 Jinlan Li1,2
Jinlan Li1,2 Jie Zhou1,2Chunbo Ji1,2Siqing Ma1,2Jianying Zhu1,2Tiejun Yang1,2Danyang Dong1,2
Jie Zhou1,2Chunbo Ji1,2Siqing Ma1,2Jianying Zhu1,2Tiejun Yang1,2Danyang Dong1,2 Yang Ping2*
Yang Ping2*- 1Clinical Medicine School of Ningxia Medical University, Yinchuan, China
- 2Department of Neurology, General Hospital of Ningxia Medical University, Yinchuan, China
This case concerns a Chinese female patient who was referred to our clinic having complained of weakness in her lower limbs. Following a series of diagnostic procedures, including electrophysiology, muscle biopsy and genetic analysis, the patient was diagnosed with limb-girdle muscular dystrophy type 2B (LGMD2B). Genetic testing revealed compound heterozygous mutations in the DYSF gene, specifically the missense mutation c.6313G>A (p.Ala2105Thr). Another variant, c.4444del (p.Glu1482Serfs*43), is a frameshift mutation. This case provides further confirmation of the LGMD2B diagnosis. It also identifies novel compound heterozygous DYSF mutations. These findings have significant implications for the diagnosis and research of genetic diseases, the management of at-risk individuals and the development of new therapies.
Introduction
Limb-girdle muscular dystrophy (LGMD) is a hereditary disorder marked by progressive skeletal muscle atrophy and weakness due to gene mutations. In the Chinese population, LGMD2B, caused by DYSF gene mutations, and LGMD2A, linked to CAPN3 mutations, account for 65.9% of cases (Lin et al., 2023). LGMD2B primarily impacts hip and shoulder muscles and is associated with the absence or dysfunction of dysferlin, a protein crucial for muscle cell membrane repair (Bouchard, 2023). This report describes the clinical case of a female patient who presented with a progressive weakness in both of her lower limbs. Initially misdiagnosed with polymyositis due to persistently elevated serum creatine kinase (CK) levels, she was eventually diagnosed with LGMD2B following muscle biopsy and genetic testing, including next-generation sequencing (NGS). Sanger sequencing revealed compound heterozygous mutations in the DYSF gene. These mutations have not been previously reported in the literature, indicating their rarity. This case highlights the importance of an accurate diagnosis in hereditary neuromuscular diseases and encourages further research into the genetic mechanisms that underlie LGMD.
Case presentation
Here we present a case of a 38-year-old unmarried woman from a kinship family in north-west China who has suffered progressive limb wasting for 23 years. Her clinical history is as follows: The patient was admitted to our hospital’s neurology department in 2001 at the age of 15 due to “bilateral lower limb weakness that had lasted for 2 years.” At that time, she could not stand up quickly after squatting and gradually lost the ability to coordinate walking and jumping. A neurological examination revealed no abnormalities in the cranial nerves. Bilateral lower limb muscle strength (including the pelvis and quadriceps primarily) was rated as grade 4 (maximum 5 points) and distal muscle strength was rated as grade 5. Both Babinski signs were negative, while the Gowers sign was positive.
Laboratory results showed serum creatine kinase levels of 27,990.83 μmol/L. Electrophysiological tests revealed chronic myogenic damage in both lower limbs. EMG showed no spontaneous potentials at rest and revealed a significantly shortened mean motor unit duration, as well as an increased number of polyphasic waves during light exercise. Polymyositis was diagnosed. The patient received intravenous methylprednisolone therapy, resulting in symptom relief and restoration of ambulation. Following treatment, the creatine kinase level fell to 3.423.7 μmol/L, after which the patient was discharged.
However, the patient was readmitted in 2005 for persistent stair-climbing weakness, having been treated with methotrexate and folic acid. Electrophysiology and a muscle biopsy confirmed damage to the gastrocnemius muscle with atrophy, hypertrophy, and fatty hyperplasia, which is consistent with LGMD 2B. Until 2024, the patient remained untreated for LGMD 2B, which resulted in severe disability and total dependence on a wheelchair. An echocardiogram showed no abnormalities, while laboratory tests revealed irregularities in the enzymes. Genetic testing clarified the subtype of limb-girdle muscular dystrophy and its genetic characteristics.
Prior to venipuncture, informed consent was obtained from the patient, her parents and control subjects. Although the patient’s parents appeared phenotypically normal, there were concerns about their consanguinity due to a family history of genetic diseases (two uncles became ill at around 20 years of age and later died in their 40s). To illustrate the genetic inheritance among the patient and her family members, a pedigree has been shown in Figure 1. (This pedigree chart is a display of the genetic inheritance of the patient and her family members. Individuals carrying the mutation are shown in black. Individuals without the mutation are shown in white. Squares represent male relatives, circles female relatives and arrows the proband).
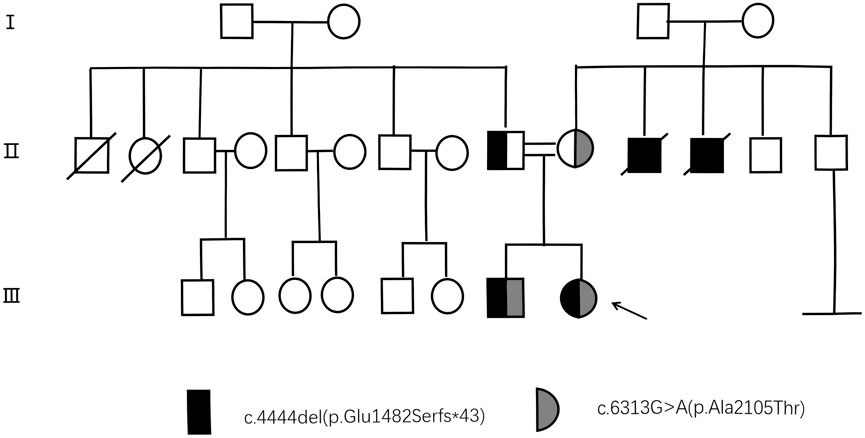
Figure 1. Pedigree chart illustrating genetic inheritance in the Patient’s family.
Genetic testing shown in Figure 2 has revealed that the individual carries the variant c.6313G>A (p.Ala2105Thr), a missense mutation in the DYSF gene affecting codon 2015. The mutation has been confirmed by Sanger sequencing and is associated with Miyoshi dystrophy/limb-girdle dystrophy, with three heterozygotes reported in the gnomAD database. Another variant is a frameshift mutation caused by a three nucleotide deletion inherited from the father, c.4444 del (p.Glu1482Serfs*43). It shows potential pathogenicity, although it is classified as a variant of uncertain significance (VUS) according to the ACMG guidelines. A novel compound heterozygous variant in the DYSF gene was identified by genetic sequencing, confirming the diagnosis of LGMD2B.
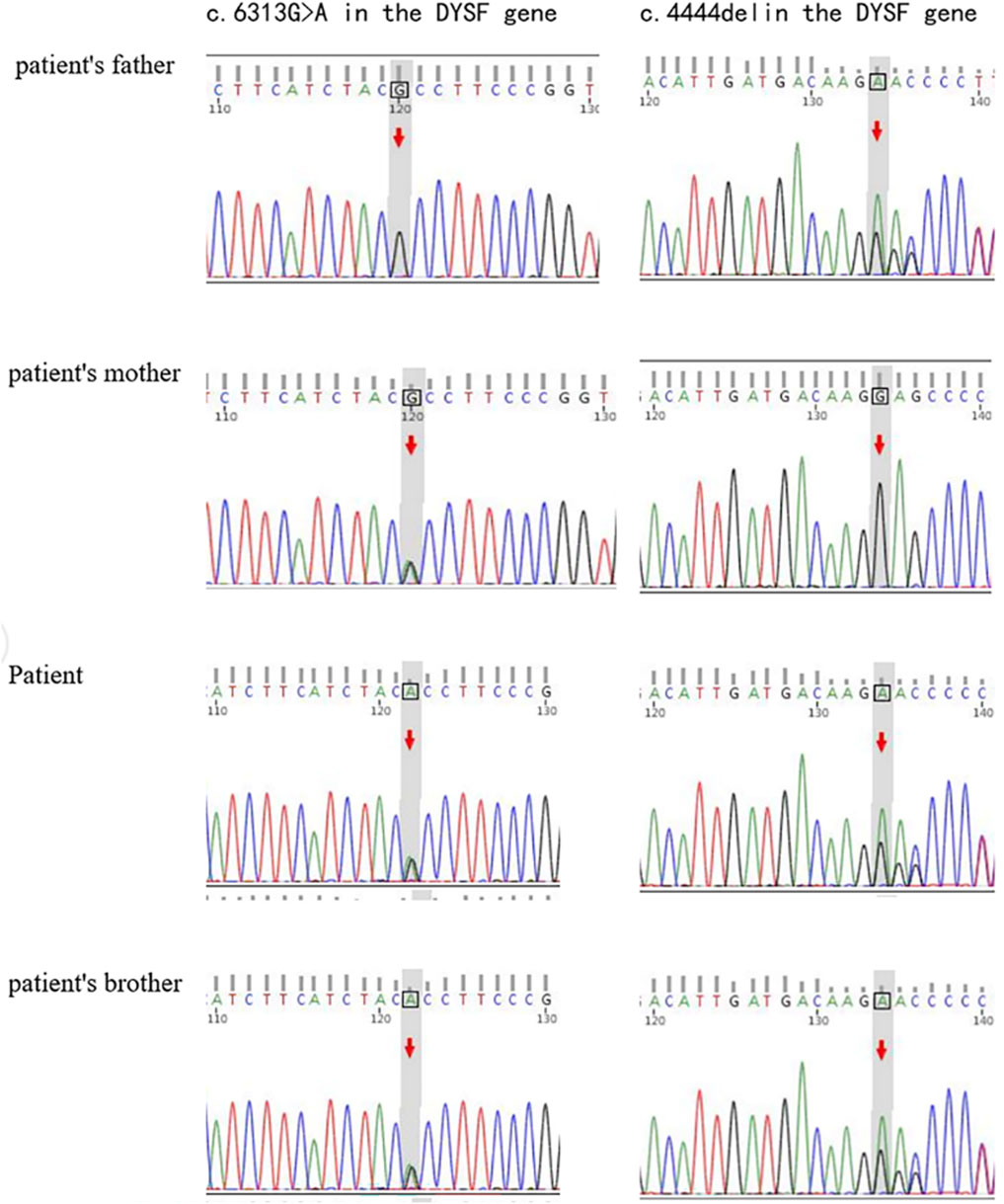
Figure 2. Genetic mutations in the DYSF gene. Sanger sequencing plot showing DYSF variable sequences in DYSF gene affected families.
Discussion
Muscle biopsies in most patients with ferritinopathies show an inflammatory response, particularly pronounced with neurological abnormalities (Angelini et al., 2018), This suggests that inflammation may manifest relatively early, as symptomatic conditions can occur even in patients exhibiting fewer clinical signs. This suggests inflammation may present early, as symptoms can arise even in patients with fewer clinical signs (Rowin et al., 1999). Additionally, dysferlin deficiency activates inflammatory vesicles in muscle, creating a pro-inflammatory environment that worsens damage (Rawat et al., 2010). Consequently, misdiagnosis of LGMD as polymyositis due to the inflammatory response of the muscle is prevalent in the early stages of the disease, and genetic testing is a pivotal discriminating factor. Such misdiagnosis can lead to decreased muscle strength, reduced bone density, and other adverse effects (Nguyen et al., 2005). Recent studies show that intermittent prednisone administration can reduce muscle damage without causing atrophy, but its impact on disease progression remains unconfirmed (Quattrocelli et al., 2017; Zelikovich et al., 2022).
We compared two mutation sites in the DYSF gene using the public databases dbSNP and ClinVar and identified the mutation c.6313G>A (p.Ala2105Thr, referred to as mtB). Three heterozygous cases were reported in the gnomAD database, but no homozygous cases were found. The literature indicates that the mtB variant is present in patients with Miyoshi myopathy and limb-girdle muscular dystrophy, often in homozygous or compound heterozygous forms (PMID: 32400077, 27066573, 21522182, 34559919, 32528171, 30028523). Based on current evidence, the clinical significance of this variant is uncertain. In contrast, the c.4444 del (p.Glu1482Serfs*43, designated mtA) variant has not been reported in the literature and is absent in gnomAD. Based on the available data, this variant is thought to be pathogenic.
Amino acid sequences of the DYSF protein from Homo sapiens, Mus musculus, rhesus monkey, chimpanzee and Drosophila melanogaster were obtained from the NCBI database. Alignments performed with Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) showed a high degree of conservation of amino acid sites corresponding to these mutations (Figure 3).
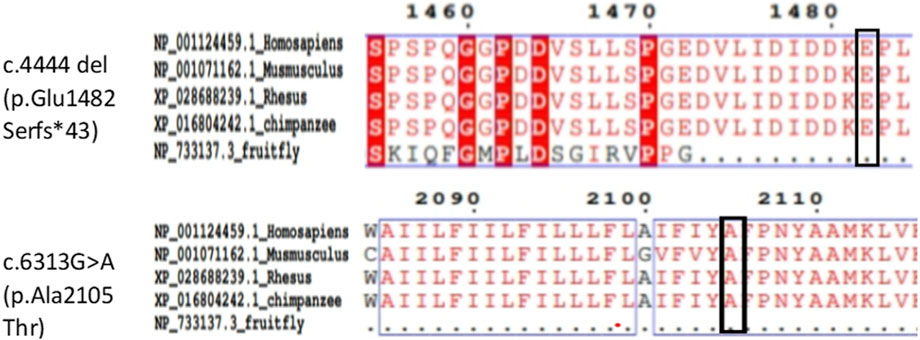
Figure 3. Amino acid sequence conservation analysis of DYSF proteins of different species.
The secondary structure of human DYSF was predicted using PSIPRED v4.0 and showed that the affected amino acids were located within irregular coil structures (Figure 4). Domain predictions from the Pfam and SMART databases indicated that mtA is outside the main domain, while mtB is within the Ferlin_C domain (Figure 5).

Figure 4. Schematic representation of the secondary structure of DYSF protein.

Figure 5. Protein structural domains of DYSF.
Three-dimensional structures of the wild-type and mutant proteins were constructed using SWISS-MODEL (http://swissmodel.expasy.org/). Analysis revealed that mt A replaces glutamic acid with serine, resulting in the loss of three hydrogen bonds. Pathogenicity of the DYSF gene: c.4444 deletion mutation (p.Glu1482Serfs*43, mtA) variant. This mutation is not located in the key functional domain or active site of the DYSF protein (as shown in Figure 5), so it may not affect enzyme activity or ligand binding. However, it may indirectly affect function by altering protein stability or interaction interfaces (as depicted in Figure 6). Therefore, we conclude that the observed phenotype in the proband is most likely caused by a frameshift mutation leading to protein truncation. while, mt B replaces alanine with threonine, resulting in the loss of one hydrogen bonds (Figure 6). These mutations change the structure of the protein from an ordered to a disordered state, affecting its functionality.
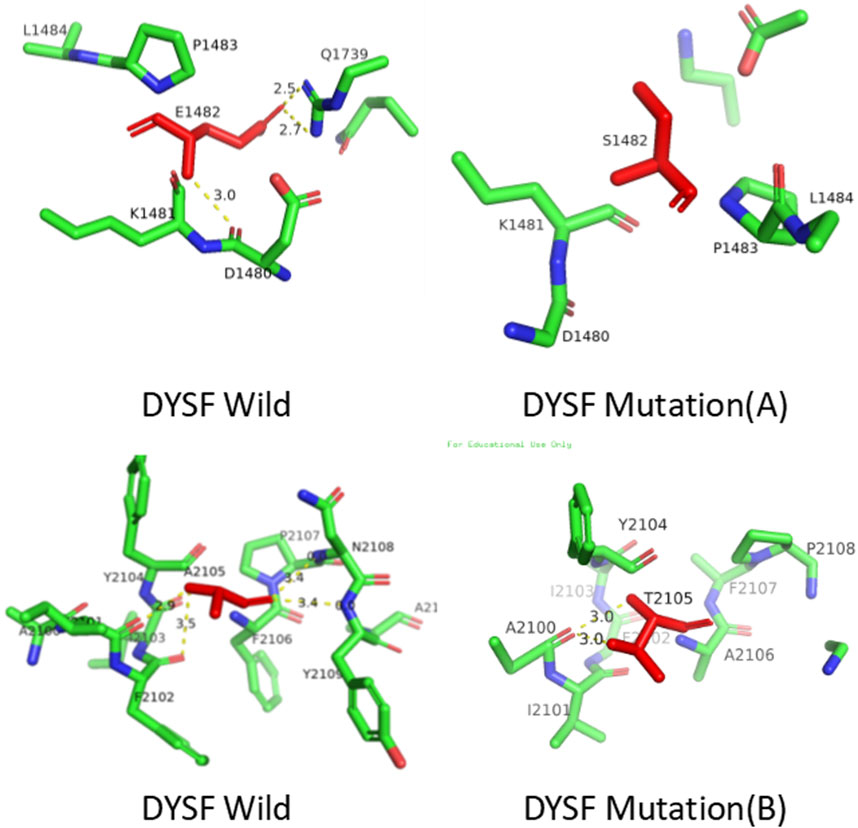
Figure 6. Schematic representation of the tertiary structure of wild type and mutant human DYSF.
This report details a significant case of limb-girdle muscular dystrophy type 2B (LGMD2B) in a female patient, emphasising the crucial diagnostic steps involved, such as muscle biopsy, advanced gene sequencing, and bioinformatics analysis. The discovery of previously unreported DYSF mutations not only facilitates further investigation into their pathogenicity but also broadens our understanding of hereditary neuromuscular diseases. By exploring the genetic complexities of these disorders, we enhance the potential for earlier diagnosis and improved patient management.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ningxia Medical University (approval number: KYLL-2024-1441). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. The animal studies were approved by Ningxia Medical University (approval number: KYLL-2024-1441). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JL: Writing – original draft, Investigation. JZ: Conceptualization, Software, Writing – review and editing. CJ: Data curation, Writing – original draft. SM: Methodology, Writing – original draft. JZ: Supervision, Writing – original draft. TY: Validation, Writing – original draft. DD: Formal Analysis, Writing – original draft. YP: Funding acquisition, Resources, Visualization, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Ningxia Key Research and Development Program for Special Projects 2021BEG03032.
Acknowledgments
The authors gratefully acknowledge the patient for permitting us to disclose details relating to this case. The authors thank the family members for participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Angelini, C., Giaretta, L., and Marozzo, R. (2018). An update on diagnostic options and considerations in limb-girdle dystrophies. Expert Rev. Neurother. 18, 693–703. doi:10.1080/14737175.2018.1508997
Bouchard, C. (2023). Diagnosis and development of therapy. J. Clin. Med., 12. doi:10.3390/jcm12186011
Lin, F., Yang, K., Lin, X., Chen, L., Zheng, F. Z., et al. Clinical features (2023). Clinical features, imaging findings and molecular data of limb-girdle muscular dystrophies in a cohort of Chinese patients. Orphanet J. Rare Dis. 18, 356. doi:10.1186/s13023-023-02897-x
Nguyen, K., Bassez, G., Bernard, R., Krahn, M., Labelle, V., Figarella-Branger, D., et al. (2005). Dysferlin mutations in LGMD2B, miyoshi myopathy, and atypical dysferlinopathies. Hum. Mutat. 26, 165. doi:10.1002/humu.9355
Quattrocelli, M., Salamone, I. M., Page, P. G., Warner, J. L., Demonbreun, A. R., and McNally, E. M. (2017). Intermittent glucocorticoid dosing improves muscle repair and function in mice with limb-girdle muscular dystrophy. Am. J. Pathology 187, 2520–2535. doi:10.1016/j.ajpath.2017.07.017
Rawat, R., Cohen, T. V., Ampong, B., Francia, D., Henriques-Pons, A., Hoffman, E. P., et al. (2010). Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathology 176, 2891–2900. doi:10.2353/ajpath.2010.090058
Rowin, J., Meriggioli, M. N., Cochran, E. J., and Sanders, D. B. (1999). Prominent inflammatory changes on muscle biopsy in patients with miyoshi myopathy. Neuromuscul. Disord. NMD 9, 417–420. doi:10.1016/s0960-8966(99)00041-3
Keywords: variants, DYSF, dysferlin, limb-girdle recessive muscular dystrophy 2, limb-girdle muscular dystrophy type 2B
Citation: Li J, Zhou J, Ji C, Ma S, Zhu J, Yang T, Dong D and Ping Y (2025) Uncovering compound heterozygous DYSF variants in a Chinese family affected by limb-girdle muscular dystrophy type 2B. Front. Genet. 16:1664086. doi: 10.3389/fgene.2025.1664086
Received: 11 July 2025; Accepted: 09 September 2025;
Published: 17 September 2025.
Edited by:
Anjana Munshi, Central University of Punjab, IndiaReviewed by:
Hicham Mansour, Saint George Hospital University Medical Center, LebanonYuqing Xu, Sichuan University, China
Copyright © 2025 Li, Zhou, Ji, Ma, Zhu, Yang, Dong and Ping. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Ping, eWFuZ3BpbmcxOTk5Lmdvb2RAMTYzLmNvbQ==