Abstract
Background:
Prader–Willi syndrome (PWS) represents a paradigm of genomic imprinting disorders. Given the severe lifelong complications of PWS, prenatal diagnosis is crucial for early intervention and genetic counseling.
Methods:
Noninvasive prenatal testing (NIPT) indicated a high risk for fetal trisomy 15 (T15), prompting confirmatory invasive testing. Amniocentesis was performed, and amniotic fluid was analyzed by karyotyping, chromosomal microarray analysis (CMA), trio-based whole-exome sequencing (trio-WES), and short tandem repeat (STR) linkage analysis to investigate the genetic etiology. Post-termination, placental tissue was analyzed by copy number variant sequencing (CNVseq) to evaluate potential mosaicism.
Results:
NIPT indicated a suspected T15 (Z-score: 16.4). Subsequent invasive testing confirmed the following: a 13.16 Mb region of homozygosity on chromosome 15q25.1q26.1 and a 273 kb Duchenne muscular dystrophy (DMD) gene deletion on chromosome Xp21.1, both identified by CMA. Trio-WES and STR linkage analysis revealed maternal segmental uniparental disomy of chromosome 15 (UPD15), confirming the genetic basis of PWS. Post-termination, CNVseq further demonstrated confined placental mosaicism (CPM) for T15.
Conclusion:
When NIPT suggests a high risk of T15, clinicians should maintain a high suspicion for the “trisomy rescue” mechanism, where an initially trisomic zygote undergoes mitotic correction, ultimately forming UPD15 with CPM. The potential discordance between NIPT and the actual fetal genetic status necessitates definitive prenatal diagnosis, which has critical implications for subsequent pregnancy management. Therefore, the concomitant findings of PWS and DMD carrier status require comprehensive prognostic evaluation and recurrence risk assessment.
1 Introduction
PWS is a rare disorder with an estimated incidence of 1/10,000–1/30,000. A recent study screening 16,579 newborns for PWS in Australia found a birth incidence of 1:8,290 (Godler et al., 2022). It is pathologically characterized by the loss of paternal gene expression in the 15q11.2-q13 region. The primary pathogenic mechanisms include gene deletion (65%–75%), maternal UPD (20%–30%), and imprinting defect (1%–3%) (Cassidy et al., 2012; Beygo et al., 2019). The syndrome is clinically defined by a characteristic triad of manifestations: neonatal hypotonia, hyperphagia-induced obesity during infancy, and developmental delay. These core features are frequently accompanied by hypogonadism, cognitive impairment, and distinctive facial characteristics. Patients with deletion are typically the most severely affected, while those with UPD or imprinting defects exhibit a less severe phenotype (Lossie et al., 2001). Differences in body mass index, head circumference, and seizure activity are the most pronounced among the classes (Mahmoud et al., 2021). When PWS coincides with DMD carrier status, additional surveillance for cardiomyopathy and muscular dystrophy is warranted. Prenatal manifestations may include reduced fetal movements and low birth weight, although definitive prenatal diagnosis remains challenging and critically important (Angelis et al., 2015).
The NIPT was based on low-pass genome-wide sequencing. This approach can detect not only autosomal aneuploidies but also copy number variations (CNVs) exceeding 1 Mb in size (Dungan et al., 2023). While NIPT can detect PWS caused by 15q11.2 deletion, it has a high false-positive rate and cannot identify UPD or imprinting defects (Butler, 2017). The definitive diagnosis of PWS requires CMA to detect deletions and STR linkage analysis to distinguish isodisomy and heterodisomy UPD. However, this combination cannot detect imprinting center defects. Trio-WES combined with STR linkage analysis can detect not only UPD but also imprinting center defects. The co-occurrence of PWS and DMD carrier status demands dual-pathway counseling that addresses both the imprinting disorder and the X-linked inheritance pattern.
2 Materials and methods
2.1 Subjects
This study involved a pregnant woman who received routine prenatal screening at the Prenatal Diagnostic Center of the Seventh Affiliated Hospital of Sun Yat-sen University. NIPT indicated a high-risk result, prompting subsequent invasive prenatal diagnosis. The pregnant woman was 33 years old (G4P3A0), and her current husband was 37 years old. Neither had a significant family genetic history (data collected in 2024). All participants in the present study provided written informed consent. Ethical approval for the study was obtained from the hospital’s ethics committee (No:KY-2025-382-01). Fetal ultrasound was performed at 27 weeks of gestation and estimated that the fetal size was equivalent to 24 weeks gestation. The findings suggested symmetrical intrauterine growth restriction (IUGR) accompanied by decreased fetal movements and oligohydramnios. Placental parenchyma was slightly thicker and more echogenic. Maternal–fetal hemodynamic testing revealed abnormalities: the fetal middle cerebral artery (MCA) peak systolic velocity was elevated (1.35–1.4 MoM). There was significantly increased umbilical artery resistance, resulting in a low cerebroplacental (CP) ratio. These findings were indicative of placental insufficiency. The uterine artery Doppler spectra showed significantly increased resistance bilaterally. Based on these hemodynamic and placental findings, the pregnant woman was assessed to be at a very high risk of eclampsia.
2.2 Methods
2.2.1 NIPT
Maternal peripheral blood (5 mL) was collected in Streck Cell-Free DNA BCT® blood collection tubes (Streck, La Vista, NE, United States) at a gestational age of greater than 12 weeks. Cell-free DNA (cfDNA) was extracted from 200 µL of maternal plasma. Library preparation included end repair, adapter ligation, and PCR. Amplified double-stranded DNA was thermally denatured into single-stranded DNA, cyclized, and converted into DNA nanoballs (DNBs). The DNBs were loaded onto sequencing chips and sequenced on the MGISEQ-2000 platform (BGI, Shenzhen, China) at 0.1× average coverage. Raw reads were aligned with genome version GRCh37/hg19 using BWA. Uniquely mapped reads were selected using SAM tags, and PCR duplicates were removed. Z-scores were calculated to detect fetal chromosomal aneuploidies and CNVs.
2.2.2 CMA
Approximately 10 mL of amniotic fluid was collected and centrifuged at 3000 rpm for 10 min to obtain the cell pellet. Genomic DNA (gDNA) for CMA, trio-WES, and STR linkage analysis was then extracted from the pellet using the QIAGEN DNA Mini Kit (Cat. 51306; QIAGEN, Hilden, Germany) per the manufacturer’s instructions. The gDNA concentration was 61.8 ng/μL; 5 µL (309 ng) was used for the CytoScan 750K array (Thermo Fisher Scientific, Waltham, MA, USA), exceeding the optimal 250 ng input. In clinical scenarios with suspected maternal blood contamination, short-term amniocyte culture can be employed to enrich adherent fetal epithelial-like cells and deplete non-adherent maternal lymphocytes; culture was not required in this case due to adequate yield and quality from uncultured cells. Extracted gDNA was amplified, labeled, and hybridized to the Affymetrix CytoScan 750K array following the manufacturer’s protocol. Raw data were analyzed in the Chromosome Analysis Suite (ChAS) and annotated to GRCh37/hg19. CNV calls required ≥50 contiguous probes and a minimum size of ≥100 kb. Runs of homozygosity (ROH) were reported and interpreted in clinical context; large ROH (>10 Mb), particularly when restricted to imprinting chromosomes (Angelis et al., 2015; Dungan et al., 2023; McKenna et al., 2010; Zhang et al., 2020; Hu et al., 2022; Benn and Grati, 2018), prompted evaluation for possible UPD. Detected CNVs were interpreted with reference to the scientific literature and public databases.
2.2.3 Whole-exome sequencing (WES)
Trio-WES was performed on gDNA from the amniotic fluid pellet (which was shared with CMA) and on parental blood gDNA. Libraries were prepared, captured by hybridization (Roche NimbleGen), and sequenced as paired-end 100-bp reads on MGISEQ-2000. Reads were processed with SOAPnuke (Chen et al., 2018) and aligned to GRCh37/hg19 using BWA (Li and Durbin, 2009); GATK was used for SNV/indel calling (McKenna et al., 2010), followed by annotation and filtering against population and disease databases. Variant interpretation followed ACMG/AMP guidelines (Richards et al., 2015) with trio-based segregation, and exome-derived SNP/ROH information was leveraged to assess segmental UPD.
2.2.4 STR linkage analysis
UPD testing was performed on gDNA from the amniotic fluid pellet (which was shared with CMA) and on parental blood gDNA using polymorphic STR markers within the imprinted 15q11–q13 region (Beygo et al., 2019). Multiplex fluorescent PCR was followed by capillary electrophoresis on an ABI 3500 Dx Genetic Analyzer (Thermo Fisher Scientific, United States), and electropherograms were analyzed in GeneMapper v6.0. UPD was inferred by trio segregation across informative loci: isodisomy was defined as a single maternal allele at informative loci, and heterodisomy as non-Mendelian inheritance of two maternal alleles with the absence of paternal contribution; mixed patterns were interpreted as segmental UPD. Quality control included standard allele-calling thresholds (minimum peak height and stutter filters) according to laboratory validation and manufacturer recommendations.
2.2.5 CNVseq
CNVseq was performed on gDNA from the umbilical cord and placental tissues (separately sampled from the fetal/chorionic plate and maternal/basal plate) using the MagPure Buffy Coat DNA Midi KF Kit following the manufacturer’s instructions. Low-pass whole-genome libraries were prepared and sequenced on the MGISEQ-2000 at approximately 0.5× coverage. Reads were aligned to GRCh37/hg19 using BWA, and read-depth-based copy-number profiles were generated. Mosaic trisomy 15 was assessed from normalized copy-number ratios across chromosome 15 and summarized as a percent mosaic fraction for each biopsy and pooled sample. CNVs were interpreted in clinical context with reference to the scientific literature and public databases; analyses met internal QC criteria for mapping performance and coverage uniformity.
3 Results
3.1 NIPT
At 16 weeks of gestation, the fetal NIPT result indicated a high risk for T15. Several quality control metrics were used to evaluate the sequencing data. The fetal DNA fraction was 11.38%. The Q30 score was 95.18%, indicating the proportion of bases with a quality score exceeding 90%. The number of unique reads was 6.63 million, and the Z-score for T15 was 16.395 (Figure 1).
FIGURE 1
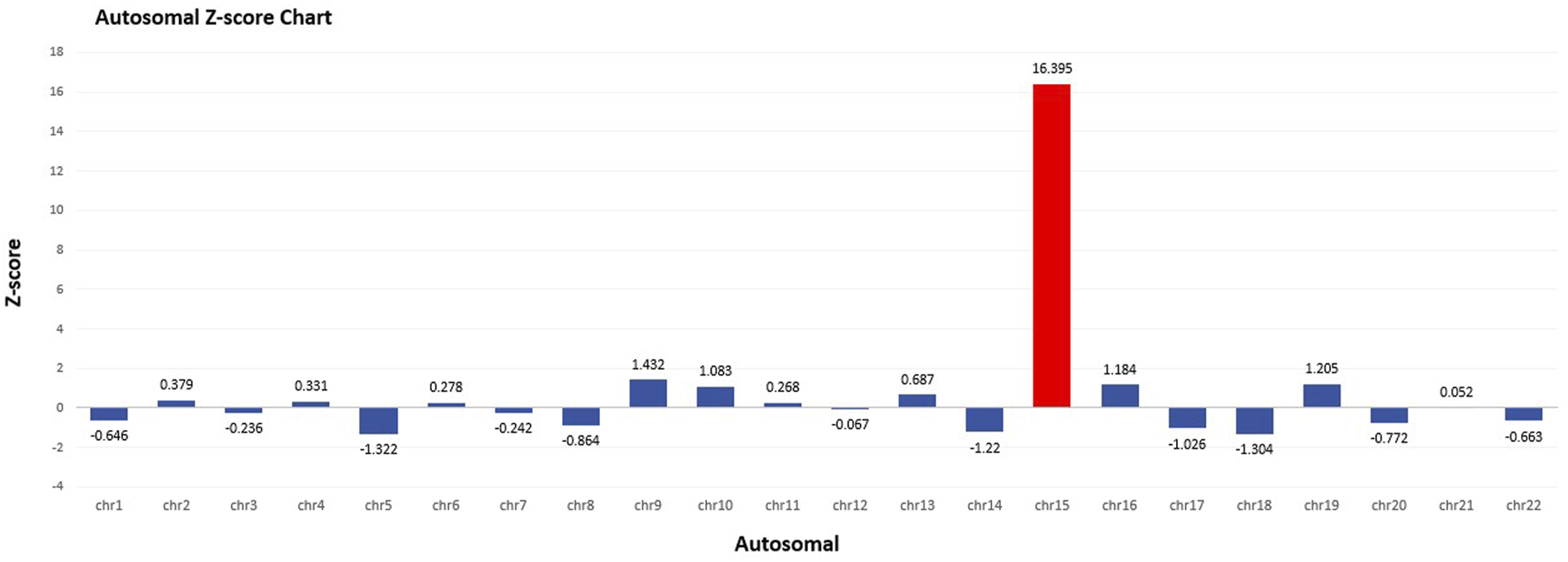
Autosomal Z-score chart in NIPT. Fetal fraction (FF): 11.38%. Z-score for T15 was 16.395.
3.2 CMA
The Affymetrix CytoScan 750K CMA identified a heterozygous pathogenic deletion CNV on arr [GRCH37] Xp21.1 (g.31784724_32057482, 272.8 kb) and a region of homozygosity (ROH) on arr [GRCH37] 15q25.1q26.1 (g.81006170_94168517, 13.2 Mb) (Figure 2).
FIGURE 2

CNV and ROH results in Affymetrix CytoScan 750K CMA. (A) Chromosome results. (B) Hemizygote deletion pathogenic CNV on Xp21.1, range 31,784,724–32,057,482, fragment length 272.8 kb. (C) ROH on 15q25.1q26.1, range 81,006,170–94,168,517, fragment length 13.2 Mb.
3.3 Trio-WES
Trio-WES strongly suggests the presence of maternal UPD15, encompassing regions of both isodisomy and heterodisomy. The isodisomy region contained a ∼10.29-Mb ROH spanning 15q25.2q26.1 (GRCH37: g.83328542_93616975). Two heterodisomy regions were identified at 15q11q25.1 and 15q26.1q26.3 (Figure 3A). These findings were consistent with four breaks and two crossover events on chromosome 15 during maternal meiosis Ⅰ (Figure 3B). Additionally, a heterozygous deletion encompassing exons 45–51 of DMD (NM_004006.2: g.31792078_31986631, 194.6 kb) was classified as pathogenic based on the criteria of the ACMG. Maternal testing confirmed that the mother is a carrier of this deletion. Furthermore, this deletion was independently validated by quantitative fluorescent PCR (QF-PCR) (Supplementary Figure S1). No pathogenic or likely pathogenic SNVs were identified.
FIGURE 3
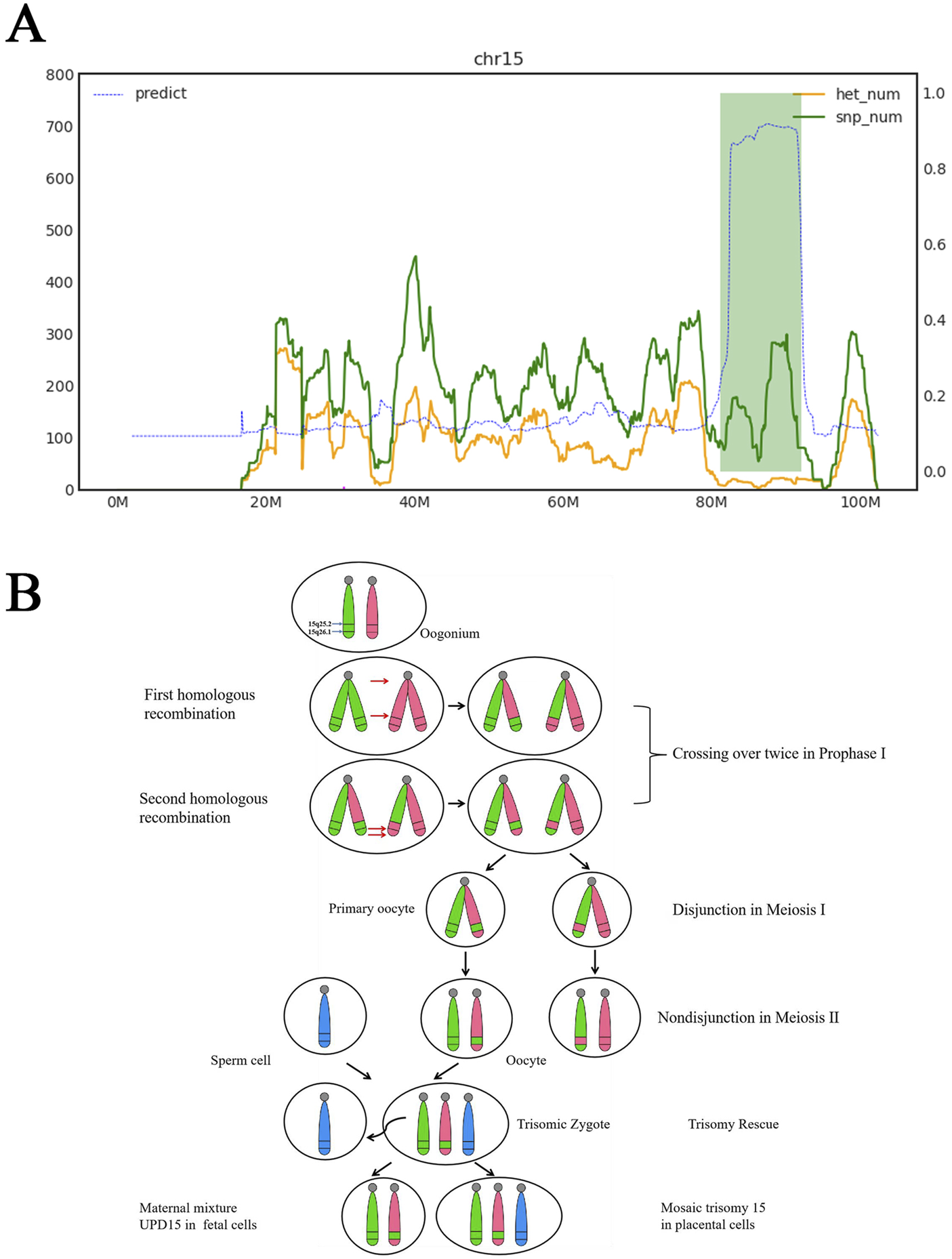
Trio-WES results and the mechanism of segmental maternal UPD15. (A) Horizontal axis represents genomic coordinates on chromosome 15. Green curve indicates that single-nucleotide polymorphism (SNP) counts at corresponding positions. Yellow curve displays heterozygous SNP counts at corresponding positions. Left vertical axis denotes gene quantities for green and yellow curves. Right vertical axis is the blue curve, which was the result of calculating ROH, where values approaching 1 indicate regions statistically identified as ROH based on pedigree SNP analysis. Chromosome 15 segment 15q11.1-q25.1 was maternal hetUPD. Chromosome 15 segment 15q25.1-q26.1 was maternal isoUPD, range 81100000–92000000, fragment length 10.9 Mb. Chromosome 15 segment 15q26.1-q26.3 was maternal hetUPD. (B) Partial isoUPD was caused by nondisjunction in meiosis Ⅰ after two crossings over, resulting in sections of isodisomy and heterodisomy on the UPD chromosome 15. IsoUPD: isodisomy uniparental disomy, hetUPD: heterodisomy uniparental disomy.
3.4 STR linkage analysis
STR genotyping of amniotic fluid demonstrated concordance with maternal alleles at all informative 15q11–q13 markers, including one locus with biallelic maternal signals, and no paternal contribution across tested loci. In conjunction with CMA and trio-WES, these segregation patterns confirm maternal segmental UPD15 comprising regions of isodisomy and heterodisomy. Placental STR analysis corroborated the maternal origin of the T15 mosaicism (Figure 4).
FIGURE 4

STR linkage analysis results. (A) Paternal STR analysis, M15-1 (17,19), M15-2 (37,40), M15-3 (11,13), M15-4 (8,16). (B) Maternal STR analysis, M15-1 (17,18), M15-2 (34,37), M15-3 (13), M15-4 (16). (C) Placenta ABCDE mix STR analysis, M15-1 (17,18,19), M15-2 (34,37,40), M15-3 (11,13), M15-4 (8,16). These confirmed mosaic T15 as of maternal origin in the maternal surface of the placenta. (D) Placenta abcde mix STR analysis, M15-1 (17,18), M15-2 (34,37), M15-3 (13), M15-4 (16); all four STR loci genotypes were fully consistent with maternal origin, with one locus exhibiting biallelic signals. These confirmed maternal hetUPD in the fetal surface of the placenta. (E) Amniotic fluid STR analysis, M15-1 (17,18), M15-2 (34,37), M15-3 (13), M15-4 (16); all four STR loci genotypes were fully consistent with maternal origin, with one locus exhibiting biallelic signals. These confirmed maternal hetUPD in the fetus.
3.5 CNVseq
CNVseq of umbilical cord tissue identified the heterozygous Xp21.1 deletion and no detectable T15 mosaicism. In contrast, placental sampling demonstrated CPM for T15 with a regional gradient—higher mosaic fraction on the basal (maternal) surface and lower levels on the chorionic (fetal) surface. The Xp21.1 deletion was consistently observed across all tissues. This basal–chorionic gradient accounts for the positive NIPT call and, together with the ultrasound/Doppler evidence of placental insufficiency, supports CPM of likely meiotic origin with post-zygotic trisomy rescue (Supplementary Figure S2; Supplementary Table S1).
4 Discussion
Genome-wide NIPT can flag rare autosomal trisomies (A meta-analysis) but shows variable positive predictive value, in part due to fetus–placenta discordance and CPM; therefore, invasive confirmation is recommended when RAT is detected (Zhang et al., 2020; Hu et al., 2022; Taglauer et al., 2014; Konya et al., 2024). On imprinting chromosomes such as 15, trisomy rescue may result in fetal UPD even when the karyotype appears normal (Liehr, 2022; Okuda et al., 2022; Benn and Grati, 2018). Indeed, cases with normal amniotic karyotypes have subsequently been confirmed as maternal UPD15 by methylation/targeted assays, underscoring the need to evaluate UPD despite a normal karyotype (Hong et al., 2023). Although precise estimates vary by chromosome and ascertainment, population-scale studies suggest that UPD arising from trisomy rescue is rare in live-borns (on the order of ∼1 in 2,000–3,500), reinforcing the clinical salience of such presentations (Nakka et al., 2019).
In this study, diagnoses were systematically validated through amniotic fluid analysis combined with a multimodal diagnostic approach encompassing karyotyping, CMA, prenatal ultrasonography, and trio-WES. UPD15 is associated with PWS/AS syndrome, which represents the most classic example of genomic imprinting disorders in humans. In our case, a ROH was initially detected by CMA, which can suggest uniparental isodisomy. However, as the threshold of ROH detection is typically set at 3–10 Mb, smaller regions may be overlooked (Xu et al., 2024). Moreover, ROH may also originate from identity by descent (IBD) rather than from UPD. Furthermore, CMA cannot directly detect uniparental heterodisomy. Trio-WES can interrogate both SNVs/indels and UPD mechanisms (Dharmadhikari et al., 2019). Subsequent Trio-WES revealed segmental UPD15 in the fetus. Additionally, pathogenic variant analysis in the homozygous regions identified no disease-causing mutations. STR linkage analysis further confirmed that both copies of chromosome 15 originated from the pregnant woman. It was hypothesized that this segmental UPD15 resulted from a meiotic nondisjunction event in meiosis Ⅰ followed by two crossover events, leading to a segmental UPD with isodisomy and heterodisomy. Post-fertilization, the zygote exhibited trisomy 15, with trisomy rescue as the likely mechanism by which the aneuploid zygote reverted to euploidy. Placental testing revealed trisomy 15 in the placenta, indicating that during the trisomy rescue process, only the fetal chromosomes were successfully corrected to euploidy, forming CPM (Figure 3B). To validate placental mosaicism, we collected five tissue samples each from the maternal and fetal surfaces of the placenta for CNVseq. The results revealed varying levels of T15 mosaicism across placental regions, with a significantly higher mosaic ratio observed on the maternal than on the fetal surface. This placental mosaicism likely explains why the NIPT indicated T15.
In early pregnancy, if a trisomy 15 rescue event occurs, miscarriage may be averted. Placental abnormalities associated with trisomy 15 may manifest as placental enlargement, structural disorganization, or focal cystic changes. Placental ischemia and hypoxia in such cases could contribute to maternal hypertension and proteinuria, potentially triggering the onset of preeclampsia. Furthermore, insufficient placental perfusion may impair nutrient and oxygen delivery to the fetus, leading to FGR (Eggenhuizen et al., 2021).
In addition to genetic mechanisms, the prenatal clinical features of PWS require special consideration. More than 95% of Prader–Willi syndrome (PWS) cases occur sporadically and lack characteristic fetal structural anomalies (Muthusamy et al., 2020). Gross et al. (2015) conducted interviews with mothers of 106 individuals with PWS and reviewed obstetric records for 47 cases under 10 years of age. They compared prenatal data from PWS pregnancies with those of sibling controls and the general population. The study revealed significantly higher rates of the following in PWS pregnancies compared to controls (p < 0.0001): reduced fetal movements (88%), small for gestational age (SGA) (65%), asymmetrical intrauterine growth (elevated head-to-abdominal circumference ratio, 43%), and polyhydramnios (34%). No major congenital malformations were observed in any PWS cases. Notably, 27%, 29%, and 24% of cases exhibited combinations of two, three, and four of these abnormalities, respectively.
Therefore, when NIPT indicates a high risk for T15 and concurrent ultrasound findings reveal reduced fetal movements and IUGR, raising clinical suspicion for possible PWS due to maternal UPD15, prompt genetic diagnostic testing is strongly recommended. A total of 90 protein coding genes is located within the ROH, spanning approximately 10.3 Mb on chromosome 15. There was an anticipated risk of having a second genetic condition besides PWS (Muthusamy et al., 2020). Trio-WES revealed no pathogenic or likely pathogenic variants within the ROH on 15q25.2q26.1. However, both Trio-WES and CMA consistently identified a hemizygous deletion spanning exons 45–51 of the DMD gene, which was confirmed to be maternally inherited. This discovery underscored that multi-technique integration can detect complex genetic comorbidities, enabling the identification of PWS arising from UPD15 alongside the DMD deletion as a distinct genetic alteration. PWS primarily arises from the loss of paternally expressed genes in the 15q11-q13 region, while DMD is an X-linked recessive disorder that predominantly affects men, with women typically serving as asymptomatic carriers (Kumar et al., 2020). Given the carrier frequency of DMD (1/4,000) (Mo and ser, 1984) and the incidence of PWS (1/8,290), the theoretical co-occurrence probability is 3.02 × 10−8. This rare comorbidity underscores the necessity for multidisciplinary collaboration among the disciplines of neurology, endocrinology, and genetics, necessitating comprehensive genetic counseling to address overlapping phenotypes and optimize therapeutic strategies. In this case, the fetus was of female gender with PWS, so she was expected to be a carrier rather than clinically affected by DMD. However, had the fetus been of male gender with PWS, the characteristic neonatal hypotonia of PWS could mask early neuromuscular manifestations of DMD, while progressive muscle weakness caused by the DMD deletion would exacerbate motor dysfunction during infancy, potentially delaying the recognition of both conditions (Brogna et al., 2019). We agree that a maternal X-linked deletion of ∼200–300 kb can be detectable by low-coverage WGS (lc-WGS) NIPT under appropriate binning, GC correction, segmentation, and quality control, as shown by reports of maternal incidental CNVs and maternal DMD CNVs identified on lc-WGS NIPT (Brison et al., 2019; Wang et al., 2024; Brison et al., 2017). In our setting, however, the assay validation and the laboratory reporting policy did not include routine analysis or disclosure of incidental maternal single-gene CNVs at this size range. Specifically, reporting was limited to a predefined set of maternal or maternofetal CNVs linked to specified conditions (Supplementary Table S2); otherwise, maternal incidental CNVs were disclosed only when they exceeded 5 Mb (Cox et al., 2025). Thus, the maternal DMD deletion of exons 45–51 was not analyzed/reported because of validation and policy scope, rather than technical impossibility. For context, haplotype-based single-gene NIPT represents a distinct workflow primarily intended for fetal interrogation of X-linked monogenic disease and is not required for the initial recognition of maternal carrier status on lc-WGS NIPT when such analysis is validated and reportable. In addition, the X-chromosomal cfDNA signal is predominantly maternal-dominated—particularly with a female fetus—limiting reliable fetal inference even when a maternal CNV is detectable; moreover, many laboratories do not routinely analyze or report maternal incidental CNVs in lc-WGS NIPT (Tang et al., 2022). From a long-term management standpoint, women with PWS who are also DMD carriers should receive cardiomyopathy surveillance in accordance with carrier care recommendations (e.g., baseline and periodic assessment with echocardiography and/or cardiac MRI) alongside standard PWS management and reproductive counseling (Birnkrant et al., 2018; Feingold et al., 2017). PWS in this case occurred sporadically, which confers a recurrence risk of less than 1% for future siblings. The mother was confirmed to be a carrier of the maternally inherited pathogenic deletion in the DMD gene, there is a 50% probability for each male offspring to inherit the deletion, and a 50% probability for each female offspring to be a carrier. Given these risks, combined with the technical limitations of routine NIPT, which cannot reliably detect fetal DMD deletion due to insufficient sequencing depth, direct prenatal diagnosis through invasive procedures such as amniocentesis or chorionic villus sampling is strongly recommended in future pregnancies. Pre-implantation genetic testing (PGT) may be considered as an alternative strategy to reduce the transmission risk of both disorders.
5 Conclusion
Our study demonstrates how trisomy rescue can result in UPD15 with CPM, underscoring the necessity for invasive diagnostic confirmation when NIPT indicates T15. An integrated multimodal approach utilizing karyotyping, CMA, trio-WES, STR analysis, and CNVseq successfully resolved the discordant findings and identified both PWS and the DMD carrier status. These findings emphasize the critical importance of comprehensive genetic testing in guiding accurate diagnosis and genetic counseling. Clinically, this dual diagnosis requires individualized risk assessment and family-centered management strategies. Despite the inherent limitations of NIPT as a standalone screening tool, our findings strongly support the implementation of integrated genomic diagnostic approaches in contemporary prenatal care.
Statements
Data availability statement
The original contributions presented in the study are publicly available. Raw data have been deposited to the National Center for Biotechnology Information (NCBI) under the BioProject with accession number PRJNA1348100, the data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1348100. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Seventh Affiliated Hospital of Sun Yat-sen University ethics committee. The studies were conducted in accordance with local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YY: Formal Analysis, Writing – original draft. YF: Validation, Writing – review and editing. ZZ: Software, Writing – review and editing. HN: Methodology, Writing – review and editing. FT: Methodology, Writing – review and editing. XW: Funding acquisition, Writing – review and editing. QF: Visualization, Writing – review and editing. ZC: Validation, Writing – review and editing. XH: Resources, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Shenzhen Science and Technology Program (Grant No. RCBS20231211090700001 to XW).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1675663/full#supplementary-material
SUPPLEMENTARY FIGURE S1QF-PCR results for exons 45 and 51 of the DMD gene. The paternal DMD gene exons 45 and 51 are consistent with normal male control, indicating a normal result. The maternal DMD gene exons 45 and 51 show approximately 50% of the level in normal female control, therefore demonstrating heterozygosity deletion. The fetal DMD gene exons 45 and 51 are consistent with the maternal profile, confirming heterozygosity deletion. NC(F): normal female control, NC(M): normal male control.
SUPPLEMENTARY FIGURE S2Placenta samples in CNVseq. Ⅰ. Placental marking A, B, C, D, and E refer to tissues on the fetal surface of the placenta. Ⅱ. Placental marking a, b, c, d, and e refer to tissues on the maternal surface of the placenta.
SUPPLEMENTARY TABLE S1CNVseq results of fetal tissue and placental tissue. Placental side A, B, C, D, and E refer to tissues on the surface of the fetal placenta. Placental side a, b, c, d, and e refer to tissues on the maternal surface of the placenta. The placenta ABCDE mix consists of equal proportions of DNA from the five positions of placenta A, B, C, D, and E. The placenta abcde mix consists of equal proportions of DNA from the five positions of placenta a, b, c, d, and e.
SUPPLEMENTARY TABLE S2Reporting framework for maternal and maternofetal CNVs in lc-WGS NIPT (laboratory validation and policy scope).
References
1
Angelis A. Tordrup D. Kanavos P. (2015). Socio-economic burden of rare diseases: a systematic review of cost of illness evidence. Health Policy119 (7), 964–979. 10.1016/j.healthpol.2014.12.016
2
Benn P. Grati F. R. (2018). Genome-wide non-invasive prenatal screening for all cytogenetically visible imbalances. Ultrasound Obst Gyn51 (4), 429–433. 10.1002/uog.19014
3
Beygo J. Buiting K. Ramsden S. C. Ellis R. Clayton-Smith J. Kanber D. (2019). Update of the EMQN/ACGS best practice guidelines for molecular analysis of prader-willi and angelman syndromes. Eur. J. Hum. Genet.27 (9), 1326–1340. 10.1038/s41431-019-0435-0
4
Birnkrant D. J. Bushby K. Bann C. M. Alman B. A. Apkon S. D. Blackwell A. et al (2018). Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol.17 (4), 347–361. 10.1016/S1474-4422(18)30025-5
5
Brison N. Van Den Bogaert K. Dehaspe L. van den Oever J. M. Janssens K. Blaumeiser B. et al (2017). Accuracy and clinical value of maternal incidental findings during noninvasive prenatal testing for fetal aneuploidies. Genet. Med.19 (3), 306–313. 10.1038/gim.2016.113
6
Brison N. Storms J. Villela D. Claeys K. G. Dehaspe L. de Ravel T. et al (2019). Maternal copy-number variations in the DMD gene as secondary findings in noninvasive prenatal screening. Genet. Med.21 (12), 2774–2780. 10.1038/s41436-019-0564-4
7
Brogna C. Coratti G. Pane M. Ricotti V. Messina S. D'Amico A. et al (2019). Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. Plos One14 (6), e0218683. 10.1371/journal.pone.0218683
8
Butler M. G. (2017). Benefits and limitations of prenatal screening for Prader-Willi syndrome. Prenat. Diag37 (1), 81–94. 10.1002/pd.4914
9
Cassidy S. B. Schwartz S. Miller J. L. Driscoll D. J. (2012). Prader-Willi syndrome. Genet. Med.14 (1), 10–26. 10.1038/gim.0b013e31822bead0
10
Chen Y. Chen Y. Shi C. Huang Z. Zhang Y. Li S. et al (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience7 (1), 1–6. 10.1093/gigascience/gix120
11
Cox S. G. Acevedo A. Ahuja A. LaBreche H. G. Alfaro M. P. Pierson S. et al (2025). Curation and reporting of pathogenic genome-wide copy-number variants in a prenatal cell-free DNA screen. Genet. Med.27 (1), 101223. 10.1016/j.gim.2024.101223
12
Dharmadhikari A. V. Ghosh R. Yuan B. Liu P. Dai H. Al M. S. et al (2019). Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Med.11 (1), 30. 10.1186/s13073-019-0639-5
13
Dungan J. S. Klugman S. Darilek S. Malinowski J. Akkari Y. Monaghan K. G. et al (2023). Noninvasive prenatal screening (NIPS) for fetal chromosome abnormalities in a general-risk population: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med.25 (2), 100336. 10.1016/j.gim.2022.11.004
14
Eggenhuizen G. M. Go A. Koster M. Baart E. B. Galjaard R. J. (2021). Confined placental mosaicism and the association with pregnancy outcome and fetal growth: a review of the literature. Hum. Reprod. Update27 (5), 885–903. 10.1093/humupd/dmab009
15
Feingold B. Mahle W. T. Auerbach S. Clemens P. Domenighetti A. A. Jefferies J. L. et al (2017). Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American heart Association. Circulation136 (13), e200–e231. 10.1161/CIR.0000000000000526
16
Godler D. E. Ling L. Gamage D. Baker E. K. Bui M. Field M. J. et al (2022). Feasibility of screening for chromosome 15 imprinting disorders in 16 579 newborns by using a novel genomic workflow. JAMA Netw. Open. 5 (1), e2141911. 10.1001/jamanetworkopen.2021.41911
17
Gross N. Rabinowitz R. Gross-Tsur V. Hirsch H. J. Eldar-Geva T. (2015). Prader-Willi syndrome can be diagnosed prenatally. Am. J. Med. Genet.A (1), 80–85. 10.1002/ajmg.a.36812
18
Hong D. K. Park J. E. Kang K. M. Shim S. H. Shim S. H. Han Y. J. et al (2023). Prenatal diagnosis of uniparental disomy in cases of rare autosomal trisomies detected using noninvasive prenatal Test: a case of Prader-Willi Syndrome. Diagnostics13 (4), 580. 10.3390/diagnostics13040580
19
Hu T. Wang J. Zhu Q. Zhang Z. Hu R. Xiao L. et al (2022). Clinical experience of noninvasive prenatal testing for rare chromosome abnormalities in singleton pregnancies. Front. Genet.13, 955694. 10.3389/fgene.2022.955694
20
Konya M. Czimbalmos A. Loczi L. Koi T. Turan C. Nagy R. et al (2024). Genome-Wide, Non-Invasive Prenatal Testing for rare chromosomal abnormalities: a systematic review and meta-analysis of diagnostic test accuracy. Plos One19 (11), e0308008. 10.1371/journal.pone.0308008
21
Kumar S. H. Athimoolam K. Suraj M. Das C. D. M. Muralidharan A. Jeyam D. et al (2020). Comprehensive genetic analysis of 961 unrelated Duchenne Muscular Dystrophy patients: focus on diagnosis, prevention and therapeutic possibilities. Plos One.15 (6), e0232654. 10.1371/journal.pone.0232654
22
Li H. Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics25 (14), 1754–1760. 10.1093/bioinformatics/btp324
23
Liehr T. (2022). Uniparental disomy is a chromosomic disorder in the first place. Mol. Cytogenet15 (1), 5. 10.1186/s13039-022-00585-2
24
Lossie A. C. Whitney M. M. Amidon D. Dong H. J. Chen P. Theriaque D. et al (2001). Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet.38 (12), 834–845. 10.1136/jmg.38.12.834
25
Mahmoud R. Leonenko A. Butler M. G. Flodman P. Gold J. A. Miller J. L. et al (2021). Influence of molecular classes and growth hormone treatment on growth and dysmorphology in Prader-Willi syndrome: a multicenter study. Clin. Genet.100 (1), 29–39. 10.1111/cge.13947
26
McKenna A. Hanna M. Banks E. Sivachenko A. Cibulskis K. Kernytsky A. et al (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. GENOME Res.20 (9), 1297–1303. 10.1101/gr.107524.110
27
Moser H. (1984). Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum. Genet.66 (1), 17–40. 10.1007/BF00275183
28
Muthusamy K. Macke E. L. Klee E. W. Tebben P. J. Hand J. L. Hasadsri L. et al (2020). Congenital ichthyosis in Prader-Willi syndrome associated with maternal chromosome 15 uniparental disomy: case report and review of autosomal recessive conditions unmasked by UPD. Am. J. Med. Genet. A182 (10), 2442–2449. 10.1002/ajmg.a.61792
29
Nakka P. Pattillo S. S. O'Donnell-Luria A. H. McManus K. F. Mountain J. L. Ramachandran S. et al (2019). Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet.105 (5), 921–932. 10.1016/j.ajhg.2019.09.016
30
Okuda T. Moroto M. Yamamoto T. (2022). Noninvasive prenatal testing suggesting an abnormality in chromosome 15 confirmed to be a case of Prader-Willi syndrome caused by trisomy rescue in the neonatal period. J. Obstet. Gynaecol.48 (8), 2214–2218. 10.1111/jog.15236
31
Richards S. Aziz N. Bale S. Bick D. Das S. Gastier-Foster J. et al (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med.17 (5), 405–424. 10.1038/gim.2015.30
32
Taglauer E. S. Wilkins-Haug L. Bianchi D. W. (2014). Review: cell-free fetal DNA in the maternal circulation as an indication of placental health and disease. Placenta. 35 Suppl (Suppl), S64–8. 10.1016/j.placenta.2013.11.014
33
Tang X. Wang Z. Yang S. Chen M. Zhang Y. Zhang F. et al (2022). Maternal Xp22.31 copy-number variations detected in non-invasive prenatal screening effectively guide the prenatal diagnosis of X-linked ichthyosis. Front. Genet. J. Artic.13, 934952. 10.3389/fgene.2022.934952
34
Wang Y. Sun Y. Meng L. He Q. Zhao J. Zhou R. et al (2024). A new strategy for prenatal genetic screening of copy number variations in the DMD gene: a large cohort study based on NIPT analysis. Clin. Transl. Med.14 (5), e1706. 10.1002/ctm2.1706
35
Xu C. Li M. Gu T. Xie F. Zhang Y. Wang D. et al (2024). Chromosomal microarray analysis for prenatal diagnosis of uniparental disomy: a retrospective study. Mol. Cytogenet17 (1), 3. 10.1186/s13039-023-00668-8
36
Zhang Y. Yan L. Liu Y. Li H. (2020). Clinical value of routine non-invasive prenatal testing for the screening of non-target chromosomal abnormalities. Zhonghua Yi Xue Yi Chuan Xue Za Zhi37 (6), 621–626. 10.3760/cma.j.issn.1003-9406.2020.06.006
Summary
Keywords
T15, UPD15, Prader–Willi syndrome, prenatal diagnosis, confined placental mosaicism, Duchenne muscular dystrophy
Citation
Ye Y, Fu Y, Zhang Z, Ning H, Tao F, Wang X, Fang Q, Chen Z and Hao X (2025) Prenatal diagnosis of Prader–Willi syndrome via maternal UPD15 with placental mosaicism: incidental discovery of fetal DMD carrier status. Front. Genet. 16:1675663. doi: 10.3389/fgene.2025.1675663
Received
29 July 2025
Accepted
18 September 2025
Published
30 October 2025
Volume
16 - 2025
Edited by
Wen-Bin He, Hunan Normal University, China
Reviewed by
Meizi Zhang, Tianjin First Central Hospital, China
Michaela Hyblova, Trisomy s.r.o., Slovakia
Updates
Copyright
© 2025 Ye, Fu, Zhang, Ning, Tao, Wang, Fang, Chen and Hao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiulan Hao, haoxiulan@sysush.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.