiGeneR 3.0 has a higher classification accuracy of 93% and can manage imbalanced data because of its streamlined model architecture. Furthermore, the computing time required for both the learning phase and the prediction of resistant strains in aiGeneR 3.0 is significantly lower compared to previously examined DL models. Furthermore, the validation of aiGeneR 3.0 establishes it as a strong and versatile model for classifying antibiotic-resistant strains and predicting multidrug resistance.
10 Conclusion
In this work, we used double-mutated E. coli NGS WGS data to show how effective aiGeneR 3.0 is in identifying bacteria that are resistant to antibiotics. Additionally, it presents the multi-drug-resistance patterns identified in the resistant strains. aiGeneR 3.0 is a hybrid computational method that employs advanced LSTM architecture and NGS data to discern resistant and susceptible strains within small and highly unbalanced datasets. Primarily, our aiGeneR 3.0 model enhances the accuracy of classification and prediction compared to earlier investigated models. The remarkable performance of our proposed pipeline is evidenced by aiGeneR 3.0, achieving a 92% accuracy, with a sensitivity of 91% and a specificity of 95% in finding resistant strains. aiGeneR 3.0 attains a 98% prediction accuracy in multi-drug identification, accompanied by a minimal MSE of 0.00054 and an RMSE of 0.02327.
Our study emphasizes the promise of predictive modelling utilizing NGS data and DL techniques to tackle the escalating problem of antibiotic resistance, perhaps leading to the creation of novel therapies. The ability of aiGeneR 3.0 to consistently and extensively generate models indicates its prospective usefulness in AMR research moving forward. Antibiotic resistance is emerging as a critical concern in the field of infectious diseases. Our research enhances our comprehension of the issue, enables us to predict its future trajectories, and eventually aids in addressing it. Due to the numerous constraints in identifying resistance patterns across different strains resulting from the limited number of strains, we want to employ deep learning models on whole genome sequencing with various augmentation techniques in our future research to find resistant strains.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
DN: Conceptualization, Data curation, Formal Analysis, Methodology, Project administration, Validation, Writing – original draft. AbP: Conceptualization, Data curation, Investigation, Software, Validation, Writing – original draft. AmP: Conceptualization, Data curation, Project administration, Resources, Supervision, Writing – original draft, Validation. MK: Methodology, Writing – review and editing, Formal Analysis, Supervision. BA: Data curation, Methodology, Validation, Writing – original draft, Writing – review and editing, Conceptualization, Investigation. SS: Data curation, Formal Analysis, Project administration, Writing – original draft, Writing – review and editing, Methodology, Validation. BS: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – original draft, Writing – review and editing, Project administration, Software. AA: Conceptualization, Data curation, Formal Analysis, Supervision, Writing – review and editing, Investigation, Validation, Writing – original draft. SM: Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review and editing. TS: Conceptualization, Data curation, Formal Analysis, Methodology, Supervision, Visualization, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R440), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Almaghrabi, R. S., Macori, G., Sheridan, F., McCarthy, S. C., Floss-Jones, A., Fanning, S., et al. (2024). Whole genome sequencing of resistance and virulence genes in multi-drug resistant Pseudomonas Aeruginosa. J. Infect. Public Health 17 (2), 299–307. doi:10.1016/j.jiph.2023.12.012
Arango-Argoty, G., Garner, E., Pruden, A., Heath, L. S., Vikesland, P., and Zhang, L. (2018). Deeparg: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 6, 23–15. doi:10.1186/s40168-018-0401-z
Bellazzi, R., and Zupan, B. (2008). Predictive data mining in clinical medicine: current issues and guidelines. Int. J. Med. Inf. 77 (2), 81–97. doi:10.1016/j.ijmedinf.2006.11.006
Boolchandani, M., D’Souza, A. W., and Dantas, G. (2019). Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 20 (6), 356–370. doi:10.1038/s41576-019-0108-4
Bryce, A., Hay, A. D., Lane, I. F., Thornton, H. V., Wootton, M., and Costelloe, C. (2016). Global prevalence of antibiotic resistance in paediatric urinary tract infections caused by Escherichia coli and association with routine use of antibiotics in primary care: systematic review and meta-analysis. BMJ 352, i939. doi:10.1136/bmj.i939
Chandra, P., Mk, U., Ke, V., Mukhopadhyay, C., Dinesh Acharya, U., Rajesh, V., et al. (2021). Antimicrobial resistance and the post antibiotic era: better late than never effort. Expert Opin. Drug Saf. 20 (11), 1375–1390. doi:10.1080/14740338.2021.1928633
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one fastq preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Dahouda, M. K., and Joe, I. (2021). A deep-learned embedding technique for categorical features encoding. IEEE Access 9, 114381–114391. doi:10.1109/access.2021.3104357
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and vcftools. Bioinformatics 27 (15), 2156–2158. doi:10.1093/bioinformatics/btr330
Das, B., Mittal, N., Goswami, R., Adhana, D., and Rathore, N. (2018). Prevalence of multidrug resistance (Mdr) and extended spectrum beta-lactamases (Esbls) among uropathogenic Escherichia coli isolates from female patients in a tertiary care hospital in North India. Int. J. Reproduction, Contracept. Obstetrics Gynecol. 7 (12), 5031–5037. doi:10.18203/2320-1770.ijrcog20184961
dos Santos, C. F. G., and Papa, J. P. (2022). Avoiding overfitting: a survey on regularization methods for convolutional neural networks. ACM Comput. Surv. (CSUR) 54 (10s), 1–25. doi:10.1145/3510413
Eskofier, B. M., Lee, S. I., Daneault, J.-F., Golabchi, F. N., Ferreira-Carvalho, G., Vergara-Diaz, G., et al. (2016). “Recent machine learning advancements in sensor-based mobility analysis: deep learning for Parkinson's disease assessment,” in Paper presented at the 2016 38th annual international conference of the IEEE Engineering in Medicine and Biology Society (EMBC), 655–658. doi:10.1109/EMBC.2016.7590787
Florensa, A. F., Sommer Kaas, R., Clausen, P. T. L. C., Aytan-Aktug, D., and Aarestrup, F. M. (2022). ResFinder–an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. genomics 8 (1), 000748. doi:10.1099/mgen.0.000748
Gao, Y., Li, H., Zhao, C., Li, S., and Wang, H. (2024). Machine learning and feature extraction for rapid antimicrobial resistance prediction of Acinetobacter Baumannii from whole-genome sequencing data. Front. Microbiol. 14, 1320312. doi:10.3389/fmicb.2023.1320312
GitHub (2025). Deep transfer learning enables robust prediction of antimicrobial resistance for novel antibiotics. Available online at: https://github.com/YunxiaoRen/deep_transfer_learning_AMR (Accessed December 12, 2025).
Green, A. G., Yoon, C.Ho, Chen, M. L., Ektefaie, Y., Fina, M., Freschi, L., et al. (2022). A convolutional neural network highlights mutations relevant to antimicrobial resistance in Mycobacterium tuberculosis. Nat. Commun. 13 (1), 3817. doi:10.1038/s41467-022-31236-0
Grey, B., Upton, M., and Joshi, L. T. (2023). Urinary tract infections: a review of the current diagnostics landscape. J. Med. Microbiol. 72 (11), 001780. doi:10.1099/jmm.0.001780
Gunasekaran, H., Ramalakshmi, K., Rex Macedo Arokiaraj, A., Deepa Kanmani, S., Venkatesan, C., and Dhas, C. S. G. (2021). Analysis of DNA sequence classification using CNN and hybrid models. Comput. Math. Methods Med. 2021, 1835056. doi:10.1155/2021/1835056
Hu, M., Shu, X., Yu, G., Wu, X., Välimäki, M., and Feng, H. (2021). A risk prediction model based on machine learning for cognitive impairment among Chinese community-dwelling elderly people with normal cognition: development and validation study. J. Med. Internet Res. 23 (2), e20298. doi:10.2196/20298
Jafri, S. A., Qasim, M., Masoud, M. S., Izhar, M., Kazmi, S., and Kazmi, S. (2014). Antibiotic resistance of E. coli isolates from urine samples of urinary tract infection (UTI) patients in Pakistan. Bioinformation 10 (7), 419–422. doi:10.6026/97320630010419
Jamthikar, A., Gupta, D., Khanna, N. N., Saba, L., Araki, T., Viskovic, K., et al. (2019). A low-cost machine learning-based cardiovascular/stroke risk assessment system: integration of conventional factors with image phenotypes. Cardiovasc. diagnosis Ther. 9 (5), 420–430. doi:10.21037/cdt.2019.09.03
Jamthikar, A., Gupta, D., Khanna, N. N., Saba, L., Laird, J. R., and Suri, J. S. (2020). Cardiovascular/stroke risk prevention: a new machine learning framework integrating carotid ultrasound image-based phenotypes and its harmonics with conventional risk factors. Indian Heart J. 72 (4), 258–264. doi:10.1016/j.ihj.2020.06.004
Jin, C., Jia, C., Hu, W., Xu, H., Shen, Y., and Yue, M. (2024). Predicting antimicrobial resistance in E. coli with discriminative position fused deep learning classifier. Comput. Struct. Biotechnol. J. 23, 559–565. doi:10.1016/j.csbj.2023.12.041
Kavvas, E. S., Catoiu, E., Mih, N., Yurkovich, J. T., Seif, Y., Dillon, N., et al. (2018). Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat. Commun. 9 (1), 4306. doi:10.1038/s41467-018-06634-y
Kuang, Q., and Zhao, L. (2009). “A practical Gpu based Knn algorithm,” in Paper presented at the Proceedings. The 2009 International Symposium on Computer Science and Computational Technology (ISCSCI 2009).
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, C., Sutherland, D., Austin Hammond, S., Yang, C., Taho, F., Bergman, L., et al. (2022). Amplify: attentive deep learning model for discovery of novel antimicrobial peptides effective against who priority pathogens. BMC Genomics 23 (1), 77. doi:10.1186/s12864-022-08310-4
Liu, Z., Deng, D., Sun, J., Ma, X., and Rong, T. (2020). Evaluation of machine learning models for predicting antimicrobial resistance of actinobacillus pleuropneumoniae from whole genome sequences. Front. Microbiol. 11, 474876. doi:10.3389/fmicb.2020.00048
Lueftinger, L., Majek, P., Beisken, S., Rattei, T., and Posch, A. E. (2021). Learning from limited data: towards best practice techniques for antimicrobial resistance prediction from whole genome sequencing data. Front. Cell. Infect. Microbiol. 11, 610348. doi:10.3389/fcimb.2021.610348
Malekian, N., Agrawal, A. A., Berendonk, T. U., Al-Fatlawi, A., and Schroeder, M. (2022). A genome-wide scan of wastewater E. coli for genes under positive selection: focusing on mechanisms of antibiotic resistance. Sci. Rep. 12 (1), 8037. doi:10.1038/s41598-022-11432-0
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57 (7), 3348–3357. doi:10.1128/AAC.00419-13
Medcalc (2025). Medcalc statistical software version. Available online at: https://www.medcalc.org/ (Accessed December 14, 2025).
Mohanty, S., Nayak, D. S. K., and Swarnkar, T. (2023). “A neural network framework for predicting adenocarcinoma cancer using high-throughput gene expression data,” in Paper presented at the AIP Conference Proceedings. doi:10.1063/5.0137033
Moradigaravand, D., Palm, M., Farewell, A., Mustonen, V., Warringer, J., and Parts, L. (2018). Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput. Biol. 14 (12), e1006258. doi:10.1371/journal.pcbi.1006258
Nayak, D. S. K., Das, J., and Swarnkar, T. (2022). “Quality control pipeline for next generation sequencing data analysis,” in Intelligent and cloud computing: proceedings of Icicc 2021 (Springer), 215–225.
Nayak, D. S. K., Mohapatra, S., Al-Dabass, D., and Swarnkar, T. (2023). “Deep learning approaches for high dimension cancer microarray data feature prediction: a review,” in Computational intelligence in cancer diagnosis, 13–41.
Nayak, D. S. K., Mahapatra, S., Routray, S. P., Sahoo, S., Sahoo, S. K., Fouda, M. M., et al. (2024). Aigener 1.0: an artificial intelligence technique for the revelation of informative and antibiotic resistant genes in Escherichia coli. Front. Bioscience-Landmark 29 (2), 82. doi:10.31083/j.fbl2902082
Niranjan, V., and Malini, A. (2014). Antimicrobial resistance pattern in Escherichia coli causing urinary tract infection among inpatients. Indian J. Med. Res. 139 (6), 945–948.
Paul, S., Maindarkar, M., Saxena, S., Saba, L., Turk, M., Kalra, M., et al. (2022). Bias investigation in artificial intelligence systems for early detection of parkinson’s disease: a narrative review. Diagnostics 12 (1), 166. doi:10.3390/diagnostics12010166
Pesesky, M. W., Tahir, H., Dantas, G., Patel, S., Andleeb, S., Burnham, C. A. D., et al. (2016). Evaluation of machine learning and rules-based approaches for predicting antimicrobial resistance profiles in gram-negative Bacilli from whole genome sequence data. Front. Microbiol. 7, 223089. doi:10.3389/fmicb.2016.01887
Python (2025). Python. Available online at: https://www.python.org/ (Accessed January 11, 2025).
Ren, Y., Chakraborty, T., Doijad, S., Falgenhauer, L., Falgenhauer, J., Goesmann, A., et al. (2022a). Multi-label classification for multi-drug resistance prediction of Escherichia coli. Comput. Struct. Biotechnol. J. 20, 1264–1270. doi:10.1016/j.csbj.2022.03.007
Ren, Y., Chakraborty, T., Doijad, S., Falgenhauer, L., Falgenhauer, J., Goesmann, A., et al. (2022b). Deep transfer learning enables robust prediction of antimicrobial resistance for novel antibiotics. Antibiotics 11 (11), 1611. doi:10.3390/antibiotics11111611
Reza Asadi, M., Habibi, M., and Bouzari, S. (2019). Urinary tract infection: pathogenicity, antibiotic resistance and development of effective vaccines against uropathogenic Escherichia coli. Mol. Immunol. 108, 56–67. doi:10.1016/j.molimm.2019.02.007
Satam, H., Joshi, K., Mangrolia, U., Waghoo, S., Zaidi, G., Rawool, S., et al. (2023). Next-generation sequencing technology: current trends and advancements. Biology 12 (7), 997. doi:10.3390/biology12070997
Sharma, S., Chauhan, A., Ranjan, A., Mathkor, D. M., Haque, S., Ramniwas, S., et al. (2024). Emerging challenges in antimicrobial resistance: implications for pathogenic microorganisms, novel antibiotics, and their impact on sustainability. Front. Microbiol. 15, 1403168. doi:10.3389/fmicb.2024.1403168
Shi, J., Yan, Y., Links, M. G., Li, L., Dillon, J.-A. R., Horsch, M., et al. (2019). Antimicrobial resistance genetic factor identification from whole-genome sequence data using deep feature selection. BMC Bioinforma. 20, 535–14. doi:10.1186/s12859-019-3054-4
Skandha, S. S., Gupta, S. K., Saba, L., Koppula, V. K., Johri, A. M., Khanna, N. N., et al. (2020). 3-D optimized classification and characterization artificial intelligence paradigm for cardiovascular/stroke risk stratification using carotid ultrasound-based delineated plaque: atheromatic™ 2.0. Comput. Biol. Med. 125, 103958. doi:10.1016/j.compbiomed.2020.103958
Stoesser, N., Batty, E. M., Eyre, D. W., Morgan, M., Wyllie, D. H., Elias, C. D. O., et al. (2013). Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella Pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 68 (10), 2234–2244. doi:10.1093/jac/dkt180
Swain, R. R., Nayak, D. S. K., and Swarnkar, T. (2023). “A comparative analysis of machine learning models for Colon cancer classification,” in Paper presented at the 2023 International Conference in Advances in Power, Signal, and Information Technology (APSIT), 1–5. doi:10.1109/apsit58554.2023.10201691
Tan, C. W., and Chlebicki, M. P. (2016). Urinary tract infections in adults. Singap. Med. J. 57 (9), 485–490. doi:10.11622/smedj.2016153
Taylor, R. A., Moore, C. L., Cheung, K.-H., and Brandt, C. (2018). Predicting urinary tract infections in the emergency department with machine learning. PloS one 13 (3), e0194085. doi:10.1371/journal.pone.0194085
Totsika, M., Moriel, D. G., Idris, A., Rogers, B. A., Wurpel, D. J., Phan, M.-D., et al. (2012). Uropathogenic Escherichia coli mediated urinary tract infection. Curr. drug targets 13 (11), 1386–1399. doi:10.2174/138945012803530206
Truswell, A., Lee, Z. Z., Stegger, M., Blinco, J., Abraham, R., Jordan, D., et al. (2023). Augmented surveillance of antimicrobial resistance with high-throughput robotics detects transnational flow of fluoroquinolone-resistant Escherichia coli strain into poultry. J. Antimicrob. Chemother. 78 (12), 2878–2885. doi:10.1093/jac/dkad323
Vasudevan, R. (2014). Urinary tract infection: an overview of the infection and the associated risk factors. J. Microbiol. Exp. 1 (2), 00008. doi:10.15406/jmen.2014.01.00008
Vermeiren, H., Van Craenenbroeck, E., Alen, P., Bacheler, L., Picchio, G., Lecocq, P., et al. (2007). Prediction of HIV-1 drug susceptibility phenotype from the viral genotype using linear regression modeling. J. virological methods 145 (1), 47–55. doi:10.1016/j.jviromet.2007.05.009
Wilson, B. A., Garud, N. R., Feder, A. F., Assaf, Z. J., and Pennings, P. S. (2016). The population genetics of drug resistance evolution in natural populations of viral, bacterial and eukaryotic pathogens. Mol. Ecol. 25 (1), 42–66. doi:10.1111/mec.13474
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67 (11), 2640–2644. doi:10.1093/jac/dks261
Keywords: deep learning, machine learning, next-generation sequencing, antimicrobial resistance, antibiotic resistance genes
Citation: Nayak DSK, Pati A, Panigrahi A, Khan M, Alabdullah B, Sahoo SK, Sahu B, Almjally A, Mallik S and Swarnkar T (2025) aiGeneR 3.0: an enhanced deep network model for resistant strain identification and multi-drug resistance prediction in Escherichia coli causing urinary tract infection using next-generation sequencing data. Front. Genet. 16:1651917. doi: 10.3389/fgene.2025.1651917
Received: 22 June 2025; Accepted: 07 October 2025;
Published: 23 October 2025.
Edited by:
Francesco Asnicar, University of Trento, ItalyReviewed by:
Vinothkumar Kolluru, Stevens Institute of Technology, United StatesVolkan Alparslan, Kocaeli University Faculty of Medicine, Türkiye
Copyright © 2025 Nayak, Pati, Panigrahi, Khan, Alabdullah, Sahoo, Sahu, Almjally, Mallik and Swarnkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saurav Mallik, c2F1cmF2bXRlY2gyQGdtYWlsLmNvbQ==, c21hbGxpa0Bhcml6b25hLmVkdQ==, c21hbGxpa0Boc3BoLmhhcnZhcmQuZWR1; Mudassir Khan, bXVkYXNzaXJraGFuMTJAZ21haWwuY29t, bWttaXlvYkBra3UuZWR1LnNh; Abhilash Pati, ZXIuYWJoaWxhc2gucGF0aUBnbWFpbC5jb20=