 Roman Zug1,2*
Roman Zug1,2*- 1Institute of Organismic and Molecular Evolution (iomE), Johannes Gutenberg University Mainz, Mainz, Germany
- 2Institute for Quantitative and Computational Biosciences (IQCB), Johannes Gutenberg University Mainz, Mainz, Germany
Maturity-onset diabetes of the young (MODY) is an autosomal dominant form of monogenic diabetes, frequently caused by heterozygous loss-of-function variants in transcription factor (TF) genes. Why are MODY variants in TF genes dominantly inherited? Here I present a systems biology-based explanation. The fact that MODY-associated TFs are master regulators of pancreatic β cell fate suggests that pathogenic variants cause defects in cell fate determination. From a systems biology perspective, cell fate defects are based on disrupted bistability, a crucial feature of dynamical systems to make binary choices. Bistability requires both positive feedback and ultrasensitivity, the latter often in the form of cooperativity. MODY-associated TFs exhibit both features, which not only allows for bistability, but also makes these TFs extremely dosage sensitive, which explains why heterozygous loss of function is sufficient to cause a disease phenotype. A review of the literature strongly supports this hypothesis. Moreover, the hypothesis also helps to explain why incomplete penetrance is such a pervasive feature of MODY-associated variants in TF genes.
Introduction
Maturity-onset diabetes of the young (MODY) is a rare inherited form of diabetes caused by mutations in a single gene and characterized by an early onset, typically before the age of 25 years. MODY represents the most common form of monogenic diabetes and is due to impaired development and function of pancreatic β cells, resulting in deficient secretion of insulin. MODY is inherited predominantly in an autosomal dominant mode, which is remarkable because other forms of monogenic diabetes do not show this inheritance pattern (Bonnefond et al., 2023). Why are most MODY variants dominantly inherited? In an attempt to address this issue, Li et al. (2022) asked: “Could it be because of some biological property of the insulin-secreting pancreatic β cells that makes them susceptible to the deleterious effects of heterozygous but not homozygous variants?” Interestingly, however, rather than giving an answer to this question, Li et al. proposed that autosomal recessive forms of MODY “are at least as common as the dominant ones, but have not been discovered yet”. In their view, the preponderance of autosomal dominant MODY does not have a biological reason, but rather reflects our inability to detect recessive variants. While this is theoretically possible, I here argue that there is indeed a plausible biological reason why MODY is mostly dominant. In the following sections I will outline the hypothesis step by step, together with empirical evidence. Finally, I discuss why the hypothesis also helps to explain incomplete penetrance of MODY variants, and I disprove the idea that homozygous variants are less deleterious.
Most MODY cases are caused by variants in master transcription factor (TF) genes
Although the protein products of MODY genes serve a variety of molecular functions, by far the largest functional group comprises transcription factors (TFs) (Figure 1). These TFs act as master regulators of pancreatic development, and of β cell differentiation and function in particular (Arda et al., 2013; Conrad et al., 2014; Dassaye et al., 2016; Wortham and Sander, 2021). Therefore, MODY cases that are due to impaired pancreas and β cell development, caused by variants in master TF genes, should be considered developmental disorders (Zug, 2022).
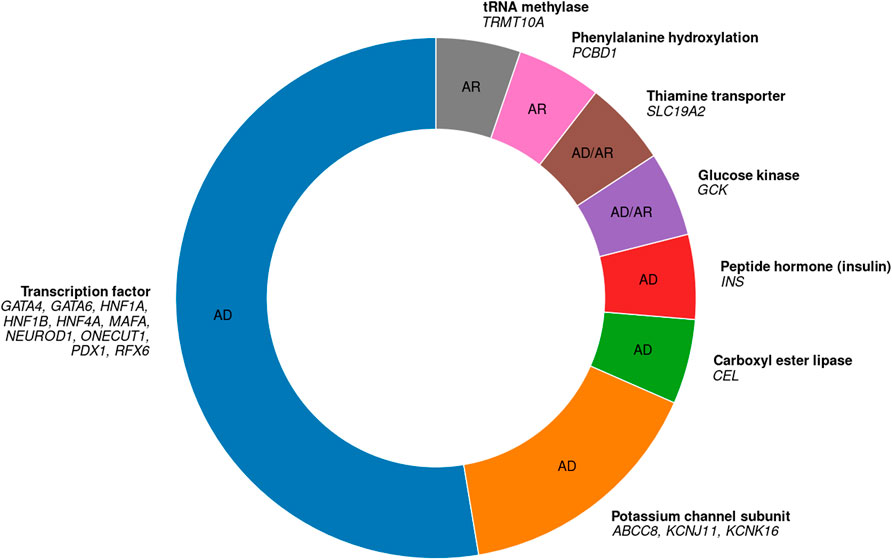
Figure 1. All 19 known autosomal MODY genes, their protein functions, and their inheritance modes. 10 genes code for transcription factors, 3 for potassium channel subunits, and one gene, respectively, for each of the other categories. AD, autosomal dominant; AR, autosomal recessive. Based on Bonnefond et al. (2023) and Sriram et al. (2025).
Out of 19 autosomal genes in which MODY-causing variants are known, 10 code for TFs (Figure 1) (Bonnefond et al., 2023). These TF genes are given in Table 1. Note that I do not include two other TF genes, KLF11 and PAX4 (nor the genes APPL1, BLK, and WFS1), because there is insufficient evidence that variants in these genes actually cause MODY (Laver et al., 2022; Sriram et al., 2025). Of the 10 known MODY-associated TF genes, the three most common ones alone are estimated to account for more than two-thirds of all MODY cases: HNF1A (52%), HNF4A (10%), and HNF1B (6%) (Shields et al., 2010).
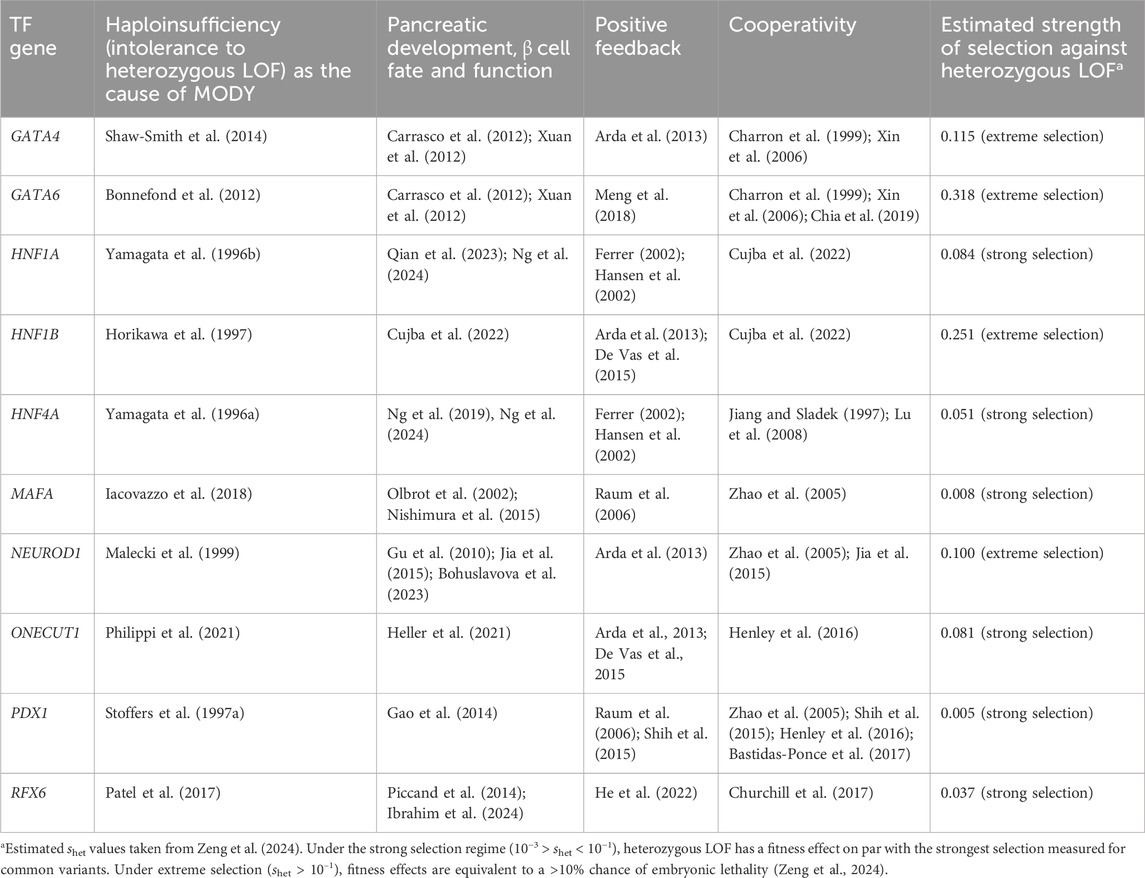
Table 1. Features of transcription factor (TF) genes in which MODY-causing variants have been identified. All variants exhibit autosomal dominant inheritance.
Haploinsufficiency of master TF genes as a cause of developmental disorders
All known MODY-causing variants in TF genes show dominant inheritance. Why is that? In order to answer this question, let us look at these variants in more detail. All MODY-associated variants in TF genes cause loss-of-function (LOF). This is not surprising, as most mutations cause LOF. What is surprising, though, is that LOF of a single allele is sufficient to cause a clinical phenotype. In other words, a 50% reduction in gene expression is not tolerated. This pronounced dosage sensitivity is called haploinsufficiency. Strikingly, all MODY-associated TF genes are haploinsufficient and hence intolerant to heterozygous LOF variants (Table 1). Haploinsufficiency is a manifestation of genetic dominance, as a phenotype is already visible in the heterozygous state (Zschocke et al., 2023). Haploinsufficiency represents a particularly strict form of gene essentiality, which can be defined as a considerable reduction in organismal fitness associated with a gene’s LOF (Bartha et al., 2018). Accordingly, in haploinsufficient genes, there is strong negative selection even against heterozygous LOF variants, reducing the frequency of such variants in the population (‘selective constraint’) (Zeng et al., 2024). Haploinsufficiency is a hallmark of master regulator TF genes and can lead to a plethora of developmental disorders, MODY being one of them (Seidman and Seidman, 2002; Zug, 2022). Most often, MODY is caused by LOF variants in the coding regions of TF genes, but it can also be due to LOF variants in the cis-regulatory elements (CREs) of these genes, as has been shown, for example, for HNF1A (Gragnoli et al., 1997) and HNF4A (Hansen et al., 2002).
Bistability and its disruption through TF haploinsufficiency
Why are heterozygous LOF variants in TF genes (or their CREs) sufficient to cause MODY? In other words, why are these TFs so dosage-sensitive? Building upon earlier work (Wilkie, 1994; Veitia, 2002; Johnson et al., 2019), I have recently proposed the hypothesis that developmental disorders caused by TF haploinsufficiency result from defects in cell fate determination, which can be traced to disrupted bistability in the underlying gene regulatory network (Zug, 2022). Bistability is a crucial feature of dynamical systems that are able to make binary choices such as cell fate decisions. Bistability means that a system can be resting in two alternative stable states but not in intermediate states, resulting in switch-like threshold effects. The threshold corresponds to an unstable steady state separating the two stable steady states. A system is able to generate bistability if its components engage both in positive feedback and ultrasensitivity (Ferrell, 2002). Positive feedback prevents the system from resting in intermediate states. Ultrasensitivity means that an increase in the input signal first has little effect, but then produces higher and higher levels of output, as represented by a steep sigmoidal curve. Ultrasensitivity filters small stimuli out of the feedback loop, allowing the system to have a stable off-state (Ferrell, 2002), and often comes in the form of cooperativity (Zhang et al., 2013). Bistable switches based on positive feedback and cooperativity are a pervasive regulatory motif underling cell fate determination. Cell fate is mainly controlled by the assembly of master regulators at cell-type-specific enhancers, often called super-enhancers (Figure 2A). Genes associated with super-enhancers include those encoding the master regulators themselves, thus establishing autoregulatory positive feedback loops. Super-enhancers assemble a high density of master regulators, allowing for extensive cooperative TF-DNA binding (e.g., via dimerization). The requirement of both positive feedback and cooperativity for proper cell fate determination helps to explain the distinct dosage sensitivity (that is, haploinsufficiency) of master regulators: it is the high level of cooperativity (an instantiation of ultrasensitivity) that makes the positive feedback loops particularly sensitive to changes in TF concentration. Therefore, heterozygous LOF variants that disrupt positive feedback or cooperativity are sufficient to interfere with proper cell fate determination and eventually lead to developmental disorders such as MODY (Figure 2B) (Zug, 2022). Although bistability has been invoked before to explain MODY etiology (in the context of the HNF1A–HNF4A positive feedback loop: Ferrer, 2002; Kaci et al., 2024), these studies ignore cooperativity, failing to account for both requirements of bistability.
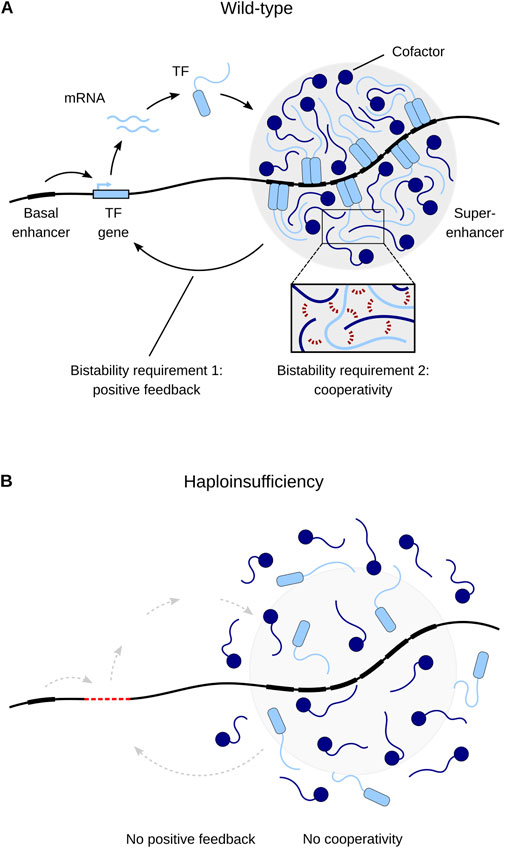
Figure 2. A model of transcriptional regulation of cell fate and its misregulation due to haploinsufficiency. (A) Master transcription factors (TFs) maintain their own expression through positive feedback by cooperatively binding to their own super-enhancers. Cooperative binding occurs through multivalent interactions between TFs and cofactors (inset, red dashed lines). (B) Haploinsufficiency (here caused by TF gene deletion) disrupts positive feedback and cooperativity and thus leads to disease. Adapted from Zug (2022).
Supporting evidence with respect to MODY
Here I collect evidence in support of the hypothesis that heterozygous LOF variants in MODY-associated TF genes disrupt positive feedback or cooperativity and thus cause the disease. As shown in Table 1, all known MODY-associated TF genes (1) are haploinsufficient, (2) are master regulators of pancreatic development, and of β cell fate and function in particular, (3) engage in positive feedback and cooperativity, and (4) exhibit strong or extreme selection against heterozygous LOF, as estimated by a powerful Empirical Bayes approach (Zeng et al., 2024). Moreover, for HNF1A and HNF4A, disruption of positive feedback (Hansen et al., 2002) and of cooperativity (Hua et al., 2000; Singh et al., 2019) has been identified as the cause of MODY. Taken together, this evidence strongly supports the hypothesis that heterozygous LOF variants in master regulators of β cell fate are sufficient to disrupt TF positive feedback or cooperativity and thus cause MODY. This explains why MODY-associated variants in TF genes are dominantly inherited.
A better understanding of incomplete penetrance of MODY-associated TF genes
The hypothesis outlined above also helps to better understand why not every individual carrying a pathogenic variant actually develops the disease, a phenomenon termed incomplete penetrance (Kingdom and Wright, 2022). Many MODY-associated TF genes show incomplete penetrance, e.g., HNF1A, HNF1B, HNF4A, NEUROD1, PDX1, and RFX6 (Mirshahi et al., 2022; Li et al., 2023; Sharp et al., 2025; Sriram et al., 2025). Even though incomplete penetrance can be caused by a range of factors, it has a strong genetic basis (Kingdom and Wright, 2022). Goldschmidt (1938) explained incomplete penetrance by assuming stochastic fluctuation in gene expression combined with some threshold effect. The stochastic nature of gene expression is now well established (Raj and van Oudenaarden, 2008). Goldschmidt’s postulated threshold effect can be readily explained as well, at least with respect to master regulator TFs: as outlined above, these TFs engage in positive feedback and cooperativity, which allows for bistability and, thus, threshold effects. Heterozygous LOF variants bring gene expression levels close to the threshold, but only those variants for which gene expression happens to lie below the threshold will elicit a disease phenotype (Figure 3). This idea explains incomplete penetrance of variants in TF genes, including those associated with MODY (Zug, 2022).
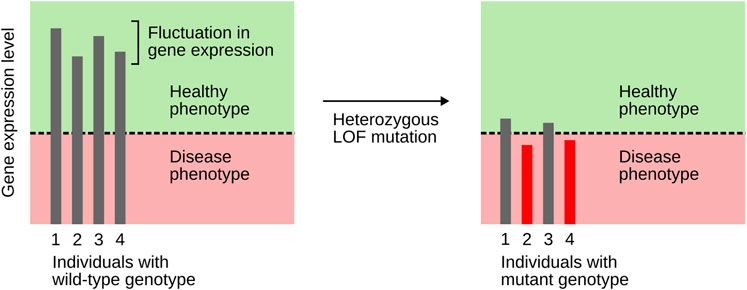
Figure 3. An explanation of incomplete penetrance, based on the combined action of stochastic fluctuation in gene expression and a threshold effect. The latter results from TF genes engaging in positive feedback and cooperativity. Heterozygous LOF mutations bring expression levels close to the threshold. Only individuals whose gene expression lies below the threshold (here, individuals 2 and 4) will show a disease phenotype. Based on Hobert (2010).
Homozygous LOF variants generally show worse outcomes than heterozygous variants
Lastly, I would like to address the assumption made by Li et al. (2022) that β cells are susceptible to heterozygous but not homozygous variants. I argue that this assumption is wrong, at least with respect to TF genes. The reason is that, in TF genes, variants in the homozygous state have generally more severe consequences than in the heterozygous state, a phenomenon termed semi-dominance (Zschocke et al., 2023). In many MODY-associated TF genes, homozygous LOF variants are generally thought to be embryonically lethal or result in early mortality, e.g., in GATA4 (Kuo et al., 1997), GATA6 (Morrisey et al., 1998), HNF1A (Harries et al., 2009), HNF1B (Barbacci et al., 1999), HNF4A (Chen et al., 1994), and ONECUT1 (Philippi et al., 2021). In other MODY-associated TF genes, homozygous variants exist but cause severe syndromic and usually neonatal diabetes, such as in MAFA (Iacovazzo et al., 2018), NEUROD1 (Rubio-Cabezas et al., 2010), PDX1 (Stoffers et al., 1997b) and RFX6 (Smith et al., 2010). Therefore, the question is not why MODY-associated TF genes are not susceptible to homozygous variants (they are very much so), but rather why homozygous variants are tolerated at all (at least in some genes), given the generally high intolerance of these genes even to heterozygous variants.
Conclusion
LOF variants in TFs controlling pancreatic β cell fate are a common cause of MODY. To understand the dominant inheritance of such variants, I have adopted a systems biology perspective. I have shown that MODY-associated TFs are involved in positive feedback and cooperativity, which makes them extremely dosage-sensitive and thus explains why even heterozygous LOF is not tolerated, resulting in dominance. The proposed hypothesis also helps to explain incomplete penetrance, which is widespread in MODY, and thus advances our understanding of the most common form of monogenic diabetes. Future studies should gather further empirical evidence showing that MODY is caused by disrupted TF cooperativity or positive feedback (beyond HNF1A and HNF4A). Another important issue for future work is to investigate what distinguishes those TF genes that are able to tolerate homozygous LOF variants. It will also be interesting to elucidate how functioning of the bistable switch motif is affected by polygenic background, which can substantially modify MODY penetrance and expressivity (Murray Leech et al., 2025).
Author contributions
RZ: Conceptualization, Investigation, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by funding from the Alexander von Humboldt Foundation (to Hanna Kokko) and from the Open Access Publication Fund of the Johannes Gutenberg University Mainz.
Acknowledgements
I thank the reviewer for their constructive comments, which improved the article. I apologize to all authors whose work I could not cite due to space constraints.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arda, H. E., Benitez, C. M., and Kim, S. K. (2013). Gene regulatory networks governing pancreas development. Dev. Cell 25, 5–13. doi:10.1016/j.devcel.2013.03.016
Barbacci, E., Reber, M., Ott, M.-O., Breillat, C., Huetz, F., and Cereghini, S. (1999). Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. Development 126, 4795–4805. doi:10.1242/dev.126.21.4795
Bartha, I., di Iulio, J., Venter, J. C., and Telenti, A. (2018). Human gene essentiality. Nat. Rev. Genet. 19, 51–62. doi:10.1038/nrg.2017.75
Bastidas-Ponce, A., Roscioni, S. S., Burtscher, I., Bader, E., Sterr, M., Bakhti, M., et al. (2017). Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic β-cells. Mol. Metab. 6, 524–534. doi:10.1016/j.molmet.2017.03.007
Bohuslavova, R., Fabriciova, V., Smolik, O., Lebrón-Mora, L., Abaffy, P., Benesova, S., et al. (2023). NEUROD1 reinforces endocrine cell fate acquisition in pancreatic development. Nat. Commun. 14, 5554. doi:10.1038/s41467-023-41306-6
Bonnefond, A., Sand, O., Guerin, B., Durand, E., De Graeve, F., Huyvaert, M., et al. (2012). GATA6 inactivating mutations are associated with heart defects and, inconsistently, with pancreatic agenesis and diabetes. Diabetologia 55, 2845–2847. doi:10.1007/s00125-012-2645-7
Bonnefond, A., Unnikrishnan, R., Doria, A., Vaxillaire, M., Kulkarni, R. N., Mohan, V., et al. (2023). Monogenic diabetes. Nat. Rev. Dis. Prim. 9, 12. doi:10.1038/s41572-023-00421-w
Carrasco, M., Delgado, I., Soria, B., Martín, F., and Rojas, A. (2012). GATA4 and GATA6 control mouse pancreas organogenesis. J. Clin. Invest. 122, 3504–3515. doi:10.1172/JCI63240
Charron, F., Paradis, P., Bronchain, O., Nemer, G., and Nemer, M. (1999). Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol. Cell. Biol. 19, 4355–4365. doi:10.1128/MCB.19.6.4355
Chen, W. S., Manova, K., Weinstein, D. C., Duncan, S. A., Plump, A. S., Prezioso, V. R., et al. (1994). Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 8, 2466–2477. doi:10.1101/gad.8.20.2466
Chia, C. Y., Madrigal, P., Denil, S. L. I. J., Martinez, I., Garcia-Bernardo, J., El-Khairi, R., et al. (2019). GATA6 cooperates with EOMES/SMAD2/3 to deploy the gene regulatory network governing human definitive endoderm and pancreas formation. Stem Cell Rep. 12, 57–70. doi:10.1016/j.stemcr.2018.12.003
Churchill, A. J., Dominguez Gutiérrez, G., Singer, R. A., Lorberbaum, D. S., Fischer, K. A., and Sussel, L. (2017). Genetic evidence that Nkx2.2 acts primarily downstream of Neurog3 in pancreatic endocrine lineage development. eLife 6, e20010. doi:10.7554/eLife.20010
Conrad, E., Stein, R., and Hunter, C. S. (2014). Revealing transcription factors during human pancreatic β cell development. Trends Endocrinol. Metab. 25, 407–414. doi:10.1016/j.tem.2014.03.013
Cujba, A.-M., Alvarez-Fallas, M. E., Pedraza-Arevalo, S., Laddach, A., Shepherd, M. H., Hattersley, A. T., et al. (2022). An HNF1α truncation associated with maturity-onset diabetes of the young impairs pancreatic progenitor differentiation by antagonizing HNF1β function. Cell Rep. 38, 110425. doi:10.1016/j.celrep.2022.110425
Dassaye, R., Naidoo, S., and Cerf, M. E. (2016). Transcription factor regulation of pancreatic organogenesis, differentiation and maturation. Islets 8, 13–34. doi:10.1080/19382014.2015.1075687
De Vas, M. G., Kopp, J. L., Heliot, C., Sander, M., Cereghini, S., and Haumaitre, C. (2015). Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development 142, 871–882. doi:10.1242/dev.110759
Ferrell, J. E. (2002). Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr. Opin. Chem. Biol. 6, 140–148. doi:10.1016/s0955-0674(02)00314-9
Ferrer, J. (2002). A genetic switch in pancreatic β-cells: implications for differentiation and haploinsufficiency. Diabetes 51, 2355–2362. doi:10.2337/diabetes.51.8.2355
Gao, T., McKenna, B., Li, C., Reichert, M., Nguyen, J., Singh, T., et al. (2014). Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 19, 259–271. doi:10.1016/j.cmet.2013.12.002
Gragnoli, C., Lindner, T., Cockburn, B. N., Kaisaki, P. J., Gragnoli, F., Marozzi, G., et al. (1997). Maturity-onset diabetes of the young due to a mutation in the hepatocyte nuclear factor-4α binding site in the promoter of the hepatocyte nuclear factor-1α gene. Diabetes 46, 1648–1651. doi:10.2337/diacare.46.10.1648
Gu, C., Stein, G. H., Pan, N., Goebbels, S., Hörnberg, H., Nave, K.-A., et al. (2010). Pancreatic β cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 11, 298–310. doi:10.1016/j.cmet.2010.03.006
Hansen, S. K., Párrizas, M., Jensen, M. L., Pruhova, S., Ek, J., Boj, S. F., et al. (2002). Genetic evidence that HNF-1alpha-dependent transcriptional control of HNF-4alpha is essential for human pancreatic beta cell function. J. Clin. Invest. 110, 827–833. doi:10.1172/JCI15085
Harries, L. W., Brown, J. E., and Gloyn, A. L. (2009). Species-specific differences in the expression of the HNF1A, HNF1B and HNF4A genes. PLoS ONE 4, e7855. doi:10.1371/journal.pone.0007855
He, P., Lim, K., Sun, D., Pett, J. P., Jeng, Q., Polanski, K., et al. (2022). A human fetal lung cell atlas uncovers proximal-distal gradients of differentiation and key regulators of epithelial fates. Cell 185, 4841–4860.e25. doi:10.1016/j.cell.2022.11.005
Heller, S., Li, Z., Lin, Q., Geusz, R., Breunig, M., Hohwieler, M., et al. (2021). Transcriptional changes and the role of ONECUT1 in hPSC pancreatic differentiation. Commun. Biol. 4, 1298. doi:10.1038/s42003-021-02818-3
Henley, K. D., Stanescu, D. E., Kropp, P. A., Wright, C. V. E., Won, K.-J., Stoffers, D. A., et al. (2016). Threshold-dependent cooperativity of Pdx1 and Oc1 in pancreatic progenitors establishes competency for endocrine differentiation and β-cell function. Cell Rep. 15, 2637–2650. doi:10.1016/j.celrep.2016.05.040
Hobert, O. (2010). Gene regulation: enhancers stepping out of the shadow. Curr. Biol. 20, R697–R699. doi:10.1016/j.cub.2010.07.035
Horikawa, Y., Iwasaki, N., Hara, M., Furuta, H., Hinokio, Y., Cockburn, B. N., et al. (1997). Mutation in hepatocyte nuclear factor-1β gene (TCF2) associated with MODY. Nat. Genet. 17, 384–385. doi:10.1038/ng1297-384
Hua, Q.-X., Zhao, M., Narayana, N., Nakagawa, S. H., Jia, W., and Weiss, M. A. (2000). Diabetes-associated mutations in a β-cell transcription factor destabilize an antiparallel “mini-zipper” in a dimerization interface. Proc. Natl. Acad. Sci. U. S. A. 97, 1999–2004. doi:10.1073/pnas.97.5.1999
Iacovazzo, D., Flanagan, S. E., Walker, E., Quezado, R., de Sousa Barros, F. A., Caswell, R., et al. (2018). MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc. Natl. Acad. Sci. U. S. A. 115, 1027–1032. doi:10.1073/pnas.1712262115
Ibrahim, H., Balboa, D., Saarimäki-Vire, J., Montaser, H., Dyachok, O., Lund, P.-E., et al. (2024). RFX6 haploinsufficiency predisposes to diabetes through impaired beta cell function. Diabetologia 67, 1642–1662. doi:10.1007/s00125-024-06163-y
Jia, S., Ivanov, A., Blasevic, D., Müller, T., Purfürst, B., Sun, W., et al. (2015). Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic β-cell function. EMBO J. 34, 1417–1433. doi:10.15252/embj.201490819
Jiang, G., and Sladek, F. M. (1997). The DNA binding domain of hepatocyte nuclear factor 4 mediates cooperative, specific binding to DNA and heterodimerization with the retinoid X receptor alpha. J. Biol. Chem. 272, 1218–1225. doi:10.1074/jbc.272.2.1218
Johnson, A. F., Nguyen, H. T., and Veitia, R. A. (2019). Causes and effects of haploinsufficiency. Biol. Rev. 94, 1774–1785. doi:10.1111/brv.12527
Kaci, A., Solheim, M. H., Silgjerd, T., Hjaltadottir, J., Hornnes, L. H., Molnes, J., et al. (2024). Functional characterization of HNF4A gene variants identify promoter and cell line specific transactivation effects. Hum. Mol. Genet. 33, 894–904. doi:10.1093/hmg/ddae027
Kingdom, R., and Wright, C. F. (2022). Incomplete penetrance and variable expressivity: from clinical studies to population cohorts. Front. Genet. 13, 920390. doi:10.3389/fgene.2022.920390
Kuo, C. T., Morrisey, E. E., Anandappa, R., Sigrist, K., Lu, M. M., Parmacek, M. S., et al. (1997). GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 11, 1048–1060. doi:10.1101/gad.11.8.1048
Laver, T. W., Wakeling, M. N., Knox, O., Colclough, K., Wright, C. F., Ellard, S., et al. (2022). Evaluation of evidence for pathogenicity demonstrates that BLK, KLF11, and PAX4 should not be included in diagnostic testing for MODY. Diabetes 71, 1128–1136. doi:10.2337/db21-0844
Li, M., Rivière, J.-B., and Polychronakos, C. (2022). Why all MODY variants are dominantly inherited: a hypothesis. Trends Genet. 38, 321–324. doi:10.1016/j.tig.2021.10.001
Li, M., Popovic, N., Wang, Y., Chen, C., and Polychronakos, C. (2023). Incomplete penetrance and variable expressivity in monogenic diabetes; a challenge but also an opportunity. Rev. Endocr. Metab. Disord. 24, 673–684. doi:10.1007/s11154-023-09809-1
Lu, P., Rha, G. B., Melikishvili, M., Wu, G., Adkins, B. C., Fried, M. G., et al. (2008). Structural basis of natural promoter recognition by a unique nuclear receptor, HNF4alpha. Diabetes gene product. J. Biol. Chem. 283, 33685–33697. doi:10.1074/jbc.M806213200
Malecki, M. T., Jhala, U. S., Antonellis, A., Fields, L., Doria, A., Orban, T., et al. (1999). Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat. Genet. 23, 323–328. doi:10.1038/15500
Meng, Y., Moore, R., Tao, W., Smith, E. R., Tse, J. D., Caslini, C., et al. (2018). GATA6 phosphorylation by Erk1/2 propels exit from pluripotency and commitment to primitive endoderm. Dev. Biol. 436, 55–65. doi:10.1016/j.ydbio.2018.02.007
Mirshahi, U. L., Colclough, K., Wright, C. F., Wood, A. R., Beaumont, R. N., Tyrrell, J., et al. (2022). Reduced penetrance of MODY-associated HNF1A/HNF4A variants but not GCK variants in clinically unselected cohorts. Am. J. Hum. Genet. 109, 2018–2028. doi:10.1016/j.ajhg.2022.09.014
Morrisey, E. E., Tang, Z., Sigrist, K., Lu, M. M., Jiang, F., Ip, H. S., et al. (1998). GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 12, 3579–3590. doi:10.1101/gad.12.22.3579
Murray Leech, J., Beaumont, R. N., Arni, A. M., Chundru, V. K., Sharp, L. N., Colcough, K., et al. (2025). Common genetic variants modify disease risk and clinical presentation in monogenic diabetes. Nat. Metab. 7, 1819–1829. doi:10.1038/s42255-025-01372-0
Ng, N. H. J., Jasmen, J. B., Lim, C. S., Lau, H. H., Krishnan, V. G., Kadiwala, J., et al. (2019). HNF4A haploinsufficiency in MODY1 abrogates liver and pancreas differentiation from patient-derived induced pluripotent stem cells. iScience 16, 192–205. doi:10.1016/j.isci.2019.05.032
Ng, N. H. J., Ghosh, S., Bok, C. M., Ching, C., Low, B. S. J., Chen, J. T., et al. (2024). HNF4A and HNF1A exhibit tissue specific target gene regulation in pancreatic beta cells and hepatocytes. Nat. Commun. 15, 4288. doi:10.1038/s41467-024-48647-w
Nishimura, W., Takahashi, S., and Yasuda, K. (2015). MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 58, 566–574. doi:10.1007/s00125-014-3464-9
Olbrot, M., Rud, J., Moss, L. G., and Sharma, A. (2002). Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. 99, 6737–6742. doi:10.1073/pnas.102168499
Patel, K. A., Kettunen, J., Laakso, M., Stančáková, A., Laver, T. W., Colclough, K., et al. (2017). Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nat. Commun. 8, 888. doi:10.1038/s41467-017-00895-9
Philippi, A., Heller, S., Costa, I. G., Senée, V., Breunig, M., Li, Z., et al. (2021). Mutations and variants of ONECUT1 in diabetes. Nat. Med. 27, 1928–1940. doi:10.1038/s41591-021-01502-7
Piccand, J., Strasser, P., Hodson, D. J., Meunier, A., Ye, T., Keime, C., et al. (2014). Rfx6 maintains the functional identity of adult pancreatic β cells. Cell Rep. 9, 2219–2232. doi:10.1016/j.celrep.2014.11.033
Qian, M. F., Bevacqua, R. J., Coykendall, V. M. N., Liu, X., Zhao, W., Chang, C. A., et al. (2023). HNF1α maintains pancreatic α and β cell functions in primary human islets. JCI Insight 8, e170884. doi:10.1172/jci.insight.170884
Raj, A., and van Oudenaarden, A. (2008). Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. doi:10.1016/j.cell.2008.09.050
Raum, J. C., Gerrish, K., Artner, I., Henderson, E., Guo, M., Sussel, L., et al. (2006). FoxA2, Nkx2.2, and PDX-1 regulate islet β-cell-specific mafA expression through conserved sequences located between base pairs -8118 and -7750 upstream from the transcription start site. Mol. Cell. Biol. 26, 5735–5743. doi:10.1128/MCB.00249-06
Rubio-Cabezas, O., Minton, J. A. L., Kantor, I., Williams, D., Ellard, S., and Hattersley, A. T. (2010). Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes 59, 2326–2331. doi:10.2337/db10-0011
Seidman, J. G., and Seidman, C. (2002). Transcription factor haploinsufficiency: when half a loaf is not enough. J. Clin. Invest. 109, 451–455. doi:10.1172/JCI15043
Sharp, L. N., Colclough, K., Murray Leech, J., Cannon, S. J., Laver, T. W., Hattersley, A. T., et al. (2025). Population prevalence, penetrance, and mortality for genetically confirmed MODY. doi:10.1210/clinem/dgaf599
Shaw-Smith, C., De Franco, E., Lango Allen, H., Batlle, M., Flanagan, S. E., Borowiec, M., et al. (2014). GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes 63, 2888–2894. doi:10.2337/db14-0061
Shields, B. M., Hicks, S., Shepherd, M. H., Colclough, K., Hattersley, A. T., and Ellard, S. (2010). Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53, 2504–2508. doi:10.1007/s00125-010-1799-4
Shih, H. P., Seymour, P. A., Patel, N. A., Xie, R., Wang, A., Liu, P. P., et al. (2015). A gene regulatory network cooperatively controlled by Pdx1 and Sox9 governs lineage allocation of foregut progenitor cells. Cell Rep. 13, 326–336. doi:10.1016/j.celrep.2015.08.082
Singh, P., Tung, S.-P., Han, E. E., Lee, I.-K., and Chi, Y.-I. (2019). Dimerization defective MODY mutations of hepatocyte nuclear factor 4α. Mutat. Res. Fund. Mol. Mech. Mutagen 814, 1–6. doi:10.1016/j.mrfmmm.2019.01.002
Smith, S. B., Qu, H.-Q., Taleb, N., Kishimoto, N. Y., Scheel, D. W., Lu, Y., et al. (2010). Rfx6 directs islet formation and insulin production in mice and humans. Nature 463, 775–780. doi:10.1038/nature08748
Sriram, A., Wakeling, M. N., Hattersley, A. T., Weedon, M. N., Colclough, K., Laver, T. W., et al. (2025). Rare variants in NEUROD1 and PDX1 are low-penetrance causes of MODY, whereas those in APPL1 and WFS1 are not associated with MODY. Diabetes 74, 2123–2131. doi:10.2337/db25-0442
Stoffers, D. A., Ferrer, J., Clarke, W. L., and Habener, J. F. (1997a). Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet. 17, 138–139. doi:10.1038/ng1097-138
Stoffers, D. A., Zinkin, N. T., Stanojevic, V., Clarke, W. L., and Habener, L. F. (1997b). Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 15, 106–110. doi:10.1038/ng0197-106
Veitia, R. A. (2002). Exploring the etiology of haploinsufficiency. BioEssays 24, 175–184. doi:10.1002/bies.10023
Wilkie, A. O. M. (1994). The molecular basis of genetic dominance. J. Med. Genet. 31, 89–98. doi:10.1136/jmg.31.2.89
Wortham, M., and Sander, M. (2021). Transcriptional mechanisms of pancreatic β-cell maturation and functional adaptation. Trends Endocrinol. Metab. 32, 474–487. doi:10.1016/j.tem.2021.04.011
Xin, M., Davis, C. A., Molkentin, J. D., Lien, C.-L., Duncan, S. A., Richardson, J. A., et al. (2006). A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc. Natl. Acad. Sci. U. S. A. 103, 11189–11194. doi:10.1073/pnas.0604604103
Xuan, S., Borok, M. J., Decker, K. J., Battle, M. A., Duncan, S. A., Hale, M. A., et al. (2012). Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J. Clin. Invest. 122, 3516–3528. doi:10.1172/JCI63352
Yamagata, K., Furuta, H., Oda, N., Kaisaki, P. J., Menzel, S., Cox, N. J., et al. (1996a). Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 384, 458–460. doi:10.1038/384458a0
Yamagata, K., Oda, N., Kaisaki, P. J., Menzel, S., Furuta, H., Vaxillaire, M., et al. (1996b). Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 384, 455–458. doi:10.1038/384455a0
Zeng, T., Spence, J. P., Mostafavi, H., and Pritchard, J. K. (2024). Bayesian estimation of gene constraint from an evolutionary model with gene features. Nat. Genet. 56, 1632–1643. doi:10.1038/s41588-024-01820-9
Zhang, Q., Bhattacharya, S., and Andersen, M. E. (2013). Ultrasensitive response motifs: basic amplifiers in molecular signalling networks. Open Biol. 3, 130031. doi:10.1098/rsob.130031
Zhao, L., Guo, M., Matsuoka, T., Hagman, D. K., Parazzoli, S. D., Poitout, V., et al. (2005). The islet β cell-enriched MafA activator is a key regulator of insulin gene transcription. J. Biol. Chem. 280, 11887–11894. doi:10.1074/jbc.M409475200
Zschocke, J., Byers, P. H., and Wilkie, A. O. M. (2023). Mendelian inheritance revisited: dominance and recessiveness in medical genetics. Nat. Rev. Genet. 24, 442–463. doi:10.1038/s41576-023-00574-0
Keywords: monogenic diabetes, cell fate, haploinsufficiency, dosage sensitivity, positive feedback, cooperativity, bistability, incomplete penetrance
Citation: Zug R (2025) Why all MODY variants in transcription factor genes are dominantly inherited. Front. Genet. 16:1690468. doi: 10.3389/fgene.2025.1690468
Received: 21 August 2025; Accepted: 20 October 2025;
Published: 20 November 2025.
Edited by:
Amin Gasmi, Francophone Society of Nutritherapy and Applied Nutrigenetics, France, FranceReviewed by:
Ning Chen, Fudan University (Xiamen Branch), ChinaCopyright © 2025 Zug. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roman Zug, cm9tYW4uenVnQHVuaS1tYWluei5kZQ==