 Ekaterina Nuzhnaya1*
Ekaterina Nuzhnaya1* Tatiana Cherevatova1Ekaterina Lotnik1Aleksandra Shagiazdanova2Zhanna Markova1Ekaterina Filimonova2
Tatiana Cherevatova1Ekaterina Lotnik1Aleksandra Shagiazdanova2Zhanna Markova1Ekaterina Filimonova2 Olga Parshina3
Olga Parshina3 Anastasiia Buianova3Anastasia Bobreshova1
Anastasiia Buianova3Anastasia Bobreshova1 Andrey Marakhonov1Natalia Semenova1
Andrey Marakhonov1Natalia Semenova1- 1Research Centre for Medical Genetics, Moscow, Russian Federation, Moscow, Russia
- 2Russian Children’s Clinical Hospital, A Branch of Pirogov Russian National Research Medical University, Moscow, Russia
- 3Institute of Translational Medicine, Pirogov Russian National Research Medical University, Moscow, Russia
We report a pediatric case of cholestatic liver disease associated with two novel compound heterozygous variants in the USP53 gene: a truncating c.1219A>T (p.Lys407*) variant inherited from the father and a maternally inherited gross deletion involving exons 13–19. Despite the disruptive nature of these variants, the patient presented with a benign recurrent intrahepatic cholestasis (BRIC) characterized by episodic pruritus, jaundice and elevated bile acids with preserved liver function between episodes. Liver histology revealed fibrosis with a cholestatic component, consistent with mild progressive familial intrahepatic cholestasis (PFIC) features. Molecular diagnosis was confirmed by whole-exome sequencing (WES), chromosomal microarray, and Sanger sequencing. A systematic review of 39 published cases was conducted, revealing that USP53-related disease exhibits broad clinical variability, ranging from BRIC to PFIC7. Our findings expand the spectrum of USP53 variants, underscore the relevance of large deletions and emphasize the inclusion of USP53 in genetic panels for idiopathic low-gamma-glutamyl transferase (GGT) cholestasis.
Introduction
PFIC is a rare group of autosomal recessive liver diseases characterized by early-onset cholestasis, pruritus, and progressive liver dysfunction, often leading to transplantation (Alam et al., 2024; Zheng et al., 2023). Recently, biallelic variants in USP53 have been identified as a cause of PFIC7, expanding the spectrum of cholestatic disorders (Maddirevula et al., 2019).
Although USP53 encodes a catalytically inactive deubiquitinase, it serves an essential structural role in maintaining the integrity of tight junctions through its interactions with TJP1 (ZO-1) and TJP2 (ZO-2). Both TJP1 and TJP2 are scaffolding proteins that organize and stabilize tight junction complexes by linking transmembrane components such as claudins and occludins to the actin cytoskeleton, thereby regulating paracellular permeability and epithelial polarity. Disruption of USP53 interferes with its association with these proteins, leading to defective tight junction assembly, weakening of the canalicular membranes in hepatocytes, and subsequent cholestasis. In addition, because tight junctions formed by TJP1 and TJP2 are also critical for maintaining ionic homeostasis and barrier integrity in cochlear epithelial cells, USP53 dysfunction can impair cochlear tight junctions, providing a mechanistic explanation for the sensorineural hearing loss observed in affected individuals (Xia et al., 2024).
Clinically, USP53-related disease often presents with a milder, benign recurrent intrahepatic cholestasis (BRIC)-like phenotype, featuring episodic jaundice and pruritus with slow disease progression. Hearing loss has been inconsistently reported (Alhebbi et al., 2020; Zhang et al., 2020).
To date, only two patients have been described with сopy number variation (CNV) in USP53 (Bull et al., 2020; Cheema et al., 2023). Here, we report a unique case of a patient with BRIC-like presentation carrying a novel nonsense variant and the largest reported USP53 deletion (exons 13–19). We also review previously reported PFIC7 cases to highlight the phenotypic variability and diagnostic importance of USP53 variants.
Case presentation
A 16-year-old male patient was referred from the Department of Gastroenterology at the Russian Children’s Clinical Hospital with complaints of pruritus and abnormal blood test results.
He is the only child of his parents’ current marriage. His mother has a healthy son from a previous marriage. There are no known familial cases of similar disease, and the parents are non-consanguineous. The patient was born full term with a birth weight of 4000 g and a length of 53 cm. Apgar scores were 8 and 9. The perinatal period was unremarkable.
At 5 months of age, the patient developed pruritus and jaundice. Laboratory testing revealed hyperbilirubinemia, with total bilirubin reaching 190 μmol/L and direct bilirubin 160 μmol/L, without signs of cytolysis. Liver biopsy demonstrated features of giant cell hepatitis with fibrosis (Ishak stage 3, grade 7). A follow-up biopsy 1 year later revealed chronic hepatitis with porto-central fibrosis and progression to cirrhosis. Concomitantly, cytomegalovirus infection (CMV) was diagnosed: IgM 0.2, IgG 56, avidity 82%, and CMV DNA detected by PCR.
At the age of 1 year, during a planned hospitalization, liver biochemistry revealed the following: alanine aminotransferase (ALT) 72 U/L, aspartate aminotransferase (AST) 80 U/L, alkaline phosphatase (ALP) 352 U/L, GGT27 U/L, total bilirubin 41 μmol/L, direct bilirubin 32.5 μmol/L (78% of total), total protein 65 g/L, glucose 4.06 mmol/L, cholesterol 2.55 mmol/L, and albumin 37.36 g/L. The patient was followed with a diagnosis of chronic hepatitis, presumably of CMV etiology. From 1.5 years of age, liver function tests were monitored regularly, and transaminase and bilirubin levels remained within normal ranges. At age 3, a transient episode of hyperbilirubinemia (total 43 μmol/L, direct 16 μmol/L) resolved spontaneously. From that point until age 14, he was monitored annually by a gastroenterologist and no evidence of cytolysis or cholestasis.
At age 14, following an episode of acute respiratory viral infection, the patient experienced recurrence of pruritus, jaundice, pale stools, dark urine, and weight loss. Biochemical testing showed a progressive increase in bilirubin levels (92, 133, 270, and 355 μmol/L), predominantly direct (81, 113, 221, and 292 μmol/L). Transaminase levels were mildly elevated (ALT 41 U/L, AST 49 U/L); GGT remained normal (27 U/L), ALP was 339 U/L, international normalized ratio (INR) was 1.2, and bile salts were elevated to 147 μmol/L. Abdominal ultrasound revealed hepatomegaly, cholecysto cholangitis, enlargement of the pancreatic head and splenomegaly. Magnetic resonance imaging (MRI) demonstrated hepatosplenomegaly, hypoplasia of the gallbladder, and a small cyst in the right kidney. Liver biopsy showed morphological features of fibrosis with a cholestatic component (F4 according to METAVIR). Administration of hepatoprotective therapy with ursodeoxycholic acid resulted in clinical improvement, with normalization of liver function tests and a decrease in bilirubin levels (total 35 μmol/L, direct 17.3 μmol/L).
No specific drug–disease interactions have been reported in patients with USP53-related cholestasis, and ursodeoxycholic acid is generally considered safe and well tolerated in cholestatic liver disease.
During the current hospitalization, viral and autoimmune hepatitis, alpha-1 antitrypsin deficiency, and Wilson’s disease were excluded. Ultrasound revealed hepatosplenomegaly, diffuse parenchymal changes in the liver, and signs of portal hypertension. Neurological consultation established a diagnosis of Adie-Holmes syndrome. Audiological testing showed normal hearing.
The patient was discharged with the following diagnoses: cryptogenic (congenital) cholestatic hepatitis, liver cirrhosis (F2, METAVIR), and portal hypertension syndrome.
On physical examination, the patient appeared asthenic. His height was 186 cm (standard deviation score (SDS +1.92), and his weight was 61 kg (SDS +0.01). Kyphosis was noted. Scleral icterus was present.
WES identified a previously unreported variant in exon 14 of the USP53 gene in a homozygous or hemizygous state: NM_001371395.1:c.1219A>T, resulting in a premature stop codon p. (Lys407Ter) in exon 14 as it is shown in Figures 1A,B (Robinson et al., 2022). This variant was inherited from the father and was not present in the Genome Aggregation Database (gnomAD v4.1.0). Coverage analysis of the sequencing data suggested the presence of a heterozygous deletion in USP53 on chromosome 4.

Figure 1. The pedigree and genetic testing of the proband and the results of sanger sequencing. (A) Pedigree showing the affected proband and their unaffected parents, both of whom are heterozygous carriers. (B) Sanger sequencing results demonstrating a hemizygous truncating USP53 variant in the proband inherited from the father, in whom it is present in the heterozygous state. (C) CMA showing a maternally inherited heterozygous 275 kb deletion at 4q26 involving exons 13–19 of USP53 and the terminal exon of PDE5A.
To confirm this deletion, chromosomal microarray analysis (CMA) was performed and revealed a microdeletion at 4q26 (GRCh38): chr4:119,265,361–119,541,176 (275 kb). This deletion encompasses exons 13–19 of the USP53 gene. It was maternally inherited, as confirmed by Sanger sequencing and pedigree analysis, as shown in Figure 1C. Large deletions removing the last exons, including the natural stop codon, are known to trigger non-stop decay (NSD) rather than nonsense-mediated decay (NMD), as the resulting transcript lacks a proper termination signal. In our case, the deletion removes the terminal exons without introducing a premature termination codon (PTC) upstream, resulting in an mRNA that lacks an in-frame stop codon. During translation, ribosomes read through into the 3′untranslated region (UTR) or beyond and stall at the mRNA’s end, which activates the NSD surveillance pathway to degrade such “non-stop” mRNAs and prevent the production of aberrant proteins. Importantly, this deletion also affects the USP53 catalytic domain, which is essential for its deubiquitinase activity. To date, this is the only CNV in the USP53 gene reported in the literature.
Notably, the deletion also includes the terminal exon 21 of the adjacent PDE5A gene, which encodes a cGMP-specific phosphodiesterase involved in smooth muscle regulation and intracellular signal transduction. Although PDE5A is broadly expressed and pharmacologically relevant, it has not been conclusively associated with any monogenic human disorder.
According to the gnomAD v4.1.0 database, PDE5A has a low pLI score of 0.01 and a moderately reduced observed/expected ratio for loss-of-function variants (o/e = 0.49, 95% CI: 0.35–0.70), indicating tolerance to heterozygous loss-of-function events. This suggests that PDE5A is unlikely to cause autosomal dominant disease when disrupted in a single allele.
According to the ACMG/AMP guidelines (Richards et al., 2015), the c.1219A>T variant in USP53 meets criteria PVS1 and PM2. Based on this evidence, it is classified as a pathogenic. This variant has been submitted to ClinVar as pathogenic by the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Pirogov Russian National Research Medical University on 13 April 2025 (ClinVar accession: RCV005002072.2; https://www.ncbi.nlm.nih.gov/clinvar/RCV005002072.2).
A maternally inherited heterozygous 275 kb deletion at 4q26 (GRCh38): chr4:119,265,361–119,541,176, affecting the second USP53 allele meets criteria PVS1 and PM2 and is classified as likely pathogenic. This deletion was also submitted to ClinVar by the Research Centre for Medical Genetics on 19 July 2025 (ClinVar accession: VCV004057271.1, https://www.ncbi.nlm.nih.gov/clinvar/variation/4057271/).
Together, two variants—USP53 c.1219A>T and the 4q26 deletion—located on opposite alleles and consistent with autosomal recessive inheritance, are considered the cause of the disease and support the diagnosis of PFIC 7.
Discussion
USP53 has been predicted to be a catalytically inactive deubiquitinase based on its sequence similarity to other members of the USP family. Structural studies have demonstrated that USP53 lacks a critical histidine residue within the catalytic triad, classifying it as a non-protease homolog within the USP family. Despite this predicted inactivity, experimental evidence indicates that USP53 performs important non-enzymatic functions. Studies in mouse models have revealed that USP53 regulates metabolism, bone remodeling, liver function, and hearing. In particular, USP53 contributes to the maintenance of tight junction integrity through interactions with TJP1 and TJP2, and its loss leads to canalicular membrane instability and cholestasis. Recent work by Xia et al. further summarized emerging evidence that, although catalytically inactive, USP53 exerts significant physiological and pathological roles across multiple tissues, acting as a novel regulatory protein and a potential diagnostic and therapeutic target in metabolic, skeletal, hepatic, and neoplastic disorders (Xia et al., 2024).
In this study we describe a patient with compound heterozygous USP53 variants, including a novel nonsense variant (p.Lys407Ter) and a large deletion encompassing exons 13–19. Despite the presumed pathogenic impact of these disruptive variants, the patient demonstrated a mild BRIC-like phenotype, characterized by intermittent episodes of pruritus and hyperbilirubinemia with stable liver function and no progression to hepatic failure. Histological evaluation revealed fibrosis, consistent with PFIC, but without inflammation or hepatocellular damage. Although CMV infection was detected serologically and by PCR, the serological profile indicated a past infection, and the liver histology lacked features typical of CMV hepatitis. Therefore, CMV was considered an incidental finding with no significant contribution to the liver pathology.
Given the unexpectedly benign course in this case, we conducted a comprehensive literature review to contextualize the variability in genotype and phenotype of USP53-related cholestasis. The review was performed in accordance with PRISMA guidelines in a simplified form. The PubMed database was systematically searched using the keywords “USP53” and “cholestasis”, covering all years available up to December 2024. Titles and abstracts were screened to identify reports describing human patients with pathogenic or likely pathogenic variants in USP53. Duplicates, review articles, experimental or animal studies, and papers lacking clinical or genetic data were excluded. After applying these criteria, 39 published cases were included for analysis. Clinically, cholestasis and hepatomegaly were nearly universal features across cases as it is shown in Figure 2B. In terms of disease course, BRIC-like episodes were described in 17.7% (6/34) of patients, while intermittent pruritus with mildly elevated transaminases was noted in 26.5% (9/34). The majority (76.5%, 26/34) maintained normal or only mildly elevated liver enzyme levels during follow-up. Only one patient (2.9%) progressed to hepatic failure and another died 2 years after liver transplantation performed for intractable pruritus. Histological findings in USP53-related cholestasis commonly show canalicular cholestasis, ductular proliferation and fibrosis without inflammation. Detailed clinical outcomes and patient-level data are provided in the Table 1.
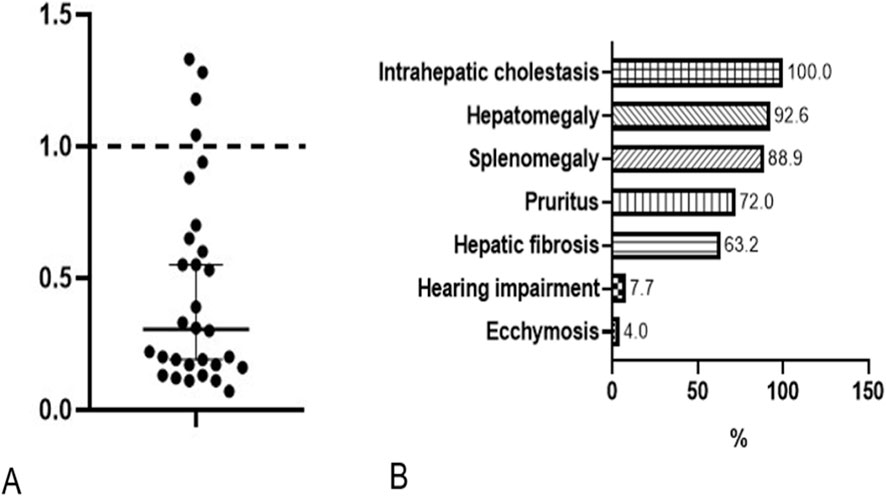
Figure 2. GGT ratio in all published patients (A). Frequency of phenotypic abnormalities, HPO terminology (B).

Table 1. Summary of clinical, biochemical, histological, imaging, genetic, and outcome data from 39 individuals with biallelic USP53 variants reported in the literature, including the current case. Data were extracted from 12 published sources and grouped to illustrate the phenotypic spectrum ranging from BRIC to PFIC-like disease.
PFIC7 is typically associated with normal or mildly elevated GGT, a key biochemical marker. To account for age- and sex-specific reference ranges, we calculated a GGT ratio for each individual by dividing the measured GGT value by the upper limit of normal for that person’s age and sex. Among 30 patients with USP53-related disease, 26 had GGT ratios ≤1.0 (normal), while 4 showed mildly elevated ratios not exceeding 1.5. These findings support the consistent low- or normal-GGT biochemical profile characteristic of this disorder (Figure 2A).
The mutation spectrum highlights that most reported variants are loss-of-function, including frameshift, nonsense, splicing defects, and gross deletions as shown in Figure 3. Missense variants were less common. Notably, gross deletions like the one identified in our case remain rare, documented in only a few reports to date.

Figure 3. Previously reported and novel protein-level variants in the USP53 gene. Legend. Schematic representation of 32 all previously reported pathogenic and likely pathogenic protein-level variants in the USP53 gene, along with two novel variants identified in the present case: a nonsense variant in exon 14 (p.Lys407*) and a heterozygous deletion encompassing exons 13–19. The exonic structure and variant positions are shown relative to the USP catalytic domain.
Although initially classified as PFIC7 in OMIM (2019), our review of 39 cases reveals that the majority exhibit a phenotype more typical of BRIC. Although periportal fibrosis was identified on liver biopsy, the overall clinical course—marked by episodic cholestasis, long asymptomatic intervals, and preserved hepatic function—remains consistent with a BRIC-like phenotype. These findings suggest that USP53-related disease may exist along a clinical spectrum, with histological features of PFIC but functional and clinical behavior resembling BRIC.
Despite carrying a gross deletion in USP53, our patient exhibited a BRIC-like phenotype with intermittent cholestasis and preserved liver function, supporting the notion that USP53-related disease spans a clinical spectrum, and even disruptive variants may present with mild courses.
A limitation of this study is the inclusion of a single patient, which restricts the generalizability of the findings. Nevertheless, the detailed clinical, genetic, and histological characterization contributes valuable information to the understanding of USP53-related cholestasis.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Local Ethics Committee of the Research Centre for Medical Genetics (approval number 11/23.11.2021, Moscow, Russia). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) and the individual(s)’ legal guardians/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
EN: Conceptualization, Data curation, Formal Analysis, Investigation, Project administration, Visualization, Writing – original draft, Methodology. TC: Investigation, Writing – review and editing. EL: Investigation, Validation, Writing – review and editing. AS: Data curation, Writing – review and editing. ZM: Investigation, Visualization, Writing – review and editing. EF: Data curation, Writing – review and editing. OP: Investigation, Writing – review and editing. ABu: Investigation, Writing – review and editing. ABo: Validation, Visualization, Writing – review and editing. AM: Conceptualization, Formal Analysis, Supervision, Validation, Writing – review and editing. NS: Conceptualization, Data curation, Formal Analysis, Methodology, Supervision, Writing – review and editing.
Funding
The authors declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahn, S., Choi, J., and Jeong, S. H. (2023). The first Korean adult case of progressive familial intrahepatic cholestasis type 7 with novel USP53 splicing variants by next generation sequencing. Yonsei Med. J. 64 (12), 745–749. doi:10.3349/ymj.2023.0161
Alam, S., Lal, B. B., Ravindranath, A., Bavdekar, A., Dheivamani, N., Snehavardhan, P., et al. (2024). Natural course and outcomes of children with ubiquitin-specific protease 53 (USP53)-related genetic chronic cholestasis. J. Pediatr. Gastroenterology Nutr. 79 (6), 1199–1208. doi:10.1002/jpn3.12392
Alhebbi, H., Peer-Zada, A. A., Al Hussaini, A. A., Algubaisi, S., Albassami, A., AlMasri, N., et al. (2020). New paradigms of USP53 disease: normal GGT cholestasis, BRIC, cholangiopathy, and responsiveness to rifampicin. J. Hum. Genet. 66 (2), 151–159. doi:10.1038/s10038-020-0811-1
Berberoğlu Ateş, B., Ceylan, A. C., Hızal, G., Duran, F., Doğan, H. T., and Hızlı, Ş. (2023). A novel homozygous mutation in the USP53 gene as the cause of benign recurrent intrahepatic cholestasis in children: a case report. Turkish J. Pediatr. 65 (6), 1012–1017. doi:10.24953/turkjped.2023.367
Bull, L. N., Ellmers, R., Foskett, P., Strautnieks, S., Sambrotta, M., Czubkowski, P., et al. (2020). Cholestasis due to USP53 deficiency. J. Pediatr. Gastroenterology Nutr. 72 (5), 667–673. doi:10.1097/MPG.0000000000002926
Cheema, H., Bertoli-Avella, A. M., Skrahina, V., Anjum, M. N., Waheed, N., Saeed, A., et al. (2020). Genomic testing in 1019 individuals from 349 Pakistani families results in high diagnostic yield and clinical utility. Npj Genomic Med. 5 (1), 44. doi:10.1038/s41525-020-00150-z
Cheema, H. A., Waheed, N., Anjum, S., Anjum, M. N., Fayyaz, Z., and Ijaz, S. (2023). The mutational landscape of genetic cholestatic diseases in Pakistani children. J. Pak. Med. Assoc. 73 (8), 1610–1621. doi:10.47391/jpma.7069
Gezdirici, A., Kalaycik Şengül, Ö., Doğan, M., Özgüven, B. Y., and Akbulut, E. (2022). Biallelic novel USP53 splicing variant disrupting the gene function that causes cholestasis phenotype and review of the literature. Mol. Syndromol. 13 (6), 471–484. doi:10.1159/000523937
Maddirevula, S., Alhebbi, H., Al-Qahtani, A., Algoufi, T., Alsaif, H. S., Ibrahim, N., et al. (2019). Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. 21 (5), 1164–1172. doi:10.1038/s41436-018-0288-x
Porta, G., Rigo, P. S. M., Porta, A., Pugliese, R. P. S., Danesi, V. L. B., Oliveira, E., et al. (2021). Progressive familial intrahepatic cholestasis associated with USP53 gene mutation in a Brazilian child. J. Pediatr. Gastroenterology Nutr. 72 (5), 674–676. doi:10.1097/MPG.0000000000003110
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Official J. Am. Coll. Med. Genet. 17 (5), 405–424. doi:10.1038/gim.2015.30
Robinson, J. T., Thorvaldsdottir, H., Turner, D., and Mesirov, J. P. (2022). “Igv.js: an embeddable JavaScript implementation of the integrative genomics viewer (IGV),”Bioinformatics. Editor C. Alkan, 39 1. btac830 doi:10.1093/bioinformatics/btac830
Samanta, A., Parveen, N., Sen Sarma, M., Poddar, U., and Srivastava, A. (2024). Cholestatic liver disease due to novel USP53 mutations: a case series of three Indian children. J. Clin. Exp. Hepatology 14 (2), 101290. doi:10.1016/j.jceh.2023.10.001
Shatokhina, O., Semenova, N., Demina, N., Dadali, E., Polyakov, A., and Ryzhkova, O. (2021). A two-year clinical description of a patient with a rare type of Low-GGT cholestasis caused by a novel variant of USP53. Genes 12 (10), 1618. doi:10.3390/genes12101618
Xia, G., Guo, Y., Zhang, J., Han, M., Meng, X., and Lv, J. (2024). An overview of the deubiquitinase USP53: a promising diagnostic marker and therapeutic target. Curr. Protein Peptide Sci. 25 (9), 708–718. doi:10.2174/0113892037292440240518194922
Zhang, J., Yang, Y., Gong, J., Li, L., Li, J., Zhang, M., et al. (2020). Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: clinical, histological and ultrastructural characterization. Liver Int. 40 (5), 1142–1150. doi:10.1111/liv.14422
Keywords: Usp53, BRIC, PFIC7, low-GGT cholestasis, pediatric hepatology, gross deletion, compound heterozygous variants
Citation: Nuzhnaya E, Cherevatova T, Lotnik E, Shagiazdanova A, Markova Z, Filimonova E, Parshina O, Buianova A, Bobreshova A, Marakhonov A and Semenova N (2025) Case Report: Mild BRIC-like cholestasis despite a gross USP53 deletion—novel findings and literature review. Front. Genet. 16:1670664. doi: 10.3389/fgene.2025.1670664
Received: 21 July 2025; Accepted: 03 November 2025;
Published: 21 November 2025.
Edited by:
Omid Vakili, Isfahan University of Medical Sciences, IranReviewed by:
Hadla Hariri, University of Michigan, United StatesMariana B. Morais, Faculdade de Medicina da Universidade de Lisboa, Portugal
Zahra Mahmoudi, Shiraz University of Medical Sciences, Iran
Copyright © 2025 Nuzhnaya, Cherevatova, Lotnik, Shagiazdanova, Markova, Filimonova, Parshina, Buianova, Bobreshova, Marakhonov and Semenova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ekaterina Nuzhnaya, bnV6aG5heWFAbWVkLWdlbi5ydQ==