 Yuying Zhu1
Yuying Zhu1 Zhen Yang
Zhen Yang- 1Quzhou Maternal and Child Health Hospital, Quzhou, Zhejiang, China
- 2Clinic Lab, BGI Genomics, Shanghai, China
Background: Heterozygous pathogenic variants in HNRNPK cause Au-Kline syndrome (AUKS), a neurodevelopmental disorder characterized by congenital anomalies and developmental delay. Prenatal diagnosis of AUKS remains challenging due to nonspecific ultrasound findings, such as increased nuchal translucency (NT) and nuchal fold (NF), which overlap with other genetic conditions.
Methods: Whole-exome sequencing (WES) was performed on a fetus exhibiting increased NT and NF thickening, alongside parental samples. Identified variants were validated by Sanger sequencing, with structural and functional impacts predicted using bioinformatic tools.
Results: WES revealed a de novo heterozygous frameshift variant in HNRNPK (NM_031263.4: c.504_507del) in exon nine of 17, which has been deposited in the ClinVar database with accession number VCV003899365 and classified as pathogenic (P). This variant results in a truncated protein (p.Lys168AsnfsTer35): bioinformatic predictions indicate the resulting mRNA is likely subject to nonsense-mediated mRNA decay (NMD), and any escaping mRNA would produce a severely truncated protein lacking critical functional domains, rendering it nonfunctional. Sanger sequencing confirmed the variant was absent in both parental genomes. Ultrasound findings aligned with AUKS-associated nonspecific prenatal anomalies, and post-induction gross examination confirmed subtle AUKS-related craniofacial features, expanding the known prenatal phenotype of AUKS and providing phenotypic severity context.
Conclusion: Structural and functional analyses provide mechanistic insights into the variant’s pathogenicity, highlighting HNRNPK’s role in fetal development. These findings advocate for integrating genomic and phenotypic data to improve prenatal diagnosis of rare genetic syndromes.
Introduction
Prenatal detection of abnormal NT and NF thickening are critical soft markers prompting investigation for underlying genetic disorders (Gadsboll et al., 2025; Sun et al., 2023). While commonly associated with aneuploidies, these findings increasingly signal the need to explore monogenic syndromes, particularly when standard cytogenetic and microarray analyses are normal.
Neurodevelopmental disorders (NDDs) represent a heterogeneous group of conditions with complex genetic etiologies. Recent advances in genomic technologies have enabled the identification of numerous causative genes, yet diagnostic challenges persist due to phenotypic variability, overlapping clinical features, and the high prevalence of variants of uncertain significance (VUS). Among these, disorders linked to RNA-binding proteins (RBPs) have emerged as critical players in neurodevelopment, given their central roles in RNA processing, chromatin remodeling, and gene expression regulation (Yamamoto, 2021). Among these, disorders linked to RNA-binding proteins (RBPs) have emerged as critical players in neurodevelopment, given their central roles in RNA processing, chromatin remodeling, and gene expression regulation (Parra and Johnston, 2022). Heterogeneous nuclear ribonucleoprotein K (HNRNPK), a member of the RBP family, is essential for multiple cellular processes, including transcriptional regulation and post-translational modifications (Xu et al., 2019). Pathogenic variants in HNRNPK are associated with AUKS (OMIM #16580), a rare multisystem disorder characterized by developmental delay, congenital anomalies, and distinctive craniofacial features (Choufani et al., 2022).
Prenatal ultrasonography plays a pivotal role in early detection of fetal anomalies, with soft markers such as increased NT and nuchal fold thickening serving as critical red flags for underlying genetic disorders. While these markers are classically associated with aneuploidies, their presence increasingly prompts investigation for monogenic syndromes, particularly when combined with normal karyotyping and microarray results (Wang C. et al., 2022). The biological basis of abnormal nuchal fluid accumulation often relates to altered extracellular matrix composition, cardiovascular dysfunction, or lymphatic dysplasia-processes implicated in multiple neurodevelopmental disorders.
AUKS was first described in 2015 in individuals with heterozygous loss-of-function (LoF) variants in HNRNPK (Au et al., 2015). Since then, the phenotypic spectrum has expanded to include variable presentations, such as cardiac, genitourinary, and skeletal abnormalities, often overlapping with other syndromes like Kabuki syndrome (Au et al., 2018; Workalemahu et al., 2023; Zhang et al., 2024). Notably, AUKS phenotypic severity correlates with variant type: postnatal studies show frameshift/nonsense variants are associated with more severe neurodevelopmental delay and craniofacial anomalies, while missense variants often present with milder cognitive impairment and fewer structural defects (Gillentine et al., 2021; Miyake et al., 2017). Despite growing recognition of AUKS, prenatal diagnosis remains challenging due to nonspecific sonographic findings, such as increased NT or nuchal fold thickening, which are also associated with other genetic and structural anomalies. Notably, the contribution of these ultrasound markers to the prenatal suspicion of AUKS has not been systematically characterized, and their association with HNRNPK dysfunction remains unexplored. To date, most reported HNRNPK variants are de novo, including missense, frameshift, and intronic variants, complicating variant interpretation without robust functional validation (Gillentine et al., 2021; Miyake et al., 2017).
Here, we report the prenatal identification of a de novo HNRNPK variant in a fetus with increased NT and nuchal fold thickening, expanding the phenotypic spectrum of AUKS to include early sonographic markers. This case underscores the importance of integrating prenatal imaging with exome sequencing and functional assays to improve diagnostic accuracy for rare genetic syndromes, particularly for nonspecific soft markers where standard testing yields negative results. Our findings also contribute to the growing evidence of HNRNPK-related disorders and highlight the need for further research into epigenetic signatures to facilitate prenatal and postnatal diagnosis.
Materials and methods
Ethical approval and sample collection
The study protocol received ethical approval from the Institutional Review Board of Quzhou Maternal and Child Healthcare Hospital. Written informed consent was obtained from both parents prior to sample collection. Prenatal amniotic fluid samples (15–20 mL) were collected under ultrasound guidance at 18+3 weeks, while peripheral blood samples (5 mL) were drawn from each parent for comparative genomic analysis. Genomic DNA was extracted from all samples using a commercial DNA isolation kit according to the manufacturer’s protocol (QIAamp DNA Blood Mini Kit, Qiagen). DNA concentration was 50–100 ng/μL with A260/A280 ratio 1.8–2.0.
Clinical preliminary tests
To evaluate fetal genetic health, invasive prenatal testing was prioritized given the pregnant woman’s history of prior fetal demise (detailed in Case presentation). At 18+1 week of gestation, conventional G-banded karyotype analysis was performed on amniotic fluid samples collected under ultrasound guidance to exclude chromosomal aneuploidies and structural abnormalities. The analysis followed standard cytogenetic protocols: metaphase chromosomes were prepared from amniotic fluid cells, stained with Giemsa, and visualized under a microscope with a resolution of 400–550 bands (Xiao et al., 2016).
WES and data processing
WES was performed on the BGI-seq2000 platform following exome capture using the Agilent SureSelect Human All Exon V6 kit. The sequencing achieved an average depth of 263× with 98.75% of target regions covered at >20× depth. Raw sequencing data underwent stringent quality control including adapter trimming using Cutadapt (v2.10) (Martin, 2011) and quality filtering with FastQC (Brown et al., 2017). Clean reads were aligned to the GRCh37/hg19 human reference genome using BWA-MEM (v0.7.17) (Li H. and Durbin, 2009), followed by duplicate marking and local realignment using GATK (v4.1.9.0) (Van der Auwera et al., 2013). Whole-exome sequencing of the proband achieved 99.89% coverage of coding regions, encompassing 58,682,415 bp across 25,701 genes. The mean sequencing depth reached 263.37x, with 98.75% of target regions exceeding 20x coverage.
Variant calling and quality control
Variant calling was performed following GATK best practices (v4.1.9.0) (Van der Auwera et al., 2013), including HaplotypeCaller for initial variant detection and Variant Quality Score Recalibration (VQSR) for quality filtering (with training sets of HapMap 3.3 and 1000 Genomes phase 3 (Genomes Project et al., 2015). Single nucleotide variants (SNVs) and small insertions/deletions (indels) were jointly called across all samples. Variant annotation was performed using ANNOVAR (v20200608) (Wang K. et al., 2010), incorporating information from the following databases: gnomAD (v3.1.2) (Karczewski et al., 2020); 1000 Genomes Project (phase 3) (Genomes Project et al., 2015); dbSNP (v155) (Sherry et al., 2001). All databases were accessed on 2024–03–15. Variant quality was further assessed using quality metrics such as QUAL score (>30), QD (>2), FS (<60), and ReadPosRankSum (>−8.0).
Variant filtering and prioritization
A multi-step variant filtering strategy was implemented to identify potentially pathogenic variants. Common variants (minor allele frequency, MAF >0.1%) were excluded based on gnomAD (v3.1.2) (Karczewski et al., 2020) and 1000 Genomes Project (phase 3) (Genomes Project et al., 2015) population frequencies. Remaining variants were prioritized based on predicted functional impact, including: 1. Protein-truncating variants (nonsense, frameshift, canonical splice site variants, defined as ±1/2 bp of exon-intron boundaries); 2. Missense variants with CADD score >20 (Rentzsch et al., 2019); 3. Variants in genes associated with relevant phenotypes in OMIM (v2024-03) (Amberger et al., 2019) or ClinVar (v2024-03) (Landrum et al., 2018). Inheritance patterns were carefully evaluated using pedigree information, with particular attention to de novo and compound heterozygous variants.
Sanger sequencing validation
Candidate variants were validated using sanger sequencing. PCR primers were designed using Primer3 (v2.4.3) (Untergasser et al., 2012) to amplify 150–300 bp fragments encompassing each variant. Amplification was performed using Taq DNA polymerase under optimized conditions: 95 C for 5 min; 35 cycles of 94 C for 30 s, 58 C for 30 s, 72 C for 30 s; final extension at 72 C for 5 min. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) and sequenced bidirectionally on an ABI 3730xl Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA) with the BigDye Terminator v3.1 Cycle Sequencing Kit. Sequence traces were analyzed using CodonCode Aligner (v10.1.1) (CodonCode Corporation, Centerville, MA, USA) to confirm variant presence and zygosity.
Bioinformatic and functional analysis
Structural modeling was conducted using Phyre2 (v2.0) (Kelley et al., 2015) with the “intensive mode” to predict potential impacts of the variant on protein structure and function. Variant pathogenicity was classified according to the ACMG/AMP guidelines, incorporating evidence from population frequency, computational predictions, SIFT (Ng and Henikoff, 2003), functional domains, and phenotypic correlation.
Statistical analysis and variant interpretation
All statistical analyses were conducted using R (v4.0.3; Available from: https://www.R-project.org/), with ggplot2 (Maag, 2018) for visualization and vcfR (v1.12.0) (Knaus and Grunwald, 2017) for VCF file processing. Variant classification was based on five lines of evidence: 1) population frequency data; 2) computational prediction scores; 3) inheritance pattern; 4) functional domain overlap (Paysan-Lafosse et al., 2025); 5) phenotypic correlation with known diseases (OMIM v2024-03) (Amberger et al., 2019). Final variant interpretation was performed by a multidisciplinary team to comprehensively evaluate potential genetic contributors to the observed phenotype.
Protein structural analysis
Protein structural analysis of HNRNPK was conducted using the STRING database (Available from: https://cn.string-db.org/). This platform predicted three-dimensional interaction networks by integrating experimentally validated physical associations, pathway co-occurrence data, and computational binding models, identifying topologically critical binding partners and functional modules relevant to HNRNPK’s KH domain-mediated RNA recognition.
Result
Case presentation
A 30-year-old primigravida (gravida 1 para 0) with regular 30-day menstrual cycles (last menstrual period: 25 October 2024) and a history of one prior fetal demise has been confirmed pregnant following amenorrhea. During first-trimester screening, a significantly increased nuchal translucency (NT) measurement of 3.4 mm was detected (Figure 1A). Fetal biometry demonstrated appropriate growth parameters for gestational age: biparietal diameter 47 mm, head circumference 188 mm, abdominal circumference 172 mm, femur length 34 mm, and humerus length 32 mm (Figure 1B).
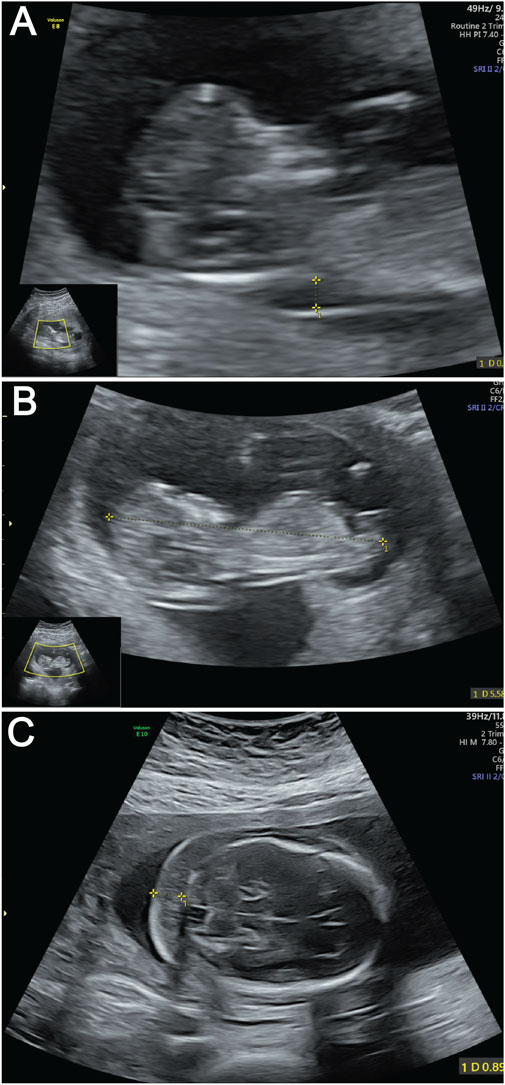
Figure 1. Ultrasound images of fetus demonstrating key anatomical views. (A) Ultrasound measurement of NT thickness. Yellow calipers mark the NT measurement points; (B) Fetal CRL measurement by ultrasound. The dotted line between yellow calipers indicates the CRL. (C) Ultrasound evaluation of NF thickness. Yellow calipers demarcate the NF measurement area.
At the current presentation, a detailed fetal anatomical survey performed at this presentation confirmed the presence of a thickened nuchal fold measuring 9 mm (Figure 1C). Additional findings included a hyperechoic intracardiac focus (EIF) in the left ventricle, a hyperechoic uterine band bridging the anterior and posterior uterine walls (noted incidentally), and a transverse cerebellar diameter and lateral ventricular width within normal limits. Comprehensive structural evaluation confirmed normal anatomy of the fetal skull, spine, lips, extremities, stomach, kidneys, bladder, and umbilical cord insertion site. Cardiac anatomy appeared balanced with a normal four-chamber view and comparable great vessel dimensions. Fetal cardiac activity was regular at 129 beats per minute, with normal fetal movements observed. The patient’s main concern was the prior fetal demise and new ultrasound findings, which prompted referral to our fetal medicine clinic for further evaluation.
The placenta was anteriorly located, with a thickness of 21 mm and Grade I maturity. Amniotic fluid volume was normal. The Amniotic Fluid Index (AFI) was calculated as follows: Right Upper Quadrant 61 mm, Left Upper Quadrant 36 mm, Right Lower Quadrant 57 mm, and Left Lower Quadrant 38 mm. Umbilical artery Doppler velocimetry showed indices within the normal range, including a Pulsatility Index of 1.29, a Resistance Index of 0.71, and a Systolic/Diastolic ratio of 3.47. Post-induction examination revealed subtle craniofacial features, such as a broad nasal bridge and micrognathia, which are consistent with postnatal AUKS phenotypes. These postnatal features are in line with the phenotypic descriptions of AUKS in previously reported cases in the literature (Au et al., 2018; Workalemahu et al., 2023; Zhang et al., 2024).
Identification of a de novo HNRNPK variant in the fetus
Given the history of prior fetal demise, serum quadruple screening was not performed; instead, invasive prenatal testing via amniotic fluid sampling was conducted at 18+1 week of gestation. Conventional G-banded karyotype analysis of the amniotic fluid sample revealed a normal 46, XY karyotype, ruling out common fetal chromosomal aneuploidies and major structural chromosomal abnormalities (Figure 2A).
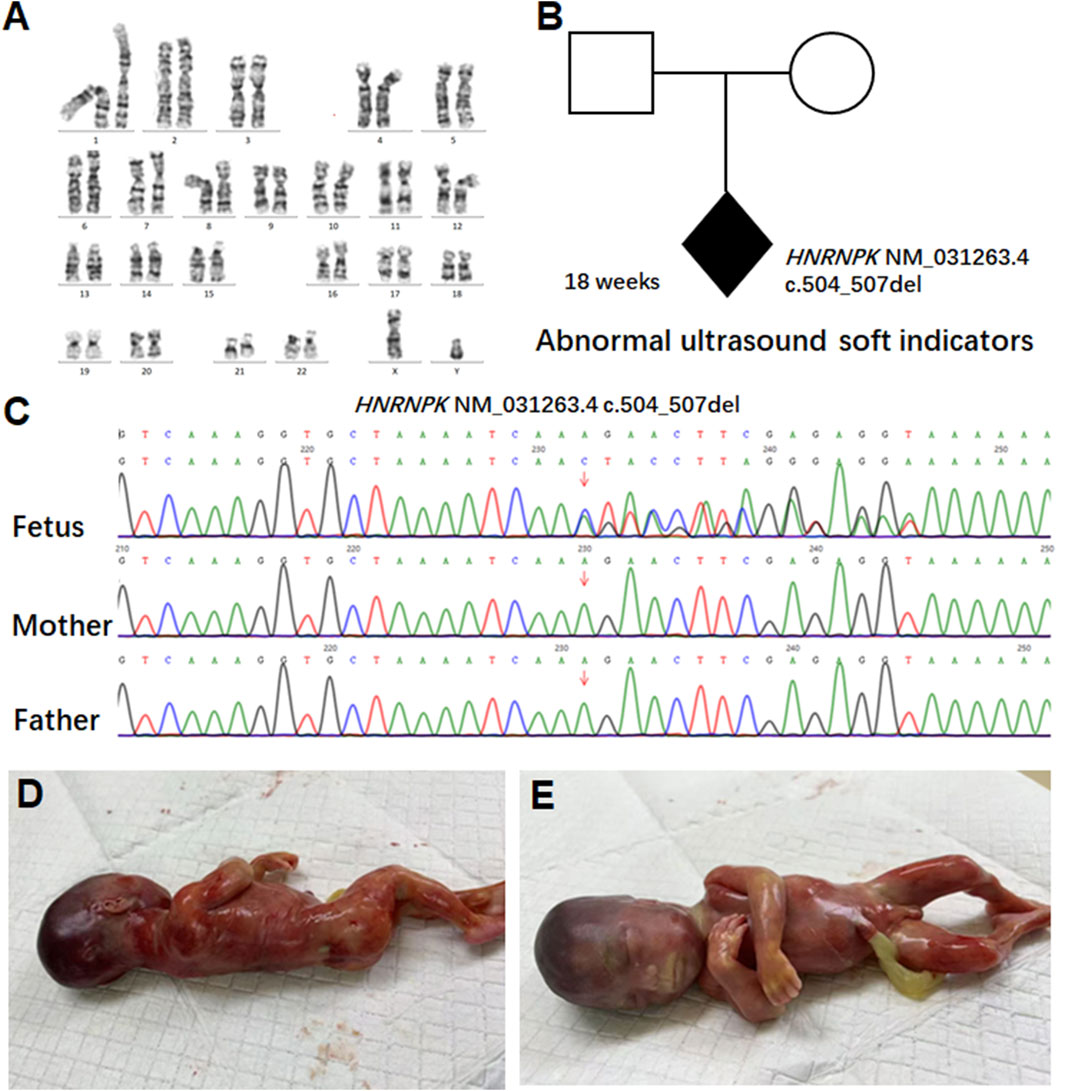
Figure 2. Pedigree analysis and genetic findings of the HNRNPK variant. (A) G-banded karyotype analysis of amniotic fluid cells from the 18-week fetus. Conventional G-banded karyotype analysis of amniotic fluid cells revealed a normal 46, XY karyotype. (B) Pedigree analysis of a family carrying an HNRNPK variant. The proband (shaded diamond, 18-week fetus) carries a heterozygous HNRNPK deletion (NM_031263.4:c.504_507del) in exon nine of 17. Parental symbols show no variant carriage. (C) Sanger sequencing validation of the HNRNPK variant. Chromatograms compare the proband (fetus) with parents at the variant site (arrow). The heterozygous 4-bp deletion appears de novo in the fetus, as both parents show wild-type sequences at this locus. (D–E) show the appearance of the 18-week-old fetus.
To explore the genetic etiology underlying the fetus’s abnormal ultrasound soft markers, we performed WES on the fetal amniotic fluid sample and peripheral blood samples of both parents. WES results identified a de novo heterozygous frameshift variant in the HNRNPK gene (NM_031263.4: c.504_507del). This variant has been deposited in the ClinVar database (accession number: VCV003899365) and is annotated as pathogenic (P) therein, though no prior clinical case reports or literature citations are associated with it in the database. This variant is localized to exon nine of the 17 exons in HNRNPK-a region with critical functional significance, as exon nine encodes the SMART KH domain (Figure 2B).
To explore the genetic etiology underlying the fetus’s abnormal ultrasound soft markers, we performed WES on the fetal amniotic fluid sample and peripheral blood samples of both parents. WES results identified a de novo heterozygous frameshift variant in the HNRNPK gene (NM_031263.4: c.504_507del). This variant has been deposited in the ClinVar database (accession number: VCV003899365) and is annotated as pathogenic (P) therein, though no prior clinical case reports or literature citations are associated with it in the database.
At the molecular level, the c.504_507del variant induces a frameshift by deleting 4 consecutive base pairs in the coding sequence of exon 9. This deletion disrupts the normal reading frame of the HNRNPK gene, starting from the lysine residue at amino acid position 168 (Lys168) of the wild-type protein, the frameshift introduces a series of non-native, abnormal amino acids into the polypeptide chain. Subsequent translation is prematurely terminated at amino acid position 202, where a premature stop codon (PTC) is generated.
Sanger sequencing was performed to validate the variant, and chromatograms confirmed that the c.504_507del variant was present exclusively in the fetal sample (Figure 2C) and absent in both parental genomes-confirming its de novo origin. Given the pathogenicity of this HNRNPK variant consistent with AUKS etiology, and the persistent abnormal ultrasound soft markers, the pregnancy was terminated at 21 weeks of gestation. Post-induction gross examination of the fetus (Figures 2D,E) revealed phenotypic features consistent with AUKS, including a broad nasal bridge and micrognathia, which further supported the pathogenicity of the identified de novo variant. Notably, this fetus lacked major structural anomalies typical of severe postnatal AUKS, representing a mild prenatal phenotypic presentation despite the severe nonsense variant, consistent with age-dependent expressivity of AUKS (Kingdom and Wright, 2022). At 4-week post-termination follow-up, parents reported reduced anxiety and no depression symptoms; they expressed satisfaction with the diagnostic timeline. In reviewing the diagnostic and clinical management pathway of this case-from initial prenatal screening to post-termination follow-up-key milestones that guided decision-making are systematically summarized in Supplementary Table S1.
Frameshift variant causes severe structural and functional defects
The p.Lys168AsnfsTer35 variant in the HNRNPK gene is a frameshift variant localized to exon nine of the 17 exons that compose the gene (Figure 3A). This frameshift event introduces a PTC at the genomic level, which activates the cellular frameshift mRNA decay pathway-a highly conserved surveillance mechanism dedicated to eliminating transcripts carrying PTCs (Figure 3B). Functional predictions based on NMD pathway characteristics indicate that approximately 99% of the mutant HNRNPK mRNAs harboring this PTC will be recognized and degraded by the NMD machinery. This efficient degradation process is expected to substantially reduce the pool of stable HNRNPK mRNA available for ribosome-mediated translation, thereby minimizing the potential for de novo protein synthesis from the mutant transcript.
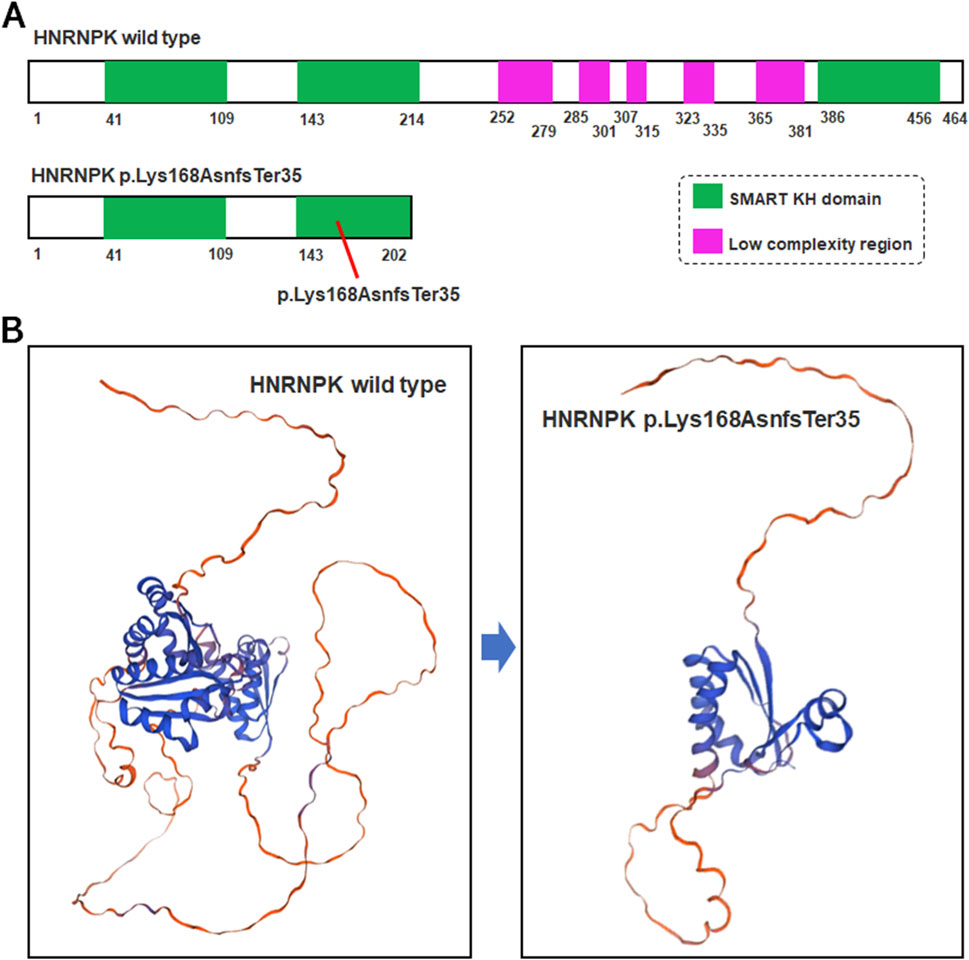
Figure 3. Structural comparison of wild-type and mutant HNRNPK protein. (A) Schematic of HNRNPK protein structure. Upper panel: Wild-type HNRNPK (464 aa) with intact SMART KH domains (green) and C-terminal low-complexity regions (purple). Lower panel: Hypothetical p.Lys168AsnfsTer35 mutant (exon nine of 17). (B) Predicted 3D structures of HNRNPK. Left panel: Wild-type with canonical fold of functional regions. Right panel: Hypothetical mutant structure-truncation at aa 202 is predicted to disrupt the SMART KH domain and lose the low-complexity region, based on the assumption of mRNA escaping NMD.
ClinVar-reported P/LP SNV variants in the HNRNPK gene
To contextualize the variants identified in our study, we analyzed the HNRNPK gene variants documented in ClinVar (Figure 4). Variants highlighted in purple represent the variant identified and reported in the current study, c.504-507del. The ClinVar-reported variants span multiple regions of the HNRNPK gene. Notably, about 15 intronic P/LP SNP variants in HNRNPK lie within ±5 bp of exon boundaries, a pattern consistent with other LoF-associated genes. These intronic variants can impact the splicing of HNRNPK pre-mRNA. As proper pre-mRNA splicing is crucial for generating mature mRNA with accurate coding information, disruptions here may interfere with the normal process of translating the HNRNPK coding DNA sequence (CDS) into a functional protein. The c.504-507del variant identified in our study is a pathogenic variant. It resides within the HNRNPK CDS, which serves as the core template for protein synthesis. Alterations in this region can directly affect the amino acid sequence of the HNRNPK protein, thereby impairing its normal function.
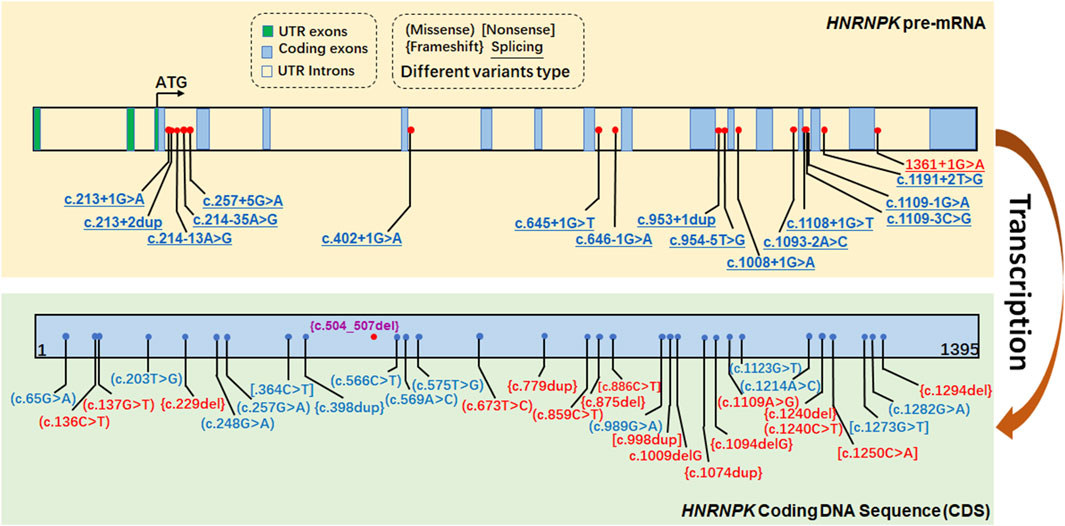
Figure 4. ClinVar-reported P/LP SNV variants in the HNRNPK gene. The figure displays variants in the HNRNPK gene documented in the ClinVar database, color-coded based on their classification: red for Pathogenic (P), and blue for Likely Pathogenic (LP). Variants highlighted in purple represent those identified and reported in the current study.
Discussion
Increased NT and NF are well-established but non-specific soft markers, commonly linked to aneuploidies like Down syndrome (Methods In Medicine CAM, 2023) and Turner syndrome (Gravholt et al., 2017; Yana et al., 2024), but also frequently reported in numerous monogenic disorders such as Noonan syndrome (Tangshewinsirikul et al., 2024), Kabuki syndrome (Sun et al., 2023), and CHARGE syndrome (Hsu et al., 2014). The primary value of this case lies in demonstrating WES as a critical tool for fetuses with nonspecific soft markers, when karyotype and CMA are normal, rather than its specificity for AUKS. This study identified a de novo heterozygous frameshift variant in HNRNPK (c.504_507del; p.Lys168AsnfsTer35) in a fetus with increased NT (3.4 mm) and NF (9 mm), representing a prenatal diagnosis of AUKS.
This study identified a de novo heterozygous frameshift variant in HNRNPK (c.504_507del; p.Lys168AsnfsTer35) in an 18 + 1 week fetus with increased NT (3.4 mm) and NF (9 mm), contributing to the prenatal phenotypic spectrum of AUKS. Prior reports have documented HNRNPK variants in prenatal cases. A study reported a HNRNPK missense variant in a 20-week fetus with NT thickening (3.2 mm) and mild cardiac hypoplasia (Zhang et al., 2024). A slightly more severe prenatal presentation than our case, which lacked cardiac anomalies, aligning with the trend that missense variants may present with earlier structural anomalies compared to frameshift variants in prenatal stages. While Workalemahu et al. identified a HNRNPK frameshift variant in a 19-week fetus with NF thickening (8 mm) but no major structural anomalies, consistent with our case’s nonspecific soft markers, though those diagnoses were finalized postnatally (Workalemahu et al., 2023). Another study reported six new cases of AUKS, with all patients harboring either de novo HNRNPK variants or de novo 9q21.32 deletions that include the HNRNPK gene (Au et al., 2018). This finding expands the prenatal phenotypic spectrum of AUKS, a neurodevelopmental disorder previously characterized primarily by postnatal features, and underscores the diagnostic value of NT and NF thickening as potential indicators of monogenic disorders like AUKS, particularly in cases with normal karyotype results.
The presence of significant NT and NF thickening in this case, in the absence of major structural anomalies typically screened for prenatally, highlights a crucial diagnostic insight. While NT ≥ 3.0 mm substantially elevates the risk for common aneuploidies (Aygun, 2018), and NF ≥ 6 mm in the second trimester is a strong marker for Down syndrome (Abou Tayoun et al., 2018), this case demonstrates that these soft markers can also herald serious monogenic conditions when aneuploidy is excluded. Our findings suggested that dysfunction of HNRNPK may underlie the abnormal nuchal fluid accumulation observed. HNRNPK is a key regulator of genes involved in diverse processes, including extracellular matrix (ECM) composition and potentially lymphatic development-pathways implicated in nuchal edema formation in other syndromes like Noonan syndrome (Li W. B. et al., 2015). This proposed mechanistic link-where HNRNPK dysfunction disrupts developmental pathways via LoF, which is further supported by the ClinGen curation for HNRNPK (https://search.clinicalgenome.org/kb/genes/HGNC:5044), which confirms that LoF and haploinsufficiency are the core drivers of AUKS. Notably, this curation also contextualizes why even a single de novo frameshift variant is sufficient to cause AUKS: haploinsufficiency of HNRNPK disrupts the delicate balance of RNA processing and gene regulation required for normal fetal development (Liu et al., 2022; Shi et al., 2023). This plausible biological basis for the ultrasound findings in this AUKS case strengthens the argument for considering HNRNPK variants in fetuses with isolated NT/NF thickening and normal karyotype/CMA.
Several limitations warrant consideration. Limitations include the single-case design, lack of postnatal neurodevelopmental data, and potential underreporting of mild AUKS cases. As a single case report, the generalizability of NT/NF thickening as specific prenatal markers for AUKS requires validation in larger cohorts. Postnatal follow-up data on neurodevelopmental outcomes for this case were unavailable, limiting our understanding of the full phenotypic correlation. Future studies should aim to define the sensitivity and specificity of these ultrasound markers for AUKS within cohorts of fetuses with isolated NT/NF thickening and negative initial testing. Furthermore, investigating the potential of HNRNPK-related epigenetic dysregulation as non-invasive prenatal biomarkers represents a promising avenue for future research. While WES proved diagnostic here, its limitations in prenatal settings include the potential for VUS, challenges in interpreting non-coding variants, incomplete detection of certain variant types, and the ethical complexities surrounding incidental findings and rapid turnaround times. Careful pre- and post-test counseling remains paramount.
The prenatal presentation observed here aligns partially with, yet also contrasts, the established postnatal AUKS phenotype. Large cohort studies report cardiac defects in 50%–60% of postnatal AUKS cases, including atrial septal defects (ASD), ventricular septal defects (VSD), and coarctation of the aorta (Au et al., 2018; Au et al., 1993). However, the fetus in this study exhibited only an isolated hyperechogenic intracardiac focus (EIF), a common finding often considered a benign variant but also associated with aneuploidy and some genetic syndromes. EIF in the left ventricle was confirmed via two orthogonal views; nuchal measurements were repeated twice by a fetal medicine specialist to ensure accuracy. This difference underscores the significant phenotypic variability and potential age-dependent expressivity of AUKS. The absence of major structural cardiac defects in utero, despite a severe LoF variant, highlights the incomplete penetrance or variable expressivity of specific features. Factors such as the precise timing of developmental disruption, genetic modifiers, or technical challenges in prenatal cardiac imaging might contribute to this observed heterogeneity (Kingdom and Wright, 2022).
Our case is distinctive for prenatal diagnosis via WES at 18+3 weeks, contrasting with prior reports where diagnosis occurred postnatally despite prenatal anomalies. The de novo nature of the variant, consistent with >95% of reported HNRNPK pathogenic variants, implies a low recurrence risk for future pregnancies (<1%), though the possibility of undetected parental germline mosaicism necessitates cautious counseling. Prenatal diagnosis enables critical anticipatory guidance and planning for potential postnatal complications commonly seen in AUKS, such as neurodevelopmental delay, hypotonia, feeding difficulties requiring gastrostomy, and craniofacial anomalies like cleft palate. Notably, most intronic pathogenic variants in HNRNPK lie within ±5 bp of exon boundaries, affecting splicing, a pattern consistent with other genes associated with loss-of-function. This clarifies that intronic variants in HNRNPK are not uncommon but follow a typical pattern of impacting splicing at exon-intron junctions.
Several limitations warrant consideration. As a single case report, the generalizability of NT/NF thickening as specific prenatal markers for AUKS requires validation in larger cohorts. Postnatal follow-up data on neurodevelopmental outcomes for this case were unavailable, limiting our understanding of the full phenotypic correlation. Future studies should aim to define the sensitivity and specificity of these ultrasound markers for AUKS within cohorts of fetuses with isolated NT/NF thickening and negative initial testing. Furthermore, investigating the potential of HNRNPK-related epigenetic dysregulation as non-invasive prenatal biomarkers represents a promising avenue for future research (Choufani et al., 2022). While WES proved diagnostic here, its limitations in prenatal settings include the potential for VUS, challenges in interpreting non-coding variants, incomplete detection of certain variant types and the ethical complexities surrounding incidental findings and rapid turnaround times. Careful pre- and post-test counseling remains paramount.
The phenotypic presentation of our prenatal case expands the clinical spectrum of AUKS, which traditionally manifests postnatally with craniofacial anomalies and cardiac defects (Nishtala et al., 2018). In contrast, our fetus exhibited only isolated nuchal thickening and EIF without major structural anomalies, highlighting that AUKS may present with soft markers alone in early gestation. This builds on prior reports of HNRNPK variants, which primarily focused on postnatal diagnoses (Miyake et al., 2017), by linking prenatal NT/NF thickening to AUKS. However, as a single case, we acknowledge limitations in establishing NT/NF as specific markers for AUKS; future studies should enroll cohorts with isolated nuchal thickening and negative CMA to determine the prevalence of HNRNPK variants. The parents reported anxiety regarding the prior fetal demise and expressed appreciation for the timely WES diagnosis, which informed their decision-making. They consented to publication to aid future prenatal counseling.
In conclusion, this report describes a prenatal diagnosis of AUKS, linking a pathogenic de novo HNRNPK frameshift variant to the isolated prenatal findings of increased nuchal translucency and nuchal fold thickening. This expands the recognized prenatal phenotype of AUKS and underscores the critical importance of considering serious monogenic disorders like AUKS in the differential diagnosis of significant soft markers, even in the absence of major structural anomalies or aneuploidy, with WES playing a central role in achieving such diagnoses.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Quzhou Maternal and Child Healthcare Hospital Ethics Review Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
YZ: Funding acquisition, Writing – original draft. ZY: Data curation, Software, Writing – review and editing. QZ: Validation, Writing – review and editing. KW: Methodology, Writing – review and editing. JH: Conceptualization, Project administration, Writing – review and editing. HZ: Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research received support from the following sources: The National Key R&D Program of China (2023YFC2705600) through its sub-project (2023YFC2705602), and Quzhou Municipal Guiding Projects for Key Technology R&D (2024ZD074).
Acknowledgments
We express our sincere gratitude to the patients who generously consented to contribute their personal data for this study and its publication.
Conflict of interest
Author ZY was employed by company BGI Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. The author(s) verify and take full responsibility for the use of generative AI in the preparation of this manuscript. Generative AI was used: to assist with English language polishing, including refining sentence structure, improving grammatical accuracy, and enhancing the clarity of expression in sections such as the Discussion and Resubmission Statement. All AI-generated language revisions were carefully reviewed and edited by the authors to ensure they accurately reflect the scientific content and intent of the original manuscript, with no substantive changes to the research data, conclusions, or scientific rationale.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1661743/full#supplementary-material
References
Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., et al. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524. doi:10.1002/humu.23626
Amberger, J. S., Bocchini, C. A., Scott, A. F., and Hamosh, A. (2019). OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 47, D1038-D1043–D1043. doi:10.1093/nar/gky1151
Au, P. Y. B., McNiven, V., Phillips, L., Innes, A. M., and Kline, A. D. (1993). “Au-Kline syndrome,” in GeneReviews((R)). Editors M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and A. Amemiya (Seattle (WA)).
Au, P. Y. B., You, J., Caluseriu, O., Schwartzentruber, J., Majewski, J., Bernier, F. P., et al. (2015). GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum. Mutat. 36, 1009–1014. doi:10.1002/humu.22837
Au, P. Y. B., Goedhart, C., Ferguson, M., Breckpot, J., Devriendt, K., Wierenga, K., et al. (2018). Phenotypic spectrum of Au-Kline syndrome: a report of six new cases and review of the literature. Eur. J. Hum. Genet. 26, 1272–1281. doi:10.1038/s41431-018-0187-2
Aygun, N. (2018). Biological and genetic features of neuroblastoma and their clinical importance. Curr. Pediatr. Rev. 14, 73–90. doi:10.2174/1573396314666180129101627
Brown, J., Pirrung, M., and McCue, L. A. (2017). FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. doi:10.1093/bioinformatics/btx373
Choufani, S., McNiven, V., Cytrynbaum, C., Jangjoo, M., Adam, M. P., Bjornsson, H. T., et al. (2022). An HNRNPK-Specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome. Am. J. Hum. Genet. 109, 1867–1884. doi:10.1016/j.ajhg.2022.08.014
Gadsboll, K., Brix, N., Sandager, P., Petersen, O. B., Souka, A. P., Nicolaides, K. H., et al. (2025). Increased nuchal translucency thickness and normal chromosomal microarray: danish nationwide cohort study. Ultrasound Obstet. Gynecol. 65, 462–469. doi:10.1002/uog.29198
Genomes Project, C., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi:10.1038/nature15393
Gillentine, M. A., Wang, T., Hoekzema, K., Rosenfeld, J., Liu, P., Guo, H., et al. (2021). Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome Med. 13, 63. doi:10.1186/s13073-021-00870-6
Gravholt, C. H., Andersen, N. H., Conway, G. S., Dekkers, O. M., Geffner, M. E., Klein, K. O., et al. (2017). Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati international Turner syndrome meeting. Eur. J. Endocrinol. 177, G1–G70. doi:10.1530/EJE-17-0430
Hsu, P., Ma, A., Wilson, M., Williams, G., Curotta, J., Munns, C. F., et al. (2014). CHARGE syndrome: a review. J. Paediatr. Child. Health 50, 504–511. doi:10.1111/jpc.12497
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi:10.1038/s41586-020-2308-7
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi:10.1038/nprot.2015.053
Kingdom, R., and Wright, C. F. (2022). Incomplete penetrance and variable expressivity: from clinical studies to population cohorts. Front. Genet. 13, 920390. doi:10.3389/fgene.2022.920390
Knaus, B. J., and Grunwald, N. J. (2017). Vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53. doi:10.1111/1755-0998.12549
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062-D1067–D1067. doi:10.1093/nar/gkx1153
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Li, W. B., Li, Y., Yu, C., and He, Y. M. (2015). Reversal of multidrug resistance by the Chinese medicine yiqi Jianpi Huaji Decoction and the mechanism of action in human gastric cancer SGC7901/VCR cells. Evid. Based Complement. Altern. Med. 2015, 390812. doi:10.1155/2015/390812
Liu, L., Li, J., Li, Y., Li, H., Yang, B., Fan, H., et al. (2022). Genetic diagnosis of common fetal renal abnormalities detected on prenatal ultrasound. Prenat. Diagn 42, 894–900. doi:10.1002/pd.6154
Maag, J. L. V. (2018). Gganatogram: an R package for modular visualisation of anatograms and tissues based on ggplot2. F1000Res 7, 1576. doi:10.12688/f1000research.16409.2
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17 (1), 10–12. doi:10.14806/ej.17.1.200
Methods In Medicine CAM (2023). Retracted: study on the effect of B-Ultrasound NT scan in early pregnancy combined with serum screening in early and middle pregnancy for Down syndrome. Comput. Math. Methods Med. 2023, 9764153. doi:10.1155/2023/9764153
Miyake, N., Inaba, M., Mizuno, S., Shiina, M., Imagawa, E., Miyatake, S., et al. (2017). A case of atypical kabuki syndrome arising from a novel missense variant in HNRNPK. Clin. Genet. 92, 554–555. doi:10.1111/cge.13023
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi:10.1093/nar/gkg509
Nishtala, A., Piers, R. J., Himali, J. J., Beiser, A. S., Davis-Plourde, K. L., Saczynski, J. S., et al. (2018). Atrial fibrillation and cognitive decline in the framingham heart study. Heart rhythm. 15, 166–172. doi:10.1016/j.hrthm.2017.09.036
Parra, A. S., and Johnston, C. A. (2022). Emerging roles of RNA-binding proteins in neurodevelopment. J. Dev. Biol. 10, 23. doi:10.3390/jdb10020023
Paysan-Lafosse, T., Andreeva, A., Blum, M., Chuguransky, S. R., Grego, T., Pinto, B. L., et al. (2025). The pfam protein families database: embracing AI/ML. Nucleic Acids Res. 53, D523–D534. doi:10.1093/nar/gkae997
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886-D894–D894. doi:10.1093/nar/gky1016
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi:10.1093/nar/29.1.308
Shi, X., Ding, H., Li, C., Liu, L., Yu, L., Zhu, J., et al. (2023). Clinical utility of chromosomal microarray analysis and whole exome sequencing in foetuses with oligohydramnios. Ann. Med. 55, 2215539. doi:10.1080/07853890.2023.2215539
Sun, Y., Liu, L., Zhi, Y., Li, Y., Tian, W., Yang, B., et al. (2023). Genetic examination for fetuses with increased nuchal translucency by exome sequencing. J. Obstet. Gynaecol. Res. 49, 530–538. doi:10.1111/jog.15482
Tangshewinsirikul, C., Wattanasirichaigoon, D., Tim-Aroon, T., Promsonthi, P., Katanyuwong, P., Diawtipsukon, S., et al. (2024). Prenatal sonographic features of Noonan syndrome: case series and literature review. J. Clin. Med. 13, 5735. doi:10.3390/jcm13195735
The mutational constraint spectrum (2021). Author correction: the mutational constraint spectrum quantified from variation in 141,456 humans. Nature 590, E53.
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3--new capabilities and interfaces. Nucleic Acids Res. 40, e115. doi:10.1093/nar/gks596
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43 (11), 10 11–11. doi:10.1002/0471250953.bi1110s43
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Wang, C., Tang, J., Tong, K., Huang, D., Tu, H., and Zhu, J. (2022). Chromosomal microarray analysis versus noninvasive prenatal testing in fetuses with increased nuchal translucency. J. Hum. Genet. 67, 533–539. doi:10.1038/s10038-022-01041-0
Workalemahu, T., Avery, C., Lopez, S., Blue, N. R., Wallace, A., Quinlan, A. R., et al. (2023). Whole-genome sequencing analysis in families with recurrent pregnancy loss: a pilot study. PLoS One 18, e0281934. doi:10.1371/journal.pone.0281934
Xiao, H., Yang, Y. L., Zhang, C. Y., Liao, E. J., Zhao, H. R., and Liao, S. X. (2016). Karyotype analysis with amniotic fluid in 12365 pregnant women with indications for genetic amniocentesis and strategies of prenatal diagnosis. J. Obstet. Gynaecol. 36, 293–296. doi:10.3109/01443615.2015.1041889
Xu, Y., Wu, W., Han, Q., Wang, Y., Li, C., Zhang, P., et al. (2019). Post-translational modification control of RNA-Binding protein hnRNPK function. Open Biol. 9, 180239. doi:10.1098/rsob.180239
Yamamoto, T. (2021). Genomic aberrations associated with the pathophysiological mechanisms of neurodevelopmental disorders. Cells 10, 2317. doi:10.3390/cells10092317
Yana, M., Kavlak, E., and Gunes, M. (2024). Combined sensory integration therapy plus neurodevelopmental therapy (NT) versus NT alone for motor and attention in children with Down syndrome: a randomized controlled trial. Int. J. Dev. Disabil. 70, 849–856. doi:10.1080/20473869.2022.2152166
Keywords: nuchal translucency, prenatal diagnosis, whole-exome sequencing, de novo variant, hnRNPK
Citation: Zhu Y, Yang Z, Zhu Q, Wu K, He J and Zhou H (2025) Prenatal diagnosis of a de novo pathogenic HNRNPK variant in a Chinese fetus with abnormal ultrasound soft markers: a case report. Front. Genet. 16:1661743. doi: 10.3389/fgene.2025.1661743
Received: 08 July 2025; Accepted: 30 September 2025;
Published: 27 October 2025.
Edited by:
Stephen J Bush, Xi’an Jiaotong University, ChinaReviewed by:
Ted Han, New York Genome Center, United StatesMaura Mingoia, National Research Council (CNR), Italy
Copyright © 2025 Zhu, Yang, Zhu, Wu, He and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei Zhou, MTUwNjcwMjI2NzZAMTYzLmNvbQ==