 Friederike Reuss1*
Friederike Reuss1* Tilman Schell2,3
Tilman Schell2,3 Haruhiko Isawa4Shinji Kasai4
Haruhiko Isawa4Shinji Kasai4 Sven Klimpel3,5,6
Sven Klimpel3,5,6 Ruth Müller7
Ruth Müller7 Markus Pfenninger5,8
Markus Pfenninger5,8 Judith Kochmann5†
Judith Kochmann5†- 1Institute of Occupational, Social and Environmental Medicine, Goethe University Frankfurt, Frankfurt am Main, Germany
- 2Senckenberg Research Institute, Frankfurt am Main, Germany
- 3LOEWE Centre for Translational Biodiversity Genomics (LOEWE-TBG), Frankfurt am Main, Germany
- 4Department of Medical Entomology, National Institute of Infectious Diseases, Japan Institute for Health Security (JIHS), Tokyo, Japan
- 5Institute for Ecology, Evolution and Diversity, Goethe-University, Frankfurt am Main, Germany
- 6Senckenberg Biodiversity and Climate Research Centre (SBiK-F), Frankfurt am Main, Germany
- 7Unit of Entomology, Institute of Tropical Medicine Antwerp, Antwerp, Belgium
- 8Institute of Organismic and Molecular Evolution (iomE), Johannes Gutenberg University, Mainz, Germany
1 Introduction
Mosquitoes (Diptera: Culicidae) are an important group of insects due to the important role played by culicid species as disease vectors. Some Aedes species are competent to vector human and veterinary relevant viruses, such as dengue, chikungunya, or Japanese encephalitis viruses. In addition, there are some highly invasive Aedes species (Lounibos, 2002). The two most widespread species globally are Aedes albopictus, native to Southeast Asia, and Aedes aegypti, native to Africa, for which genomes have been sequenced previously: Ae. aegypti AaegL5.0 (GCF_002204515.2; Matthews et al., 2018) and Ae. albopictus AalbF5 (GCF_035046485.1; Palatini et al., 2020). Globally, Ae. aegypti is the primary vector of chikungunya and dengue viruses (Sousa et al., 2012; Jansen et al., 2018). Aedes albopictus is a secondary vector to Ae. aegypti for chikungunya and dengue viruses (Jansen and Beebe, 2010; Sousa et al., 2012); however, it is the most important vector for autochthonous cases of dengue and chikungunya in Europe (Rezza et al., 2007; Gjenero-Margan et al., 2011; Succo et al., 2016). Both Ae. aegypti and Ae. albopictus are invasive species in Europe (European Centre for Disease Prevention and Control and European Food Safety Authority, 2023).
Another more recent invader to North America (Kaufman and Fonseca, 2014) and Europe is Aedes japonicus japonicus, while its sister species Aedes koreicus has established itself in Europe (European Centre for Disease Prevention and Control and European Food Safety Authority, 2023). Over the last two to three decades, Ae. j. japonicus has spread beyond its original area of distribution in East Asia via the import of used tires and trade (Kaufman and Fonseca, 2014; Koban et al., 2019) and is likely to expand its range of area distribution in the future (Cunze et al., 2020). Annotated genomes for Ae. j. japonicus and Ae. koreicus (GCA_034211315.2, GCA_024533555.2) have only recently become available (Catapano et al., 2023; Nagy et al., 2024).
Here, we describe an annotated genome and a complete mitochondrial sequence of Ae. j. japonicus from a laboratory strain in Japan (Hoshino et al., 2010). This is the first study wherein individuals from the native range of this species (Kaufman and Fonseca, 2014) were sequenced.
The mitochondrion of Ae. j. japonicus can help in constructing phylogenies. For example, the genus Aedes and the tribe of Aedini have been re-organized based on morphological analyses (reviewed in Wilkerson et al., 2015) and molecular analyses (Zadra et al., 2021). Thus, genetic datasets are highly desirable for creating a well-founded phylogeny of Aedini or Aedes (Zadra et al., 2021).
Our genome assembly can facilitate marker selection for environmental associations and genotype-to-phenotype-association studies. By doing so, the genomic basis of vector competence or invasion success can be identified within the species Ae .j. japonicus and also compared to that of other Aedes spp. More specifically, the created dataset allows conducting comparative studies regarding diapause (Kreß et al., 2016; Boyle et al., 2021), thermotolerance (Kramer et al., 2023; Couper et al., 2025), and population structure (Smitz et al., 2021), all considered potential parameters influencing invasiveness (Lahondère and Bonizzoni, 2022).
Although Ae. albopictus and Ae. aegypti are the primary vectors of dengue and chikungunya viruses, Ae. j. japonicus is only a minor vector in the transmission of disease agents, and its vector competence is largely based on laboratory competence studies (Medlock et al., 2012; Jansen et al., 2018; Wagner et al., 2018). Both Ae. j. japonicus and Ae. albopictus can undergo photoperiodic diapause (Armbruster, 2016; Krupa et al., 2021), which benefits the species’ survival in more temperate regions. In addition, this dataset provides data to study candidate genes related to not only vector competence but also insecticide resistance. It also provides genomic resources for marker identification, which can be used in eDNA approaches for a more rapid species detection in the field (Wittwer et al., 2024), genetic control measures such as gene drives, Wolbachia-based methods (Verkuijl et al., 2025; Wang et al., 2025), or RNA interference (Müller et al., 2023).
2 Methods
2.1 Origin of biological material and DNA isolation
For DNA and RNA isolation, the offspring of ten female Ae. j. japonicus were collected during the egg stage from the “Narita” laboratory strain (Hoshino et al., 2010) and raised to the desired stages (Figure 1A) for DNA and RNA isolation.
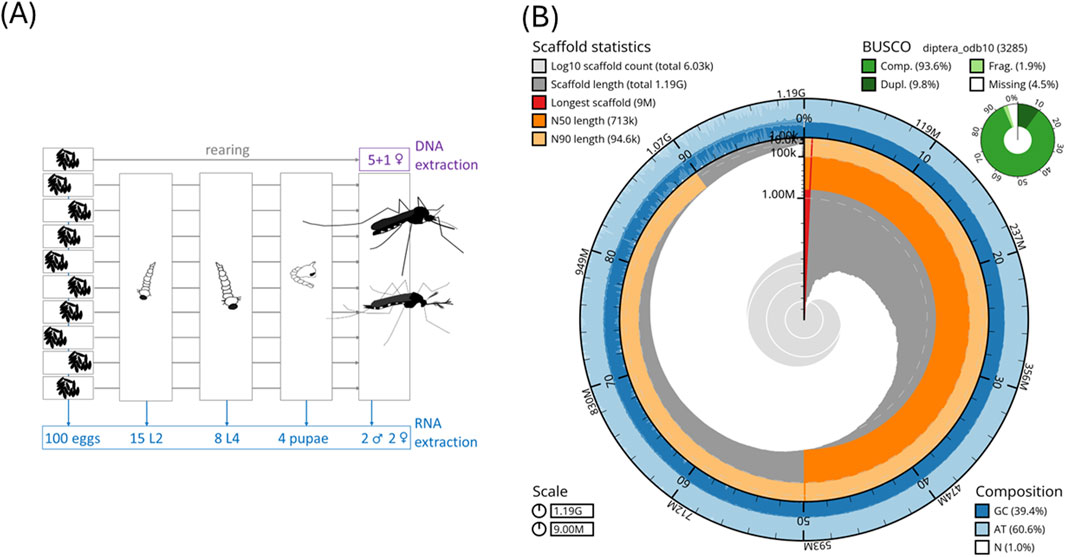
Figure 1. (A) Biological material for DNA and RNA isolation. We used closely related (offspring of one female) individuals for DNA isolation to minimize variation. (B) Snail plot of statistics of the Ae. j. japonicus assembly.
A pool of five sister species in the adult stage was used for DNA MinION long-read and Illumina short-read sequencing, while a single adult female (another sister) was used for PacBio DNA sequencing. DNA was isolated using the protocol “HMW gDNA Extraction from Single Insects” (10x Genomics, Pleasanton, CA, United States). The fragment size distributions and DNA concentrations were assessed using TapeStation (Agilent Technologies, Santa Clara, CA, United States) and Qubit Fluorometer measurements using the DNA BR kit (Thermo Fisher Scientific, Waltham, MA, United States).
2.2 DNA sequencing data
The Illumina sequencing provider (BGI Hong Kong) handed over already filtered, so-called clean reads in eight pairs. These paired-end read files were adapter-trimmed using autotrim 0.6.1 (Waldvogel et al., 2018) and its dependencies FastQC, Trimmomatic 0.39 (Bolger et al., 2014), and MultiQC (Ewels et al., 2016). After a quality-check, one file pair was additionally cropped to 140 bp in length using Trimmomatic 0.39. All trimmed reads were combined into one forward, one reverse (both paired-end), and one unpaired fastq file. Illumina reads were classified in Kraken 2 (paired-end files with the additional option-paired) using a customized database consisting of the Kraken 2 databases “bacteria,” “archaea,” “human,” and “UniVec-Core”.
MinION library preparation followed the manufacturer’s protocol for the 1D-ligation kit (SQK-LSK109) of Oxford Nanopore Technologies (ONT). In total, eight flow cells in three runs were used. ONT-basecalling from fast5 files was conducted with Guppy 3.4.5 (available via registering at https://nanoporetech.com/support) using default settings and the following specifications: the flowcell ID, the name of the kit used for library preparation (SQK-LSK109), and the device (device auto). For the single female species, one run on the PacBio Sequel II in CCS mode was performed. The Guppy-basecalling includes adapter trimming and Q-score-filtering.
2.3 RNA sequencing
For RNA extractions, 100 eggs, 15 L2 larvae, eight L4 larvae, four pupae, and two adult male and two adult female species were used (Figure 1A). Tissue samples were collected in TRIzol and extracted using the Zymo RNA Kit (Zymo Research). Eggs, larvae, and pupae were pooled for producing an immature pool. The fragment size distributions and RNA concentrations per pool were assessed using TapeStation (Agilent Technologies) and a Qubit Fluorometer with the Qubit RNA HS kit measurements (Thermo Fisher Scientific). Library construction and sequencing on a BGISEQ-500 Illumina platform were carried out at BGI Hong Kong. Raw RNA Illumina reads were quality-checked and adapter-trimmed using autotrim 0.6.1 (Waldvogel et al., 2018) and its dependencies FastQC, Trimmomatic 0.39 (Bolger et al., 2014), and MultiQC (Ewels et al., 2016). HISAT2 (Kim et al., 2019) was used to map the RNA sequencing reads to the genome assembly.
2.4 Mitochondrial genome
Raw PacBio circular consensus sequencing (CCS) reads with adapters were used in NOVOPlasty 4.2 (Dierckxsens et al., 2016) to assemble the mitochondrion of Ae. j. japonicus. For annotations, GeSeq (Tillich et al., 2017) and MITOS2 Galaxy 2.0.6 (Al Arab et al., 2017; Donath et al., 2019) were used. Using Geneious Prime 2021.2.2 (Biomatters Limited), the origin was manually set, the sequence was circularized, and the annotations were curated manually.
2.5 Genome size estimations
We used two in silico genome size estimation methods based on k-mers and read mapping. Jellyfish 2.3.0 (Marçais and Kingsford, 2011) was used to count k-mers in the Ae. j. japonicus Illumina paired-end reads processed by Kraken 2 v2.0.8 (Wood et al., 2019), which were returned as unclassified. The online version of GenomeScope 2.0 (Ranallo-Benavidez et al., 2020) was used to estimate a k-mer-based genome size (Supplementary Figure S1). backmap.pl v0.5 (Schell et al., 2017; Pfenninger et al., 2022) (dependencies: bwa 0.7.17-r1188, minimap 2 2.29-r1283, samtools 1.20, qualimap 2.2.1, bedtools 2.28.0, and multiqc 1.9) was used to estimate the fraction of the assembled reads via the mapping rate and for genome size estimation with the ModEst method (Pfenninger et al., 2022).
Flow cytometry was used as a sequencing-free method for genome size estimation. Genome sizes for Ae. j. japonicus and Ae. koreicus were estimated following a flow cytometry protocol with propidium iodide-stained nuclei (Hare and Johnston, 2012) using the modification of the method proposed by Männer et al. (2024). We included Ae. koreicus here because no flow cytometric genome size estimate exists for this species (Supplementary Table S1). One whole adult mosquito was used per suspension and chopped with a razor blade in a Petri dish. Two adults per species (one male and one female each, collected as sympatrically occurring pupae on the graveyard Wiesbaden–Kloppenheim on 27 May 2025, and lab-reared to adults) were measured on three consecutive days to minimize instrumental errors.
2.6 Genome assembly, scaffolding, and gap closing
A de novo genome was assembled with PacBio CCS reads with the Flye 2.8 assembler (Kolmogorov et al., 2019). We identified the mitochondrial sequence in the Flye assembly using blast 2.10.0 (Altschul et al., 1990), and the respective contigs (>90% target sequence identity and all blast hits per contig >70% contig length) were removed to ensure that the mitochondrion was removed but nuclear mitochondrial DNA segments (NUMTs) were retained in the nuclear genome.
Subsequently, several rounds of scaffolding and gap closing were conducted (Supplementary Figure S2): The MinION long reads were used to scaffold the Flye assembly using SLR (Luo, 2014). TGS-GapCloser 1.0.1 (Xu et al., 2020) was applied to close gaps by first using the PacBio CCS reads and then the constructed continuous long reads (“CLR” reads) together with Illumina reads. The latter were used for polishing the newly added “CLR”-gap sequence inside TGS-GapCloser. “CLR” reads are all PacBio subreads, which were not involved in the generation of a CCS read. They were filtered for the longest per zero-mode waveguide. After this sequence extension, SSPACE (Boetzer et al., 2011) was used to re-scaffold using the “CLR” reads, followed by another two-step gap closing with TGS-GapCloser using CCS reads and “CLR” and Illumina reads, as described above. This workflow allowed the incorporation of all the generated sequencing data (MinION long reads, Illumina short reads, and PacBio CCS reads) into the genome assembly (Supplementary Figure S2).
Every step of the genome assembly was evaluated regarding quality using QUAST 5.0.2 (Gurevich et al., 2013) and regarding completeness using BUSCO 5.4.6 with the diptera_odp10 gene set in the genome mode. The process of gap closing and scaffolding (Supplementary Figure S2) was checked to ensure no reduction in the quality of the resulting assembly.
2.7 Structural annotation
A reference-based annotation of the Ae. j. japonicus genome was produced using the GeMoMa 1.9 software (Keilwagen et al., 2019), own RNA sequencing data, and the Ae. albopictus and Ae. aegypti annotations for reference (GCF_035046485.1; GCF_002204515.2). The annotation of Ae. koreicus (GCA_024533555.2) was additionally included as a third reference in a second GeMoMa run (Supplementary Table S3).
In addition, an annotation with BRAKER 3.0.3 (Stanke et al., 2008; Li et al., 2009; Barnett et al., 2011; Lomsadze et al., 2014; Buchfink et al., 2015; Hoff et al., 2016; Brůna et al., 2021) with RNA sequencing data as evidence was computed.
BRAKER and GeMoMa annotations for Ae. j. japonicus were compared regarding contiguity statistics that were calculated with a custom script by author TS (named “contiguity statistics” in Table 1, Supplementary Tables S2, S3) and regarding BUSCO 5.4.6 statistics using the protein sequences as input (Supplementary Table S2). Complete and single-copy BUSCO gene IDs unique to the GeMoMa annotation were extracted and merged with the BRAKER annotation’s BUSCO IDs using gff-merge and gff3_to_fasta of the GFF3toolkit 2.1.0 (Chen et al., 2019). Since the merging did not improve the BRAKER annotation substantially (Supplementary Figure S4; Supplementary Table S2), the latter alone was used for subsequent analyses.
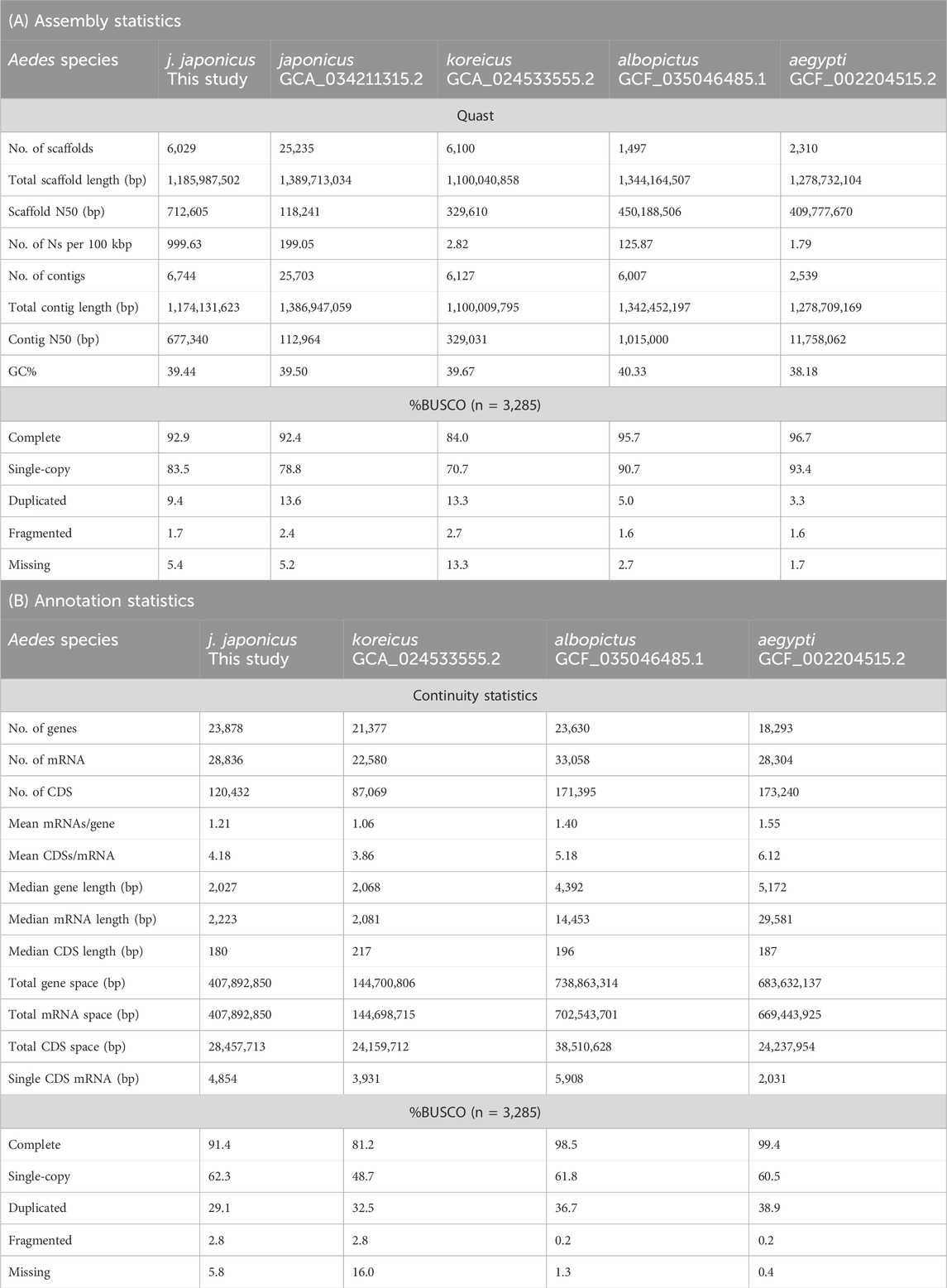
Table 1. Genome assembly (A) and annotation statistics (B) of selected Aedes spp. genomes. Calculations of contiguity statistics by a custom script. CDS: coding exon regions. Total gene space: sum of all nucleotides that are annotated as a gene. Single CDS mRNA: number of mRNAs that only have a single coding exon.
2.8 Functional annotation and detection of integrated virus sequences
InterProScan 5.61.93 (Jones et al., 2014) with the options [-f tsv -iprlookup -pa -goterms -dp -cpu 54] and blastp 2.14.0 with options [-num_threads 70 -max_hsps 1 -max_target_seqs 1 -outfmt 6] were run against the Swiss-Prot database (The UniProt Consortium et al., 2025); Pannzer2 web version (Törönen and Holm, 2022) and GhostKOALA web version (Kanehisa et al., 2016) were run to functionally annotate the amino acid file of the Ae. j. japonicus BRAKER annotation and the annotations of Ae. albopictus and Ae. aegypti for comparison (Supplementary Table S4; Supplementary Figure S5).
Integration of viral sequences was checked using a published database for endogenous viral elements (Palatini et al. 2020; their additional file 4) identified (tblastn 2.14.0 with options [-max_hsps 1 -max_target_seqs 1 -outfmt 6]; Altschul et al., 1990) in the respective Aedes amino acid files (Supplementary Table S4; Supplementary Figure S5).
3 Data analysis
3.1 Mitochondrion
The mitochondrial genome is available under the GenBank accession-number MZ566802 and NCBI accession-number NC_081591.1. The total length is 16,848 bp. As of 25 June 2025, seven additional complete mitochondrial sequences of the species are available (OP373191.1, OR668893-4.1, PQ588181.1, and PV094741-3.1), generated from mosquitoes originating from Italy, Germany, the Netherlands, and Hawaii, USA. Thus, this is the first Ae. j. japonicus mitochondrion from the species’ native range (Japan).
3.2 Assembly and genome size estimates
An Ae. j. japonicus assembly was obtained with a total length of 1.2 Gb, a contig N50 of 677 kb, a scaffold N50 of 712 kb, and 6,029 scaffolds (Figure 1B; Table 1A). The BUSCO protein set was 92.9% complete, with only 1.7% fragmented BUSCOs (Figure 1B). Flow cytometric genome size estimates were 1.3 Gb for Ae. j. japonicus as well as for Ae. koreicus (Supplementary Table S1). The latter is in line with the size of the Ae. koreicus genome (1.1 Gb; Supplementary Table S1; Nagy et al., 2024). The k-mer-based estimate of Ae. j. japonicus was 695 Mb in length, and the mapping-based estimate was the best performing, regarding peak shape, with mapped CCS reads. The mapping-based genome size estimate was 1.2 Gb (Supplementary Figure S3). This compilation of genome size estimates can facilitate calculations for genome coverage and sequencing costs for further projects.
3.3 Structural and functional annotations
The annotation with BRAKER resulted in 23,878 predicted protein-coding genes with a median length of 2,027 bp. Protein sequences of the predicted genes showed a BUSCO completeness of 91.4% (Table 1B). Among the protein-coding genes, 99% (28,458 genes) could be functionally annotated with at least one of the applied methods, but GO terms could be found for 60% of the sequences (Supplementary Table S4).
3.4 Comparisons to other Aedes genomes
The size of the nuclear genome assembly of Ae. j. japonicus is comparable to those of other genomes within Aedes (Supplementary Table S1). The Ae. j. japonicus assembly has slightly better statistics than the publicly available assembly (GCA_034211315.2) regarding continuity and BUSCO completeness (Table 1A). The GC content is the same as in the GCA_034211315.2 assembly and comparable to the sister species Ae. koreicus (Table 1A). For the three Aedes species, a comparable number (60%–70%) of integrated virus sequences could be detected (Supplementary Table S4; Supplementary Figure S5). The slightly lower number of viruses that could be recovered in the Ae. japonicus annotation is explainable by the lower quality of the scaffold-level Ae. j. japonicus genome compared to that of the chromosome-level genomes of Ae. albopictus and Ae. aegypti or the selection of the input virus database. A biological reason could be the species-specificity of viral integrations.
4 Dataset usage and availability
4.1 Dataset re-use potential
The dataset presented here can be used in subsequent analyses regarding phylogeny, evolution of diapause and invasiveness, adaptation to non-native habitats, and the search for genetic targets of vector control measures. It is the first time that individuals from the native range of Ae. j. japonicus were sequenced (nuclear and mitochondrial genomes), allowing comparative studies regarding differences between native and invasive populations of the species. Differences could occur due to the adaptation to the new environment during the invasion process. Important phenotypic traits such as diapause, heat tolerance, or insecticide resistance could be altered during invasion. The dataset presented here also fills a gap of knowledge regarding comparative studies between well-studied primary (Ae. aegypti and Ae. albopictus) and understudied secondary (Ae. j. japonicus and Ae. koreicus) vector species regarding their different competences for arboviral transmission.
Data availability statement
All datasets are available in publicly accessible repositories: This project was registered under the BioProject number PRJNA1085103 at NCBI https://www.ncbi.nlm.nih.gov/. The mitochondrial genome is available under GenBank accession-number MZ566802 https://www.ncbi.nlm.nih.gov/. The genome annotation with the corresponding assembly and the amino acid sequence file can be found at the Goethe University Data Repository (GUDe; https://gude.uni-frankfurt.de/home) under the DOI (https://doi.org/10.25716/gude.12xf-dt1*).
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because the research was carried out with samples of unregulated invertebrate animals (insects).
Author contributions
FR: Formal Analysis, Writing – original draft, Data curation, Visualization, Conceptualization, Validation, Methodology, Investigation. TS: Writing – review and editing, Supervision, Formal Analysis, Methodology, Validation, Software, Visualization, Conceptualization, Data curation. HI: Writing – review and editing, Methodology, Investigation. ShK: Supervision, Writing – review and editing, Resources. SvK: Funding acquisition, Resources, Writing – review and editing. RM: Resources, Writing – review and editing, Funding acquisition. MP: Resources, Writing – review and editing, Supervision, Funding acquisition. JK: Conceptualization, Writing – review and editing, Project administration, Visualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The present study was supported by the Center for Translational Biodiversity Genomics (LOEWE-TBG) through the program ‘LOEWE-Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz’ of Hesse’s Ministry of Higher Education, Research, and Arts. This work was supported by the Hessian Center on Climate Change and Adaptation (FZK) of the Hessian Agency for Nature Conservation, Environment, and Geology (HLNUG). The publication of this article was funded by the Open Access Publication Fund of Goethe University Frankfurt am Main.
Acknowledgments
The authors thank Charlotte Gerheim for flow cytometric measurements, Damian Baranski for help with DNA MinION long-read runs, and Carola Greve for supervision and logistical support (all associated with Senckenberg Research Institute, Lab Center for Biodiversity Genomics, Frankfurt, Germany).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1667262/full#supplementary-material
References
Al Arab, M., Höner Zu Siederdissen, C., Tout, K., Sahyoun, A. H., Stadler, P. F., and Bernt, M. (2017). Accurate annotation of protein-coding genes in mitochondrial genomes. Mol. Phylogenetics Evol. 106, 209–216. doi:10.1016/j.ympev.2016.09.024
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi:10.1016/S0022-2836(05)80360-2
Armbruster, P. A. (2016). Photoperiodic diapause and the establishment of Aedes albopictus (Diptera: culicidae) in North America. J. Med. Entomol. 53, 1013–1023. doi:10.1093/jme/tjw037
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P., and Marth, G. T. (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692. doi:10.1093/bioinformatics/btr174
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi:10.1093/bioinformatics/btq683
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi:10.1093/bioinformatics/btu170
Boyle, J. H., Rastas, P. M. A., Huang, X., Garner, A. G., Vythilingam, I., and Armbruster, P. A. (2021). A linkage-based genome assembly for the mosquito Aedes albopictus and identification of chromosomal regions affecting diapause. Insects 12, 167. doi:10.3390/insects12020167
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M., and Borodovsky, M. (2021). BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinforma. 3, lqaa108. doi:10.1093/nargab/lqaa108
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi:10.1038/nmeth.3176
Catapano, P. L., Falcinelli, M., Damiani, C., Cappelli, A., Koukouli, D., Rossi, P., et al. (2023). De novo genome assembly of the invasive mosquito species Aedes japonicus and Aedes koreicus. Parasites Vectors 16, 427. doi:10.1186/s13071-023-06048-w
Chen, M.-J. M., Lin, H., Chiang, L.-M., Childers, C. P., and Poelchau, M. F. (2019). The GFF3toolkit: QC and merge pipeline for genome annotation. Insect Genomics 1858, 75–87. doi:10.1007/978-1-4939-8775-7_7
Couper, L. I., Dodge, T. O., Hemker, J. A., Kim, B. Y., Exposito-Alonso, M., Brem, R. B., et al. (2025). Evolutionary adaptation under climate change: aedes sp. demonstrates potential to adapt to warming. Proc. Natl. Acad. Sci. U.S.A. 122, e2418199122. doi:10.1073/pnas.2418199122
Cunze, S., Kochmann, J., and Klimpel, S. (2020). Global occurrence data improve potential distribution models for Aedes japonicus japonicus in non-native regions. Pest Manag. Sci. 76, 1814–1822. doi:10.1002/ps.5710
Dierckxsens, N., Mardulyn, P., and Smits, G. (2016). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi:10.1093/nar/gkw955
Donath, A., Jühling, F., Al-Arab, M., Bernhart, S. H., Reinhardt, F., Stadler, P. F., et al. (2019). Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47, 10543–10552. doi:10.1093/nar/gkz833
European Centre for Disease Prevention and Control and European Food Safety Authority (2023). Mosquito maps. Stockholm: ECDC. Available online at: https://ecdc.europa.eu/en/disease-vectors/surveillance-and-disease-data/mosquito-maps.
Ewels, P., Magnusson, M., Lundin, S., and Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. doi:10.1093/bioinformatics/btw354
Gjenero-Margan, I., Aleraj, B., Krajcar, D., Lesnikar, V., Klobučar, A., Pem-Novosel, I., et al. (2011). Autochthonous dengue fever in Croatia, August–September 2010. Eurosurveillance 16, 19805. doi:10.2807/ese.16.09.19805-en
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi:10.1093/bioinformatics/btt086
Hare, E. E., and Johnston, J. S. (2012). “Genome size determination using flow cytometry of propidium iodide-stained nuclei,” in Molecular methods for evolutionary genetics. Editors V. Orgogozo, and M. V. Rockman (Totowa, NJ: Humana Press), 3–12. doi:10.1007/978-1-61779-228-1_1
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M., and Stanke, M. (2016). BRAKER1: unsupervised RNA-Seq-Based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769. doi:10.1093/bioinformatics/btv661
Hoshino, K., Isawa, H., Tsuda, Y., and Kobayashi, M. (2010). Laboratory colonization of Aedes japonicus japonicus (Diptera: culicidae) collected in Narita, Japan and the biological properties of the established colony. Jpn. J. Infect. Dis. 63, 401–404. doi:10.7883/yoken.63.401
Jansen, C. C., and Beebe, N. W. (2010). The dengue vector Aedes aegypti: what comes next. Microbes Infect. 12, 272–279. doi:10.1016/j.micinf.2009.12.011
Jansen, S., Heitmann, A., Lühken, R., Jöst, H., Helms, M., Vapalahti, O., et al. (2018). Experimental transmission of Zika virus by Aedes japonicus japonicus from southwestern Germany. Emerg. Microbes & Infect. 7, 192–196. doi:10.1038/s41426-018-0195-x
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi:10.1093/bioinformatics/btu031
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi:10.1016/j.jmb.2015.11.006
Kaufman, M. G., and Fonseca, D. M. (2014). Invasion biology of Aedes japonicus japonicus (Diptera: culicidae). Annu. Rev. Entomol. 59, 31–49. doi:10.1146/annurev-ento-011613-162012
Keilwagen, J., Hartung, F., and Grau, J. (2019). “GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data,” in Gene prediction. Editor M. Kollmar (New York, NY: Springer New York), 161–177. doi:10.1007/978-1-4939-9173-0_9
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi:10.1038/s41587-019-0201-4
Koban, M. B., Kampen, H., Scheuch, D. E., Frueh, L., Kuhlisch, C., Janssen, N., et al. (2019). The Asian bush mosquito Aedes japonicus japonicus (Diptera: culicidae) in Europe, 17 years after its first detection, with a focus on monitoring methods. Parasites Vectors 12, 109. doi:10.1186/s13071-019-3349-3
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi:10.1038/s41587-019-0072-8
Kramer, I. M., Pfenninger, M., Feldmeyer, B., Dhimal, M., Gautam, I., Shreshta, P., et al. (2023). Genomic profiling of climate adaptation in Aedes aegypti along an altitudinal gradient in Nepal indicates nongradual expansion of the disease vector. Mol. Ecol. 32, 350–368. doi:10.1111/mec.16752
Kreß, A., Kuch, U., Oehlmann, J., and Müller, R. (2016). Effects of diapause and cold acclimation on egg ultrastructure: new insights into the cold hardiness mechanisms of the Asian tiger mosquito Aedes (Stegomyia) albopictus. J. Vector Ecol. 41, 142–150. doi:10.1111/jvec.12206
Krupa, E., Henon, N., and Mathieu, B. (2021). Diapause characterisation and seasonality of Aedes japonicus japonicus (Diptera, Culicidae) in the northeast of France. Parasite 28, 45. doi:10.1051/parasite/2021045
Lahondère, C., and Bonizzoni, M. (2022). Thermal biology of invasive Aedes mosquitoes in the context of climate change. Curr. Opin. Insect Sci. 51, 100920. doi:10.1016/j.cois.2022.100920
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Lomsadze, A., Burns, P. D., and Borodovsky, M. (2014). Integration of mapped RNA-seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119. doi:10.1093/nar/gku557
Lounibos, L. P. (2002). Invasions by insect vectors of human disease. Annu. Rev. Entomol. 47, 233–266. doi:10.1146/annurev.ento.47.091201.145206
Luo, J. (2014). SLR. Available online at: https://github.com/luojunwei/SLR.
Männer, L., Schell, T., Spies, J., Galià-Camps, C., Baranski, D., Ben Hamadou, A., et al. (2024). Chromosome-level genome assembly of the sacoglossan sea slug Elysia timida (Risso, 1818). BMC Genomics 25, 941. doi:10.1186/s12864-024-10829-7
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi:10.1093/bioinformatics/btr011
Matthews, B. J., Dudchenko, O., Kingan, S. B., Koren, S., Antoshechkin, I., Crawford, J. E., et al. (2018). Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 563, 501–507. doi:10.1038/s41586-018-0692-z
Medlock, J. M., Hansford, K. M., Schaffner, F., Versteirt, V., Hendrickx, G., Zeller, H., et al. (2012). A review of the invasive mosquitoes in Europe: ecology, public health risks, and control options. Vector-Borne Zoonotic Dis. 12, 435–447. doi:10.1089/vbz.2011.0814
Müller, R., Bálint, M., Hardes, K., Hollert, H., Klimpel, S., Knorr, E., et al. (2023). RNA interference to combat the Asian tiger mosquito in Europe: a pathway from design of an innovative vector control tool to its application. Biotechnol. Adv. 66, 108167. doi:10.1016/j.biotechadv.2023.108167
Nagy, N. A., Tóth, G. E., Kurucz, K., Kemenesi, G., and Laczkó, L. (2024). The updated genome of the Hungarian population of Aedes koreicus. Sci. Rep. 14, 7545. doi:10.1038/s41598-024-58096-6
Palatini, U., Masri, R. A., Cosme, L. V., Koren, S., Thibaud-Nissen, F., Biedler, J. K., et al. (2020). Improved reference genome of the arboviral vector Aedes albopictus. Genome Biol. 21, 215. doi:10.1186/s13059-020-02141-w
Pfenninger, M., Schönnenbeck, P., and Schell, T. (2022). ModEst: accurate estimation of genome size from next generation sequencing data. Mol. Ecol. Resour. 22, 1454–1464. doi:10.1111/1755-0998.13570
Ranallo-Benavidez, T. R., Jaron, K. S., and Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432. doi:10.1038/s41467-020-14998-3
Rezza, G., Nicoletti, L., Angelini, R., Romi, R., Finarelli, A., Panning, M., et al. (2007). Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet 370, 1840–1846. doi:10.1016/S0140-6736(07)61779-6
Schell, T., Feldmeyer, B., Schmidt, H., Greshake, B., Tills, O., Truebano, M., et al. (2017). An annotated draft genome for radix auricularia (Gastropoda, Mollusca). Genome Biol. Evol. 9, 0–592. doi:10.1093/gbe/evx032
Smitz, N., De Wolf, K., Deblauwe, I., Kampen, H., Schaffner, F., De Witte, J., et al. (2021). Population genetic structure of the Asian bush mosquito, Aedes japonicus (Diptera, Culicidae), in Belgium suggests multiple introductions. Parasites Vectors 14, 179. doi:10.1186/s13071-021-04676-8
Sousa, C. A., Clairouin, M., Seixas, G., Viveiros, B., Novo, M. T., Silva, A. C., et al. (2012). Ongoing outbreak of dengue type 1 in the autonomous region of madeira, Portugal: preliminary report. Eurosurveillance 17, 20333. doi:10.2807/ese.17.49.20333-en
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi:10.1093/bioinformatics/btn013
Succo, T., Leparc-Goffart, I., Ferré, J.-B., Roiz, D., Broche, B., Maquart, M., et al. (2016). Autochthonous dengue outbreak in Nîmes, south of France, July to September 2015. Eurosurveillance 21. doi:10.2807/1560-7917.ES.2016.21.21.30240
The UniProt Consortium, , Bateman, A., Martin, M.-J., Orchard, S., Magrane, M., Adesina, A., et al. (2025). UniProt: the universal protein knowledgebase in 2025. Nucleic Acids Res. 53, D609–D617. doi:10.1093/nar/gkae1010
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi:10.1093/nar/gkx391
Törönen, P., and Holm, L. (2022). PANNZER —A practical tool for protein function prediction. Protein Sci. 31, 118–128. doi:10.1002/pro.4193
Verkuijl, S. A. N., Del Corsano, G., Capriotti, P., Yen, P.-S., Inghilterra, M. G., Selvaraj, P., et al. (2025). A suppression-modification gene drive for malaria control targeting the ultra-conserved RNA gene mir-184. Nat. Commun. 16, 3923. doi:10.1038/s41467-025-58954-5
Wagner, S., Mathis, A., Schönenberger, A. C., Becker, S., Schmidt-Chanasit, J., Silaghi, C., et al. (2018). Vector competence of field populations of the mosquito species Aedes japonicus japonicus and Culex pipiens from Switzerland for two west nile virus strains. Med. Vet. Entomol. 32, 121–124. doi:10.1111/mve.12273
Waldvogel, A., Wieser, A., Schell, T., Patel, S., Schmidt, H., Hankeln, T., et al. (2018). The genomic footprint of climate adaptation in Chironomus Riparius. Mol. Ecol. 27, 1439–1456. doi:10.1111/mec.14543
Wang, G.-H., Hoffmann, A., and Champer, J. (2025). Gene drive and symbiont technologies for control of mosquito-borne diseases. Annu. Rev. Entomology 70, 229–249. doi:10.1146/annurev-ento-012424-011039
Wilkerson, R. C., Linton, Y.-M., Fonseca, D. M., Schultz, T. R., Price, D. C., and Strickman, D. A. (2015). Making mosquito taxonomy useful: a stable classification of tribe aedini that balances utility with current knowledge of evolutionary relationships. PLoS ONE 10, e0133602. doi:10.1371/journal.pone.0133602
Wittwer, C., Sharif, C., Schöck, I., and Klimpel, S. (2024). Mosquitoes on a chip—environmental DNA-based detection of invasive mosquito species using high-throughput real-time PCR. PeerJ 12, e17782. doi:10.7717/peerj.17782
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. doi:10.1186/s13059-019-1891-0
Xu, M., Guo, L., Gu, S., Wang, O., Zhang, R., Peters, B. A., et al. (2020). TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9, giaa094. doi:10.1093/gigascience/giaa094
Keywords: complete mitochondrial sequence, Aedes, invasive mosquitoes, disease vector, reference genome
Citation: Reuss F, Schell T, Isawa H, Kasai S, Klimpel S, Müller R, Pfenninger M and Kochmann J (2025) Annotated genome of Aedes japonicus japonicus using a hybrid-assembly approach. Front. Genet. 16:1667262. doi: 10.3389/fgene.2025.1667262
Received: 16 July 2025; Accepted: 08 September 2025;
Published: 01 October 2025.
Edited by:
Robert M. Waterhouse, SIB Swiss Institute of Bioinformatics, SwitzerlandReviewed by:
Rohit Kumar, Helmholtz Association of German Research Centers (HZ), GermanyNikoletta Andrea Nagy, University of Debrecen, Hungary
Copyright © 2025 Reuss, Schell, Isawa, Kasai, Klimpel, Müller, Pfenninger and Kochmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friederike Reuss, Zi5yZXVzc0BtZWQudW5pLWZyYW5rZnVydC5kZQ==
†Present address: Judith Kochmann, Institute of Organismic and Molecular Evolution (iomE), Johannes Gutenberg University, Mainz, Germany