 Lupeng Zhan
Lupeng Zhan Baicheng Xu
Baicheng Xu Dujuan Lin
Dujuan Lin Ya Wang
Ya Wang Panpan Bian
Panpan Bian- Department of Otolaryngology-Head and Neck Surgery, Lanzhou University Second Hospital, Lanzhou, China
Introduction: Waardenburg syndrome type 1 (WS1) is a rare autosomal dominant disorder characterized by congenital sensorineural hearing loss and facial dysmorphisms. PAX3 mutations are a known genetic cause. This study investigated a novel PAX3 mutation in a Chinese Yugur family and assessed long-term auditory outcomes after cochlear implantation.
Methods and Results: Whole-exome and Sanger sequencing were used to identify causative variants, followed by bioinformatic analyses to predict pathogenicity. Auditory and speech rehabilitation were evaluated over a 7-year follow-up. A novel heterozygous frameshift mutation in PAX3 (c.788dup, p.Gln264ThrfsTer5) was identified and confirmed to be de novo. Structural modeling indicated disruption of a conserved domain, supporting its pathogenic role. The proband achieved excellent auditory and speech recovery and successfully integrated into mainstream education.
Discussion: This study expands the mutation spectrum of PAX3 and provides evidence supporting the pathogenicity of the c.788dup variant. It also confirms the long-term benefit of cochlear implantation and rehabilitation in WS1-related hearing loss.
1 Introduction
Waardenburg syndrome (WS) is recognized as the most common form of syndromic deafness inherited in an autosomal dominant pattern, first documented by the Dutch clinician and geneticist Petrus Johannes Waardenburg (Waardenburg, 1951). Its global prevalence is approximately one in 42,000, accounting for 2%–5% of congenital deafness cases (George et al., 2023; Tawfik et al., 2025). WS displays extensive phenotypic and genetic heterogeneity, typically involving sensorineural hearing impairment, hypertelorism, pigmentary irregularities in hair, skin, and eyes, and, less frequently, musculoskeletal defects. In 1992, Farrer et al. proposed clinical diagnostic criteria based on major and minor criteria (Farrer et al., 1992). Major criteria include: congenital hearing loss, heterochromia iridis, white frontal hair, wide-set eyes (W index >1.95), and a confirmed case among first-degree relatives; minor criteria include: skin vitiligo, fused eyebrows, excessive eyelashes, broad nasal bridge, underdeveloped nasal wings, and abnormal upper lip. A clinical diagnosis of WS1 is established when a patient fulfills either two major criteria or one major criterion in combination with at least two minor criteria. Based on WS1, WS is further classified into four subtypes according to different accompanying symptoms. WS1 is distinguished by an increased interpupillary distance (Guo et al., 2021). WS2 shares all characteristics with WS1 except for the absence of widened interpupillary distance (Han et al., 2025). WS3 presents with skeletal and muscular system abnormalities (such as upper limb deformities) in addition to WS1 (Tekin et al., 2001). WS4 combines the features of WS2 with Hirschsprung’s disease (HD), which involves congenital megacolon and intestinal atresia (Gombojav et al., 2025). Currently, researchers have identified six WS-related genes, including PAX3, MITF, SNAI2, EDN3, EDNRB, and SOX10 (Lee et al., 2022). These genes are critical in WS pathogenesis. Among them, PAX3 is predominantly associated with WS1 and WS3, while other subtypes involve different genetic mutations (Han et al., 2025).
The PAX3 gene (https://omim.org/entry/606597) codes for a transcription factor classified within the paired box family. Positioned at 2q36.1 on chromosome 2, PAX3 consists of 10 exons and encodes a protein comprising 479 amino acids. The PAX3 protein contains several evolutionarily conserved regions, including the paired box domain (PD) at the N-terminus, the octapeptide (O), and the paired-type homeodomain (HD), as well as the transcription activation domain (TAD) rich in serine-threonine-proline residues at the C-terminus. The PD and HD domains form the DNA-binding region, which collaboratively recognize and bind to the regulatory sequences of specific target genes, thereby regulating downstream target gene expression (Moore et al., 2025). These domains are crucial for the PAX3 protein’s biological functions. The integrity of the PD and HD domains ensures that PAX3 can regulate the expression of key target genes during embryonic development (Boudjadi et al., 2018). Pathogenic mutations in PAX3 disrupt its ability to regulate cell proliferation, differentiation, migration, and survival, particularly in cells derived from neural crest cells (NCCs), leading to the onset of WS1 and WS3 symptoms (Matsunaga et al., 2013).
In this study, we report a novel de novo heterozygous frameshift mutation (c.788dup, p.Gln264ThrfsTer5) in exon five of PAX3 in a 17-year-old female from a Chinese Yugur family with WS1. Clinical features included congenital bilateral sensorineural hearing loss, heterochromia iridis, and dystopia canthorum. This variant was not found in her unaffected parents and has not been previously reported. Our findings expand the PAX3 mutation spectrum and provide insight into the genetic basis of WS1.
2 Materials and methods
2.1 Clinical and hearing assessment
This study investigated a sporadic deafness family from Zhangye City, Gansu Province, China. Family history and comprehensive otologic and ophthalmologic evaluations were conducted. Audiological assessments included pure-tone audiometry (PTA), auditory brainstem response (ABR), impedance tests, and cranial imaging. Ocular and physical features were examined using fundus imaging, optical coherence tomography, and limb and pigmentation inspections. Hearing loss severity was categorized per WHO standards. Post-cochlear implantation outcomes were assessed through standardized scales such as the Categories of Auditory Performance (CAP), the Speech Intelligibility Rating (SIR), the Meaningful Auditory Integration Scale (MAIS), and the Meaningful Use of Speech Scale (MUSS). The W index was calculated using an established formula incorporating canthal and interpupillary measurements to evaluate dystopia canthorum. This study was approved by the Ethics Committee of the Second Hospital of Lanzhou University (certificate no. 2021A-027), and written informed consent was obtained from all participants.
2.2 Sample collection
2 mL of peripheral blood were collected from the patient and both parents using EDTA anticoagulant vacuum tubes. Samples were stored at −80 C and transported under cold-chain conditions to the Beijing Mygenostics Medical Testing Institute. Genomic DNA was extracted using a genomic DNA extraction kit (Mygenostics, Kangwei Century, QIAGEN). DNA concentration and purity were assessed with a Nanodrop 2000 UV spectrophotometer. Only samples with a concentration ≥30 ng/μL and an OD260/280 ratio between 1.7 and 2.0 were used for further analysis.
2.3 Whole exome sequencing
WES was performed by the Beijing Mygenostics Medical Testing Institute (Beijing, China). High-quality genomic DNA (1–3 μg) was enzymatically sheared to produce fragments averaging 150 base pairs in length. Using a standard DNA library preparation kit, the sequencing library was built through end-repair, ligation of adapters, and amplification via PCR. Whole-exome enrichment was performed with the P039-Exome probe set developed by Mygenostics, and sequencing was carried out on the Illumina NextSeq 500. Raw sequencing data were cleaned with Cutadapt v1.16 to strip adapter sequences, filter low-quality bases, remove reads with excessive N bases, and exclude those under 40 bp. Reads passing quality control were mapped to the human genome reference sequence (hg19). Post-alignment processing included filtering with SAMtools/BamTools, duplicate removal with GATK MarkDuplicates, and base quality recalibration using GATK BaseRecalibrator. Genetic variants, including single nucleotide polymorphisms (SNPs), insertions and deletions (Indels), and copy number variations (CNVs), were identified using GATK HaplotypeCaller v4.0.8.1. Variant annotation was performed with ANNOVAR. Pathogenicity interpretation followed the ACMG guidelines, integrating population databases such as gnomAD, ClinVar, and HGMD, along with prediction tools like MutationTaster.
2.4 Sanger sequencing
To validate the detected variant, Sanger sequencing was conducted. Primer pairs were designed using Primer3 software. The specific sequences were: Forward primer: 5′-CAAAGTCCTAACAATATGCATCCC-3′; Reverse primer: 5′-TGCAGTCGGAGAGAGAACTTG-3′. PCR amplification was carried out using 2X GoldStar Master Mix. The amplification protocol involved an initial heating step at 95 C for 10 min, followed by 36 repetitive cycles of denaturation (94 C, 30 s), annealing (58 C, 30 s), and extension (72 C, 45 s). An additional extension step at 72 C for 5 min was performed to complete the amplification process. PCR amplicons were checked through agarose gel electrophoresis and then isolated using magnetic bead purification. Subsequent sequencing was performed using capillary electrophoresis on an ABI 3130XL Genetic Analyzer. Using Mutation Surveyor software, the sequence data were aligned to the PAX3 reference sequence (NM_181458.4) for variant identification.
2.5 Evolutionary conservation analysis
PAX3 protein sequences in FASTA format were obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/) for representative species, including humans, mice, chimpanzees, and rhesus monkeys. The collected sequences were subsequently imported into UGENE software (version 46.0; Unipro UGENE, https://ugene.net/) for comparative analysis. Sequence alignment was conducted utilizing the Clustal W algorithm integrated within UGENE. The conservation of the mutant site, along with flanking upstream and downstream amino acid regions, was then evaluated visually.
2.6 Three-dimensional structure modeling
The wild-type PAX3 protein sequence was accessed from the UniProt database (https://www.uniprot.org/).The c.788dup mutation introduced a frameshift, generating a truncated protein product due to the introduction of an early stop codon. Both wild-type and mutant amino acid sequences were prepared in FASTA format and uploaded to the AlphaFold database (https://alphafold.ebi.ac.uk/) for structural prediction using homology modeling techniques. Default parameters were employed to obtain PDB files. The resulting models were visualized and compared using PyMOL software (v2.5, Schrödinger Inc.). The align command was applied to superimpose the wild-type and mutant structures to assess global conformational differences. A “rainbow” color scheme was used to illustrate the sequential regions of the proteins, and three-dimensional images were generated for further analysis.
3 Result
3.1 Clinical features and rehabilitation
The proband is a 17-year-old female patient with congenital bilateral deafness. She had a normal pregnancy and delivery history but failed the newborn hearing screening. Her parents are non-consanguineous, and both exhibit normal hearing and no other clinical abnormalities (Figure 1A). No additional family members have reported similar symptoms. The patient’s facial features included a broad nasal root, heterochromia iridum with a blue iris in the left eye and a normally pigmented iris in the right eye, as well as thickened eyebrows on the left side (Figure 1B). The inner canthal distance (a) was 40 mm, the interpupillary distance (b) was 66 mm, and the outer canthal distance (c) was 95 mm. The W index, calculated using the standard formula, was approximately 2.10, exceeding the diagnostic threshold of 1.95. Preoperative pure-tone audiometry revealed bilateral profound sensorineural hearing loss across all frequencies (Figures 1C,D), and ABR testing failed to elicit any waveforms. Temporal bone CT and inner ear MRI showed no structural abnormalities, such as cochlear malformations. Fundus photographs demonstrated an essentially normal fundus in the right eye (OD) with uniform pigment distribution, whereas the left eye (OS) showed markedly reduced pigmentation (Figure 2A). Optical coherence tomography (OCT) demonstrated normal foveal architecture in both eyes, with intact retinal layers in the macular region and no evident pathological changes. The average retinal nerve fiber layer (RNFL) thickness in the right eye was 98 microns, with a cup-to-disc ratio of 0.37; the average RNFL thickness in the left eye was 100 microns, with a cup-to-disc ratio of 0.31 (Figure 2B). Visual function was assessed: both direct and consensual pupillary light reflexes were present; visual acuity was normal; and there was no color-vision deficiency, headache, ocular pain, restriction of extraocular movements, or visual-field defect. In addition, the patient had no musculoskeletal abnormalities, congenital aganglionic megacolon (Hirschsprung disease), or patchy cutaneous depigmentation. Based on the diagnostic criteria for Waardenburg syndrome, the patient was clinically diagnosed with WS1.At 6 years old, the patient received unilateral cochlear implantation (right side) at our institution. Intraoperative findings showed no ossicular malformations, cochlear deformities, or ossification. Additionally, there was no perilymph or cerebrospinal fluid leakage. A CI24RE (CA) cochlear implant was successfully implanted. At the 7-year postoperative follow-up, the patient showed significant improvements in hearing and speech abilities. Detailed auditory and speech evaluation scores are presented in Table 1. Before implantation, the patient had no awareness of environmental sounds and could only respond to gestures; there was no perception of environmental or speech sounds (SIR score 1; CAP, MAIS, and MUSS all 0). By the seventh year after implantation, the patient was able to communicate by telephone with familiar individuals. The CAP and SIR scores reached levels 7 and 4, respectively, while both MAIS and MUSS achieved the maximum score of 40.
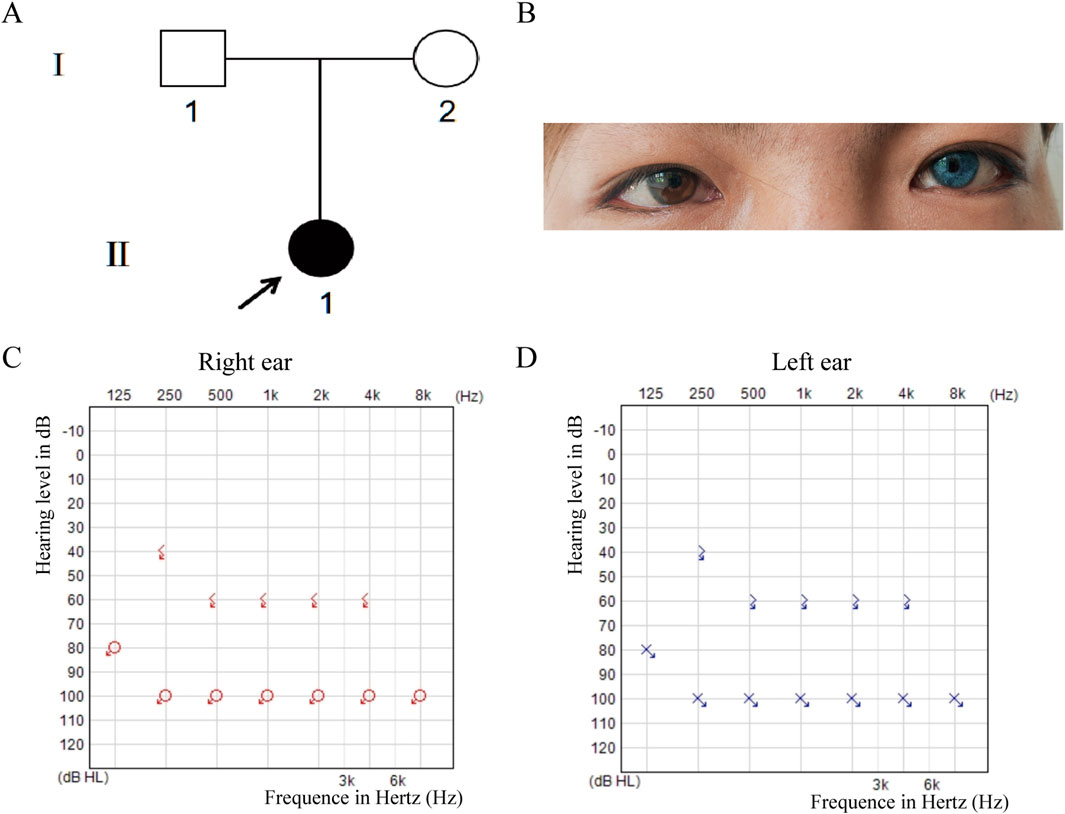
Figure 1. Pedigree, Clinical Features and Hearing Tests. (A) Pedigree shows a sporadic case with the proband (II-1, arrow) as the only affected member. (B) The proband has iris heterochromia (brown left, blue right), wide-set eyes, and a thick left eyebrow. (C,D) Preoperative pure-tone audiometry revealed profound bilateral sensorineural hearing loss at all frequencies (>90 dB HL).
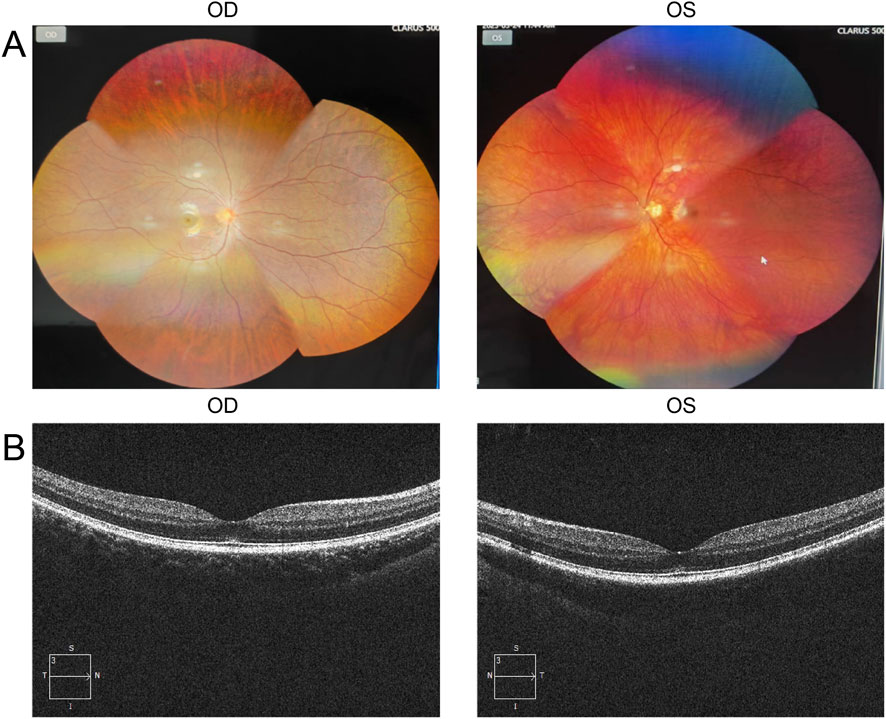
Figure 2. Ocular examination findings of the proband. (A) Color fundus photographs show an essentially normal fundus in the right eye (OD) with uniform pigment distribution, whereas the left eye (OS) exhibits markedly reduced pigmentation. (B) Optical coherence tomography (OCT) demonstrates intact retinal layers in the macular region of both eyes, with no evident pathological changes.
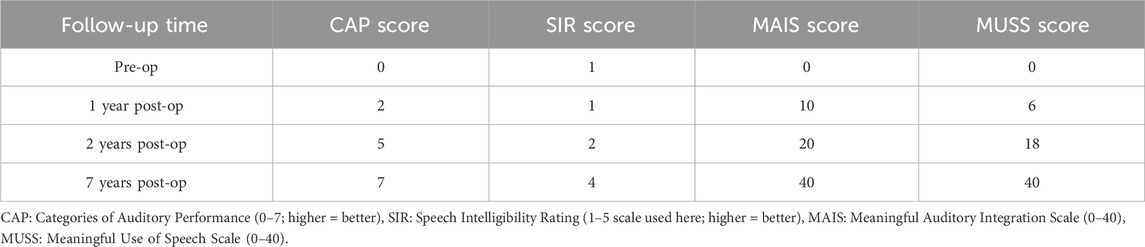
Table 1. Auditory and speech evaluation scores preoperatively and at 1, 2, and 7 years after cochlear implantation.
3.2 Whole exome sequencing result analysis
We performed target gene capture and whole-exome sequencing on the proband, covering approximately 51 Mb of target regions and over 23,000 genes. The average sequencing depth was 203.59×, with 99.49% of the target region covered at a depth greater than 20×. After aligning the sequencing data with the human reference genome (GRCh37/hg19), single nucleotide variants (SNVs) and small insertions/deletions (InDels) were identified. Variants were filtered based on three main criteria: minor allele frequency of less than 0.01 in public population databases, including the 1000 Genomes Project, the NHLBI Exome Sequencing Project (ESP6500), the Exome Aggregation Consortium (ExAC), and the Genome Aggregation Database (gnomAD); localization within exons or canonical splice sites; and functional impact, specifically focusing on non-synonymous variants. Based on variant frequency, functional impact, and phenotypic association, four candidate genes—PAX3, GJB3, DNMT1, and NLRP3—were preliminarily identified. Among these, the variants in GJB3, DNMT1, and NLRP3 were all low-frequency missense mutations lacking functional validation, phenotypic support, and familial co-segregation evidence. According to ACMG assessment, they were classified as variants of uncertain significance (VUS) and were therefore not considered pathogenic mutations (Table 2). Further analysis identified a heterozygous frameshift variant, NM_181458.4: c.788dup (p.Gln264Thrfs), in exon five of the PAX3 gene as a suspected pathogenic variant (ACMG classification: PVS1 + PM2_supporting). This variant was absent from public databases, including the Human Gene Mutation Database (HGMD), 1,000 Genomes, ESP6500, ExAC, and gnomAD, and has not been previously reported. In addition, this variant was predicted as disease-causing (probability score = 1.00) by MutationTaster.
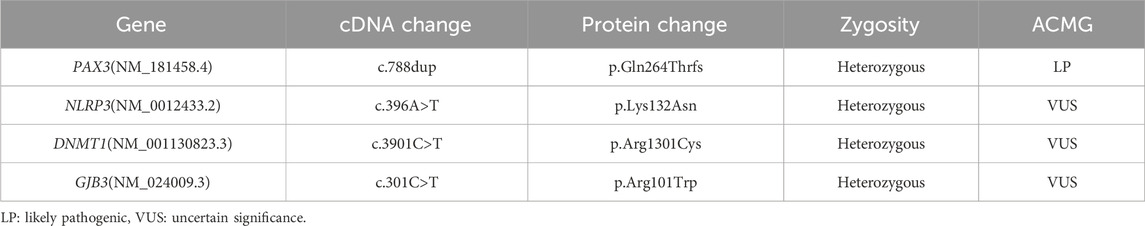
Table 2. Summary of the four candidate gene variants identified in the proband.
3.3 Sanger verification and effect
The PAX3 gene sequences obtained from the triplet samples through Sanger sequencing were compared with the reference sequence (NM_181458.4). The results showed that the proband had a clear double peak insertion pattern (heterozygous) at the c.788 site; neither parent had the insertion mutation at this site and were homozygous wild type. Therefore, this mutation is considered a de novo mutation (Figures 3A–C). Based on this, amino acid evolutionary conservation analysis indicated that the protein sequence in the mutated region is highly conserved during evolution, suggesting that this site is of significant importance for PAX3 protein function (Figure 3D). Further analysis revealed that this mutation is a repeated insertion-type frameshift mutation, resulting in the amino acid sequence of the PAX3 protein changing from QVWFSNRRAR at position 264 to TGLV* (* denotes a stop codon), i.e., the sequence changes begin at position 264 and terminate prematurely at position 268, leading to a truncated PAX3 protein and impaired function (Figure 4).
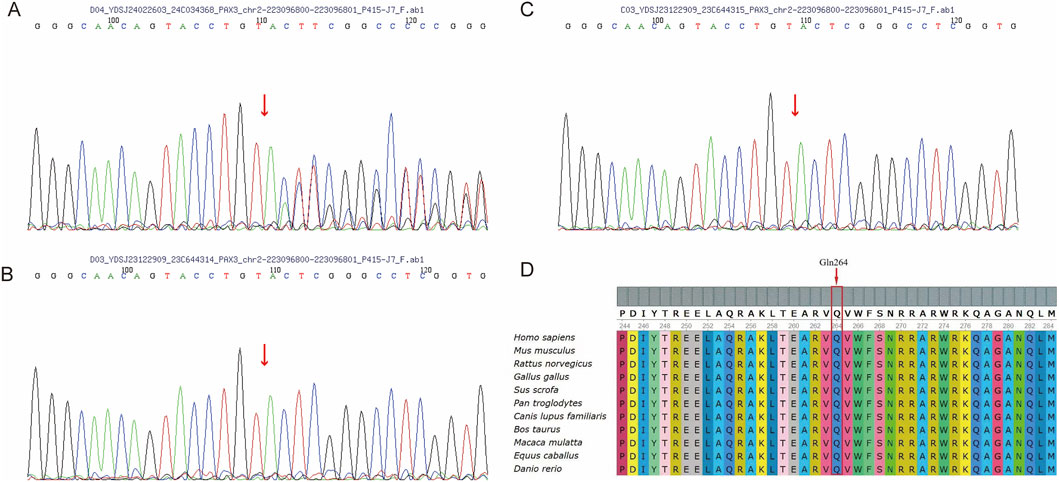
Figure 3. Sanger Sequencing and Conservation of the PAX3 (C)788dup Variant. (A) Sanger sequencing of the proband (II-1) showing a heterozygous (C)788dup variant in PAX3, indicated by a red arrow. (B,C) Sanger sequencing of the father (B, I-1) and mother (C, I-2) shows no duplication at the (C)788 position in the PAX3 gene. (D) Alignment of PAX3 protein sequences from diverse species reveals that the affected residue (Gln264) is highly conserved.
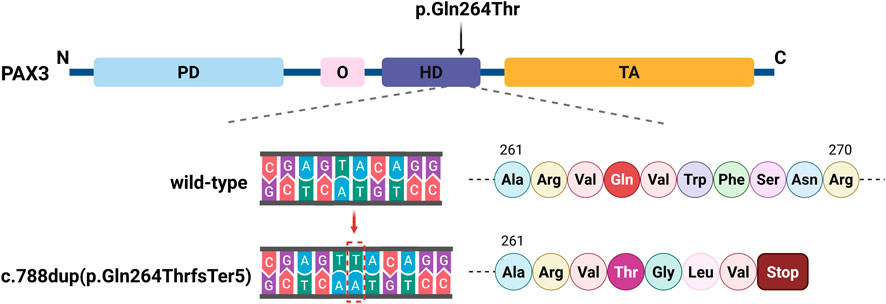
Figure 4. Schematic of the PAX3 c.788dup Mutation. The PAX3 protein includes a paired domain (PD), octapeptide (O), homeodomain (HD), and transactivation domain (TA). The c.788dup mutation causes a thymine duplication within the HD, leading to a frameshift and a premature stop codon at position 268, disrupting the downstream amino acid sequence.
3.4 Structural modeling of the PAX3 protein
To investigate how this variant might affect the structure of PAX3, structural predictions were conducted using AlphaFold to generate three-dimensional models for both the normal and altered PAX3 proteins. The modeling results showed that the wild-type protein (WT) structure remained intact, while the mutant protein (MUT) exhibited significant conformational changes at the C-terminus, resulting in the loss of the functional domain at the C-terminus of the PAX3 protein. We used PyMOL software to perform visualization analysis of the wild-type and mutant protein structures. The results indicated that the mutation significantly altered the spatial conformation of the PAX3 protein’s C-terminal segment, potentially disrupting its normal DNA binding and downstream gene regulatory functions (Figure 5).
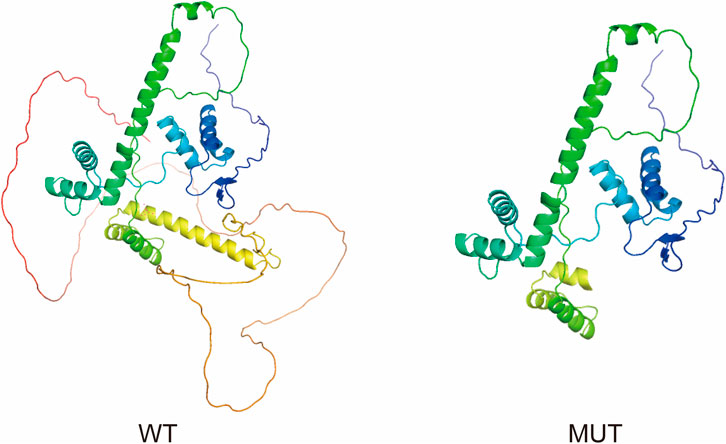
Figure 5. Structural comparison of wild-type and mutant PAX3 proteins. The proteins are displayed as ribbon diagrams with a color gradient from the N-terminus (blue) to the C-terminus (red), indicating the directionality of the polypeptide chain. Ribbon diagrams show the wild-type (WT) protein with intact secondary structures, especially at the C-terminus. The mutant (MUT) protein, affected by a frameshift, displays a truncated C-terminus and loss of several α-helices, potentially impairing structural stability and function.
4 Discussion
In this study, we identified a novel PAX3 mutation (c.788dup) in a Yugur family with WS1 and congenital deafness. This frameshift variant introduces a premature stop codon, producing a truncated protein lacking the C-terminal functional domain. Structural modeling and bioinformatic predictions support its pathogenicity. Combined with clinical features and segregation analysis, the c.788dup mutation is likely the molecular cause of WS1 in this family.
PAX3 gene encodes a critical transcription factor that is highly expressed in neural crest cells during early embryogenesis, where it promotes their differentiation into specific tissues and cell types. Under physiological conditions, PAX3 activates downstream genes, including melanocyte-inducing transcription factor (MITF), which in turn regulates the transcription of key enzymes involved in melanin biosynthesis, playing a pivotal role in melanocyte differentiation. The loss of melanocytes causes pigmentation abnormalities in the skin, hair, and eyes, along with hearing defects due to cochlear dysfunction, resulting in typical WS symptoms (Li et al., 2024). Niu et al. also identified a novel PAX3 variant (c.117C>A; p.Asn39Lys) in a four-generation Chinese family with Waardenburg syndrome type 1 (WS1). They found that this variant impaired the function of wild-type PAX3, preventing activation of the MITF promoter and ultimately leading to the WS clinical phenotype (Niu et al., 2021).
To date, a total of 226 pathogenic mutations have been identified in the PAX3 gene, including missense, nonsense, small insertions, small deletions, and splicing site mutations (https://www.hgmd.cf.ac.uk/ac/gene.php?gene=PAX3). Missense/nonsense mutations account for approximately 51% of PAX3 variants (n = 115), while small insertions represent about 6% (n = 14). Most mutations cluster between exons two and 6, affecting the PD and HD. Studies have shown that approximately 80% of WS patients have heterozygous mutations in the PAX3 gene. We have systematically summarized the PAX3 mutations reported in Chinese WS patients (Table 3). Our review found that over 60 different PAX3 mutations have been reported among Chinese WS patients, with truncating mutations accounting for about 48%, followed by missense mutations. In Chinese patients with WS, PAX3 variants are mainly concentrated in the PD and HD; the detailed distribution is shown in Figure 6. Several variants have been reported repeatedly and may represent hotspots in this population, including c.667C>T, c.117C>A, c.123del, c.127G>T, and c.452–2A>G. WS is inherited in an autosomal dominant manner. While most PAX3 variants in WS are inherited from a parent, multiple studies have also reported de novo mutations. In a cohort of 90 WS probands, Wang et al. identified PAX3 variants in 13 individuals (14.4%; 13/90), three of which were de novo (c.668C>T, c.752T>C, and c.922dupG), accounting for 23.1% (3/13) of PAX3-positive cases (Wang et al., 2022). In addition, de novo PAX3 variants reported in Chinese WS patients include c.166C>A, c.433C>T, c.456_459dupTTCC, c.592delG, c.626_627delCT, c.667C>G, c.668G>T, c.795_800delCTGGTT, c.1459C > T; none were detected in either parent, and most were truncating or missense variants. In our study, the PAX3 c.788dup variant was likewise absent in both parents, confirming it to be de novo. This variant occurs in exon five and alters the PAX3 amino acid sequence beginning at residue 264 (p.Gln264Thrfs); this region lies within the highly conserved homeodomain, which mediates DNA binding. According to MutationTaster, the variant is predicted to cause a frameshift, resulting in a premature termination codon (PTC) and likely triggering nonsense-mediated mRNA decay (NMD). Additionally, the mutation may impair both protein domain function and splicing regulation, ultimately leading to loss of PAX3 function.
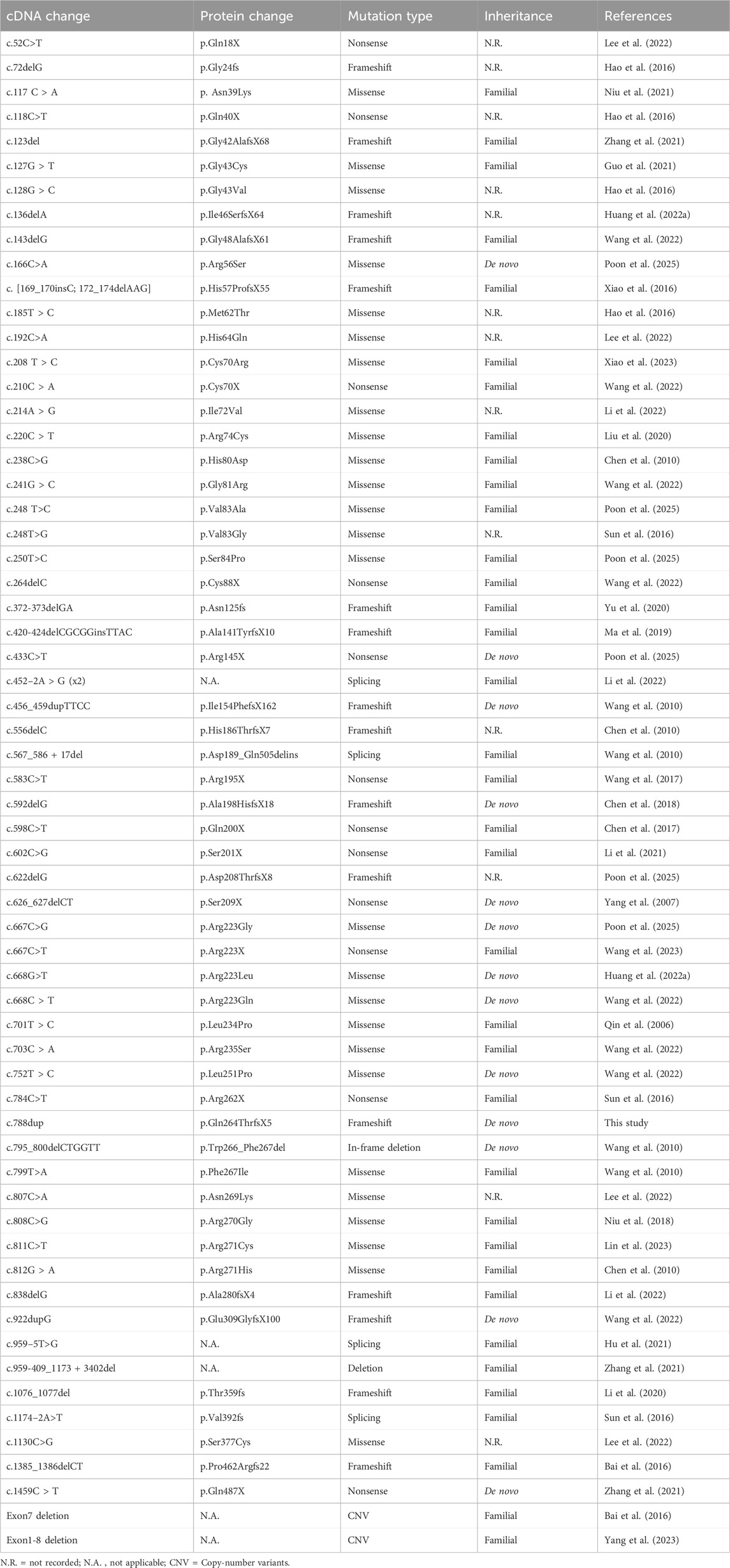
Table 3. The reported PAX3 mutations in Chinese patients with Waardenburg syndrome.
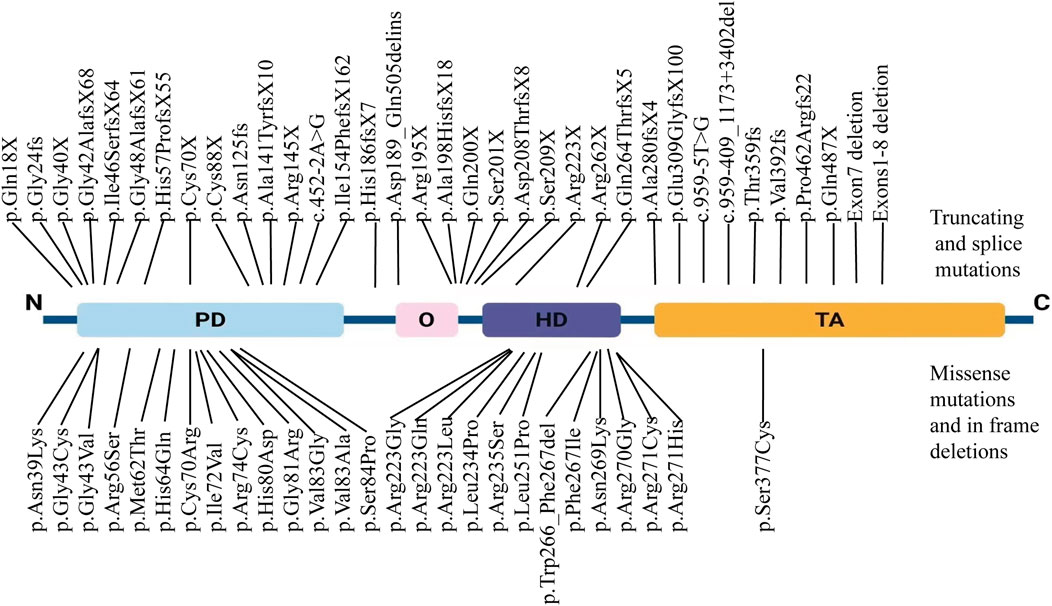
Figure 6. Distribution of PAX3 variants in Chinese patients with Waardenburg syndrome. Truncating and splice-site variants (nonsense, frameshift, and canonical splice-site variants) are shown above the schematic, whereas missense variants and in-frame indels are shown below. Variants span all domains, with notable clustering in the paired domain (PD) and homeodomain (HD).
Several scientific reports confirm that mutations in the HD region of PAX3 alter its spatial conformation, disrupting its nuclear localization and chromatin binding capacity, thereby impairing its function as a transcription factor. Corry et al. reported that HD mutations significantly compromise the dynamic localization and nuclear anchoring of PAX3, leading to aberrant regulation of downstream genes (Corry et al., 2008). Wu et al. further observed that HD-mutated PAX3 variants fail to bind chromosomes during mitosis, thereby affecting cell proliferation and differentiation (Wu et al., 2015). Additionally, HD mutations in PAX3 have been shown to prevent the activation of the MITF promoter, resulting in impaired melanocyte differentiation and survival in WS1 patients (Watanabe et al., 1998). Therefore, this mutation is likely to reduce PAX3 protein levels through mechanisms such as NMD, leading to haploinsufficiency and resulting in pathogenicity (Huang L. et al., 2022).
It is noteworthy that the PAX3 mutation identified in this study (c.788dup, p.Gln264Thrfs) is a frameshift variant, whereas the literature also reports a missense mutation at the same nucleotide position, PAX3 (c.788T>A, p.Val263Gly), both of which are associated with the typical WS1 phenotype (Hazan et al., 2013). This observation suggests that this site is critical for maintaining the structure and transcriptional activity of PAX3. Moreover, several studies have shown that even identical mutations at the same site can exhibit markedly different clinical phenotypes in Waardenburg syndrome. Guo et al. identified a novel missense mutation in exon two of PAX3 (c.127G>A) in the proband of a Han Chinese WS1 family with hearing loss; the proband had unilateral moderate hearing loss, whereas his mother carrying the same mutation exhibited bilateral profound deafness (Guo et al., 2021). Ma et al. reported a WS1 patient diagnosed with a new PAX3 mutation (c.420-424delCGCGGinsTTAC) who had bilateral congenital sensorineural hearing loss, yet the proband’s sister carrying the same mutation had normal hearing (Ma et al., 2019). Such variability may be attributed to the combined influence of epigenetic modifications and environmental factors on the penetrance and expressivity of pathogenic mutations.
Many studies have sought to clarify genotype–phenotype correlations in WS. Li et al. conducted genotype–phenotype assessments in eight Chinese WS families and identified several novel PAX3 variants (c.838delG, c.452–2A>G, and c.214A>G). They also observed that cases carrying PAX3 mutations often exhibited unilateral hearing loss and marked clinical variability (Li et al., 2022). Wang et al. reported the clinical and genetic features of 90 Chinese WS probands, among whom 13 (14.4%) harbored PAX3 mutations; that study further found that PAX3 variants were more frequently associated with asymmetric hearing loss and moderate hearing impairment (Wang et al., 2022). By enrolling 30 genetically confirmed Chinese WS patients and comparing them with other cohorts in the literature, Poon et al. identified six novel PAX3 variants (c.250T>C, c.166C>A, c.248T>C, c.667C>G, c.433C>T, and c.622delG). In that cohort, 22% of WS patients with PAX3 variants presented with asymmetric or unilateral hearing loss, and the prevalence of hearing loss reached 43%, findings similar to those of Wang et al. (Poon et al., 2025).
Currently, there is no cure for WS, and clinical management mainly relies on supportive measures such as hearing aids and cochlear implants to improve hearing (Huang S. et al., 2022). In this case, the patient achieved significant auditory and speech recovery after cochlear implantation and rehabilitation. Three years post-surgery, she successfully transferred to a regular school and is now able to communicate by phone with familiar individuals (CAP level 7), underscoring the importance of early genetic diagnosis and timely intervention in WS1 with severe hearing loss. WS involves multiple organ systems and therefore requires comprehensive, multidisciplinary management. In addition to hearing, attention should be given to patients’ optometric/visual needs, photoprotection, cosmetic concerns, and psychological wellbeing, with appropriate education and interventions provided during follow-up. During follow-up, we offered psychological support and genetic counseling to the patient and family to enhance their understanding of the condition. Moreover, for cosmetic concerns arising from differences in hair or iris pigmentation, we advised options such as appropriate hair dyeing or tinted contact lenses. For concerns about the visibility of the external cochlear-implant sound processor, we suggested physical concealment using hairstyles or accessories. With the development of gene therapy and other technologies, precise treatments targeting such loss-of-function mutations may become feasible in the future. For example, Yao et al. successfully corrected the pathogenic point mutation (MITF c.740T>C) using CRISPR-Cas9-mediated gene editing, restoring vision and hearing loss in a WS pig model (Yao et al., 2021). Additionally, Li et al. developed inner ear organoid models from induced pluripotent stem cells (iPSCs) derived from WS1 patients carrying the PAX3 c.214A>G mutation and from healthy controls. Using CRISPR/Cas9, they corrected the PAX3 mutation in patient-derived iPSCs. In the corrected organoids, they observed restoration of WNT1/β-catenin signaling, reduced apoptosis, increased organoid size, and significant reversal of cochlear developmental defects associated with WS1 (Li et al., 2024).
In summary, the novel heterozygous PAX3 mutation identified in this study is the likely genetic cause of WS1 in this family, expanding the known mutation spectrum and contributing to research in minority populations. Future efforts should focus on functional validation, animal models, and gene-editing strategies. Multi-omics approaches may clarify phenotypic variability. Clinically, early genetic screening, neonatal hearing tests, and counseling are essential. Advances in gene therapy, such as CRISPR/Cas9, hold promise for precise, targeted treatment of WS.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1345158. BioProject accession: PRJNA1345158; BioSample accession: SAMN52662672.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Second Hospital of Lanzhou University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
LZ: Investigation, Visualization, Writing – original draft, Writing – review and editing. BX: Conceptualization, Funding acquisition, Writing – review and editing. DL: Data curation, Writing – review and editing. YW: Data curation, Writing – review and editing. PB: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China Regional Projects (No. 32160149, No. 82460222), the Key Research and Development Program of Gansu Province (No. 21JR11RA093), the “Cuiying Scientific and Technological Innovation” Program of Second Hospital of Lanzhou University (No. CY2023-MS-B08), and the Longyuan Youth Talent Program (No. PR0161005).
Acknowledgments
We thank the patient and her family for their valuable participation and support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bai, Y., Liu, N., Kong, X. D., Yan, J., Qin, Z. B., and Wang, B. (2016). PAX3 gene mutation analysis for two Waardenburg syndrome type Ⅰ families and their prenatal diagnosis. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 51, 896–901. doi:10.3760/cma.j.issn.1673-0860.2016.12.004
Boudjadi, S., Chatterjee, B., Sun, W., Vemu, P., and Barr, F. G. (2018). The expression and function of PAX3 in development and disease. Gene 666, 145–157. doi:10.1016/j.gene.2018.04.087
Chen, H., Jiang, L., Xie, Z., Mei, L., He, C., Hu, Z., et al. (2010). Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochem. Biophysical Res. Commun. 397, 70–74. doi:10.1016/j.bbrc.2010.05.066
Chen, D., Zhao, N., Wang, J., Li, Z., Wu, C., Fu, J., et al. (2017). Whole-exome sequencing analysis of Waardenburg syndrome in a Chinese family. Hum. Genome Var. 4, 17027. doi:10.1038/hgv.2017.27
Chen, K., Zhan, Y., Wu, X., Zong, L., and Jiang, H. (2018). Germinal mosaicism of PAX3 mutation caused Waardenburg syndrome type I. Int. J. Pediatr. Otorhinolaryngology 104, 200–204. doi:10.1016/j.ijporl.2017.11.011
Corry, G. N., Hendzel, M. J., and Underhill, D. A. (2008). Subnuclear localization and mobility are key indicators of PAX3 dysfunction in Waardenburg syndrome. Hum. Mol. Genet. 17, 1825–1837. doi:10.1093/hmg/ddn076
Farrer, L. A., Grundfast, K. M., Amos, J., Arnos, K. S., Asher, J. H., Beighton, P., et al. (1992). Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS consortium. Am. J. Hum. Genet. 50, 902–913.
George, F. S. A., Sam, L. E., Tanwar, M., and Wall, L. (2023). Association of autism spectrum disorder with Waardenburg syndrome in a toddler. BMJ Case Rep. 16, e254741. doi:10.1136/bcr-2023-254741
Gombojav, B., Erdenechuluun, J., Batsaikhan, T., Danshiitsoodol, N., Makhbal, Z., Jargalmaa, M., et al. (2025). Waardenburg syndrome type 4 in Mongolian children: genetic and clinical characterization. IJMS 26, 6258. doi:10.3390/ijms26136258
Guo, M., Li, Q., Jiang, C., Li, S., and Ruan, B. (2021). A novel PAX3 mutation in a Chinese Han family with Waardenburg syndrome type 1. Int. J. Pediatr. Otorhinolaryngology 147, 110758. doi:10.1016/j.ijporl.2021.110758
Han, W., Yang, R., Chen, X., Chen, Y., Zhang, T., and Ma, J. (2025). A novel frameshift mutation of SOX10 identified in Waardenburg syndrome type 2. Hum. Mol. Genet. 34, 668–672. doi:10.1093/hmg/ddaf010
Hao, Z., Zhou, Y., Li, P., Zhang, Q., Li, J., Wang, P., et al. (2016). Mutation analysis of seven patients with Waardenburg syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 33, 312–315. doi:10.3760/cma.j.issn.1003-9406.2016.03.007
Hazan, F., Ozturk, A. T., Adibelli, H., Unal, N., and Tukun, A. (2013). A novel missense mutation of the paired box 3 gene in a Turkish family with Waardenburg syndrome type 1. Mol. Vis. 19, 196–202.
Hu, Q., Ma, H., Shen, J., Zhuang, Z., Li, J., Huang, X., et al. (2021). Case report: a novel PAX3 mutation associated with Waardenburg syndrome type 1. Front. Genet. 12, 609040. doi:10.3389/fgene.2021.609040
Huang, L., Guo, M., and Li, N. (2022a). Case report: exotropia in Waardenburg syndrome with novel variations. Front. Genet. 13, 969680. doi:10.3389/fgene.2022.969680
Huang, S., Song, J., He, C., Cai, X., Yuan, K., Mei, L., et al. (2022b). Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome. Gene Ther. 29, 479–497. doi:10.1038/s41434-021-00240-2
Lee, C., Lo, M., Chen, Y., Lin, P., Hsu, C., Chen, P., et al. (2022). Identification of nine novel variants across PAX3, SOX10, EDNRB, and MITF genes in Waardenburg syndrome with next-generation sequencing. Molec Gen and Gen Med 10, e2082. doi:10.1002/mgg3.2082
Li, S., Guo, M., Ruan, B., Liu, Y., Cui, X., Han, W., et al. (2020). A novel PAX3 mutation in a Chinese family with Waardenburg Syndrome type 1. Genet. Test. Mol. Biomarkers 24, 249–255. doi:10.1089/gtmb.2019.0231
Li, X., Zhao, S., Bi, X., Lou, F., Zeng, W., Gao, Y., et al. (2021). Genotype and phenotype analysis of a family with Waardenburg syndrome type Ⅰcaused by a novel mutation in PAX3 gene. Lin. Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 35, 621–626. doi:10.13201/j.issn.2096-7993.2021.07.010
Li, S., Qin, M., Mao, S., Mei, L., Cai, X., Feng, Y., et al. (2022). A comprehensive genotype–phenotype evaluation of eight Chinese probands with Waardenburg syndrome. BMC Med. Genomics 15, 230. doi:10.1186/s12920-022-01379-6
Li, S., He, C., Mei, L., Wu, X., Feng, Y., and Song, J. (2024). PAX3 mutation suppress otic progenitors proliferation and induce apoptosis by inhibiting WNT1/β-catenin signaling pathway in WS1 patient iPSC-derived inner ear organoids. Biochem. Biophysical Res. Commun. 698, 149510. doi:10.1016/j.bbrc.2024.149510
Lin, Z., Tang, J., Yun, L., Zhou, K., Huang, L., Qu, S., et al. (2023). Analysis of molecular genetics and clinical characteristics of 3 children with Waardenburg syndrome. Lin. Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 37, 107–112. doi:10.13201/j.issn.2096-7993.2023.02.006
Liu, Q., Cheng, J., Lu, Y., Zhou, J., Wang, L., Yang, C., et al. (2020). The clinical and genetic research of Waardenburg syndrome type I and II in Chinese families. Int. J. Pediatr. Otorhinolaryngology 130, 109806. doi:10.1016/j.ijporl.2019.109806
Ma, J., Lin, K., Jiang, H., Yang, Y., Zhang, Y., Yang, G., et al. (2019). A novel mutation of the PAX3 gene in a Chinese family with Waardenburg syndrome type I. Molec Gen and Gen Med 7, e00798. doi:10.1002/mgg3.798
Matsunaga, T., Mutai, H., Namba, K., Morita, N., and Masuda, S. (2013). Genetic analysis of PAX3 for diagnosis of Waardenburg syndrome type I. Acta Oto-Laryngologica 133, 345–351. doi:10.3109/00016489.2012.744470
Moore, S. P. G., Ganesh Krishnan, S., Jaswanth Kothari, R., Prince, N. B., Kenny, C., Zhang, C., et al. (2025). PAX3 regulatory signatures and gene targets in Melanoma cells. Genes 16, 577. doi:10.3390/genes16050577
Niu, Z., Li, J., Tang, F., Sun, J., Wang, X., Jiang, L., et al. (2018). Identification and functional analysis of a novel mutation in the PAX3 gene associated with Waardenburg syndrome type I. Gene 642, 362–366. doi:10.1016/j.gene.2017.11.035
Niu, Z., Mei, L., Tang, F., Li, J., Wang, X., Sun, J., et al. (2021). Identification and functional analysis of a novel missense mutation of PAX3 associated with Waardenburg syndrome type I. Eur. Arch. Otorhinolaryngol. 278, 2807–2815. doi:10.1007/s00405-020-06361-5
Poon, J. Y. Y., Yiu, W. C., Ho, S. K. L., Cheng, S. S. W., Luk, H., and Lo, I. F. M. (2025). Waardenburg Syndrome: review of genotype–phenotype relationships in 30 patients in Hong Kong. Am. J Med Genet. Pt A, e64193. doi:10.1002/ajmg.a.64193
Qin, W., Shu, A., Qian, X., Gao, J., Xing, Q., Zhang, J., et al. (2006). A novel mutation of PAX3 in a Chinese family with Waardenburg syndrome. Mol. Vis. 12, 1001–1008.
Sun, L., Li, X., Shi, J., Pang, X., Hu, Y., Wang, X., et al. (2016). Molecular etiology and genotype-phenotype correlation of Chinese Han deaf patients with type I and type II Waardenburg Syndrome. Sci. Rep. 6, 35498. doi:10.1038/srep35498
Tawfik, C. A., Essawi, M. L., Nowara, M., Mohsen, R., and Elbagoury, N. M. (2025). Concurrent novel mutations in PAX3 and CFAP410 in a patient with Waardenburg syndrome type 1 associated with retinitis pigmentosa. Ophthalmic Genet. 46, 305–312. doi:10.1080/13816810.2025.2473972
Tekin, M., Bodurtha, J., Nance, W., and Pandya, A. (2001). Waardenburg syndrome type 3 (Klein–Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3: a simple variant or a true syndrome? Clin. Genet. 60, 301–304. doi:10.1034/j.1399-0004.2001.600408.x
Waardenburg, P. J. (1951). A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am. J. Hum. Genet. 3, 195–253.
Wang, J., Li, S., Xiao, X., Wang, P., Guo, X., and Zhang, Q. (2010). PAX3 mutations and clinical characteristics in Chinese patients with Waardenburg syndrome type 1. Mol. Vis. 16, 1146–1153.
Wang, L., Qin, L., Li, T., Liu, H., Ma, L., Li, W., et al. (2017). Prenatal diagnosis and genetic counseling�for Waardenburg syndrome type�i and II in Chinese families. Mol. Med. Rep. 17, 172–178. doi:10.3892/mmr.2017.7874
Wang, G., Li, X., Gao, X., Su, Y., Han, M., Gao, B., et al. (2022). Analysis of genotype–phenotype relationships in 90 Chinese probands with Waardenburg syndrome. Hum. Genet. 141, 839–852. doi:10.1007/s00439-021-02301-3
Wang, L., Mao, L., Xu, H., Sun, S., Zuo, B., and Lu, W. (2023). Analysis of clinical phenotype and genetic variants among four Chinese pedigrees affected with Waardenburg syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 40, 661–667. doi:10.3760/cma.j.cn511374-20220727-00498
Watanabe, A., Takeda, K., Ploplis, B., and Tachibana, M. (1998). Epistatic relationship between Waardenburg Syndrome genes MITF and PAX3. Nat. Genet. 18, 283–286. doi:10.1038/ng0398-283
Wu, T.-F., Yao, Y.-L., Lai, I.-L., Lai, C.-C., Lin, P.-L., and Yang, W.-M. (2015). Loading of PAX3 to mitotic chromosomes is mediated by arginine methylation and associated with Waardenburg Syndrome. J. Biol. Chem. 290, 20556–20564. doi:10.1074/jbc.M114.607713
Xiao, Y., Luo, J., Zhang, F., Li, J., Han, Y., Zhang, D., et al. (2016). A novel mutation in PAX3 associated with Waardenburg syndrome type I in a Chinese family. Acta Oto-Laryngologica 136, 439–445. doi:10.3109/00016489.2015.1132846
Xiao, X., Huang, Y., Zhang, J., Cao, Y., and Zhang, M. (2023). Identification of two variants in PAX3 and FBN1 in a Chinese family with Waardenburg and Marfan syndrome via whole exome sequencing. Funct. Integr. Genomics 23, 114. doi:10.1007/s10142-023-01012-4
Yang, S.-Z., Cao, J.-Y., Zhang, R.-N., Liu, L.-X., Liu, X., Zhang, X., et al. (2007). Nonsense mutations in the PAX3 gene cause Waardenburg syndrome type I in two Chinese patients. Chin. Med. J. Engl. 120, 46–49. doi:10.1097/00029330-200701010-00009
Yang, S.-Z., Hou, L., Qi, X., Wang, G.-J., Huang, S.-S., Zhang, S.-S., et al. (2023). A gross deletion of the PAX3 gene in a large Chinese family with Waardenburg syndrome type I. World J. Pediatr. 19, 1203–1207. doi:10.1007/s12519-023-00746-2
Yao, J., Wang, Y., Cao, C., Song, R., Bi, D., Zhang, H., et al. (2021). CRISPR/Cas9-mediated correction of MITF homozygous point mutation in a Waardenburg syndrome 2A pig model. Mol. Ther. - Nucleic Acids 24, 986–999. doi:10.1016/j.omtn.2021.04.009
Yu, Y., Liu, W., Chen, M., Yang, Y., Yang, Y., Hong, E., et al. (2020). Two novel mutations of PAX3 and SOX10 were characterized as genetic causes of Waardenburg Syndrome. Molec Gen and Gen Med 8, e1217. doi:10.1002/mgg3.1217
Keywords: PAX3, Waardenburg syndrome, whole-exome sequencing, de novo mutation, hearing loss
Citation: Zhan L, Xu B, Lin D, Wang Y and Bian P (2025) A novel frameshift variant of PAX3 in a Chinese Yugur family with Waardenburg syndrome type 1. Front. Genet. 16:1679351. doi: 10.3389/fgene.2025.1679351
Received: 04 August 2025; Accepted: 06 October 2025;
Published: 22 October 2025.
Edited by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Simon Carlo, Ponce Health Sciences University, Puerto RicoHaoyu Li, Central South University, China
Copyright © 2025 Zhan, Xu, Lin, Wang and Bian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panpan Bian, YmlhbnBhbnBhbjY2NzMxQDE2My5jb20=