 Alastair Barraclough1†
Alastair Barraclough1† Isabel Bär2†
Isabel Bär2† Tirsa van Duijl3
Tirsa van Duijl3 Karin Fijnvandraat1
Karin Fijnvandraat1 Jeroen C. J. Eikenboom4Frank W. G. Leebeek2
Jeroen C. J. Eikenboom4Frank W. G. Leebeek2 Ruben Bierings2
Ruben Bierings2 Jan Voorberg3*Despoina Trasanidou3†
Jan Voorberg3*Despoina Trasanidou3†- 1Department of Pediatric Hematology, Emma Children’s Hospital, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Hematology, Erasmus University Medical Centre, Rotterdam, Netherlands
- 3Molecular Hematology, Sanquin Research and Landsteiner Laboratory, Amsterdam University Medical Centre, Amsterdam, Netherlands
- 4Division of Thrombosis and Hemostasis, Department of Internal Medicine, Leiden University Medical Centre, Leiden, Netherlands
In recent years gene therapy has emerged as a powerful technology for treatment of a large variety of inherited disorders. With the FDA approval of in vivo gene therapy of hemophilia A and B using AAV-mediated transgene delivery to hepatocytes, the path towards a new treatment era seemed paved. Also, CRISPR-Cas based approaches have reached the clinic, as in the ex vivo treatment of hematopoietic stem cells for sickle cell disease and thalassemia patients. The question arises whether these innovative strategies will also be suitable for patients with von Willebrand Disease (VWD). Whilst in and ex vivo delivery to endothelial cells (ECs) has been demonstrated, and CRISPR-Cas9 gene editing has been successful in ECs, there are currently no gene therapy options available for VWD. The wide variety of pathogenic VWF mutations makes development of broadly applicable, cost-effective gene therapies challenging. While delivery of von Willebrand factor (VWF) as a therapeutic transgene would be optimal, the size of VWF challenges efficient delivery. Therefore, treatment of VWD requires targeted, personalized gene therapy; for instance by using the newest CRISPR-Cas technologies which can be tailored to facilitate alteration and restoration of various pathogenic VWD variants. This review describes the inherited bleeding disorder VWD and potential gene therapy approaches for management of the disease. Thereby we are exploring different CRISPR-Cas technologies and recent developments in the field. Moreover, we will discuss the ongoing advances of in vivo delivery systems, all with the scope on ECs.
1 Introduction on von Willebrand Disease (VWD)
1.1 The heterogeneous landscape of VWD
Von Willebrand Disease (VWD) is the most commonly inherited bleeding disorder, estimated to have a worldwide prevalence of 25.6 per million people in the general population (Leebeek and Eikenboom, 2016; Stonebraker et al., 2023). Patients with VWD have qualitative and/or quantitative abnormalities with von Willebrand Factor (VWF), a large multimeric protein that is pivotal during primary hemostasis. There are different types of VWD, with ranging severities. Type 1 VWD is the most common, where patients have reduced VWF in circulation and a mild bleeding phenotype (Atiq et al., 2023). If the reduced amount of VWF is due to enhanced clearance, it is described as type 1C VWD. Type 2 VWD is characterized by a qualitative deficiency of VWF and can be further classified into various subtypes: A, B, M, and N (Table 1). The qualitative impairment spans from insufficient multimerization of VWF dimers into its hemostatically active mature VWF multimer, to impaired binding and susceptibility to other proteins involved in maintaining hemostasis (Daniel et al., 2024; Jacobi et al., 2012; Maas et al., 2021; Othman and Favaloro, 2021). Type 3 is the rarest but most severe form, where patients have close to undetectable VWF levels in circulation and experience a severe bleeding phenotype (Eikenboom, 2001; van Kwawegen and Leebeek, 2024).
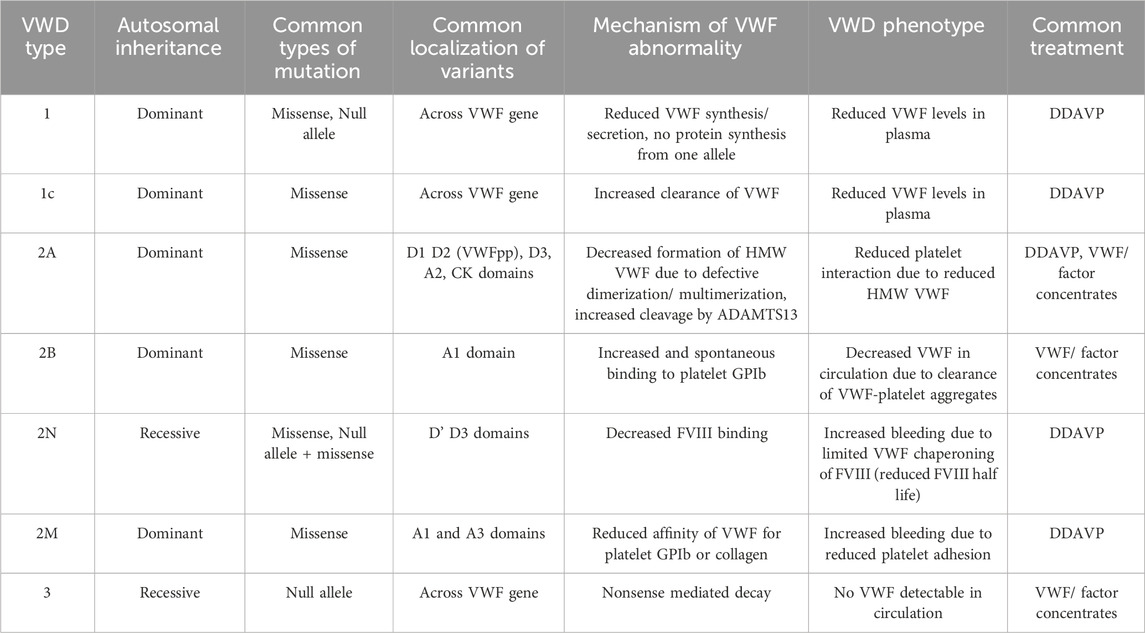
Table 1. Summary of von Willebrand disease sub-types, disease mechanism, and treatment.
The different types of VWD often arise from mutations impacting different domains of VWF (Figure 1A). Currently, more than 750 unique pathogenic VWF mutations are known, leading to a range of bleeding complications including mucocutaneous bleeding, heavy menstrual bleeding, joint bleeds, gastrointestinal bleeding and bleeding during surgery (de Jong and Eikenboom, 2017; Weyand and Veronica, 2021). The Leiden Open Variation Database (LOVD) alone reported a list of 505 VWF variants that were found in VWD patients in the clinic, of which ∼450 were missense and nonsense variants (Seidizadeh et al., 2023). Utilizing those variants from the LOVD database as an example, Figure 1B illustrates the distribution of VWF variants. Variants resulting in type 1 and 3 VWD can be found throughout VWF. In contrast, type 2 VWD variants are often located around the A1 domain, affecting the binding to platelets and/or collagen, or around the D’D3 or CK domains, impacting multimerization. More VWF variants have been reported by various studies and can be found in additional databases such as ClinVar, GnomAD, genome browser, human gene mutation database (HGMD) and more (UCSC, 2025; gnomAD, 2025; NCBI, 2025; HGMD, 2025). Although the prevalence of VWD subtypes varies per database and cohort, multiple studies have shown that type 1 VWD is by far the most common form, accounting for around 60%–85% of VWD cases. Type 2 VWD follows as the second most common with a prevalence of 15%–45% (Soucie et al., 2021; Seidizadeh et al., 2023). Orphanet estimates the prevalence of type 1 VWD worldwide to be 1–5/ 10,000, and the prevalence of type 2 VWD worldwide to be 1–9/1,000,000 (Orphanet, 2025a; Orphanet, 2025b). Type 3 VWD remains the rarest form of the disease.
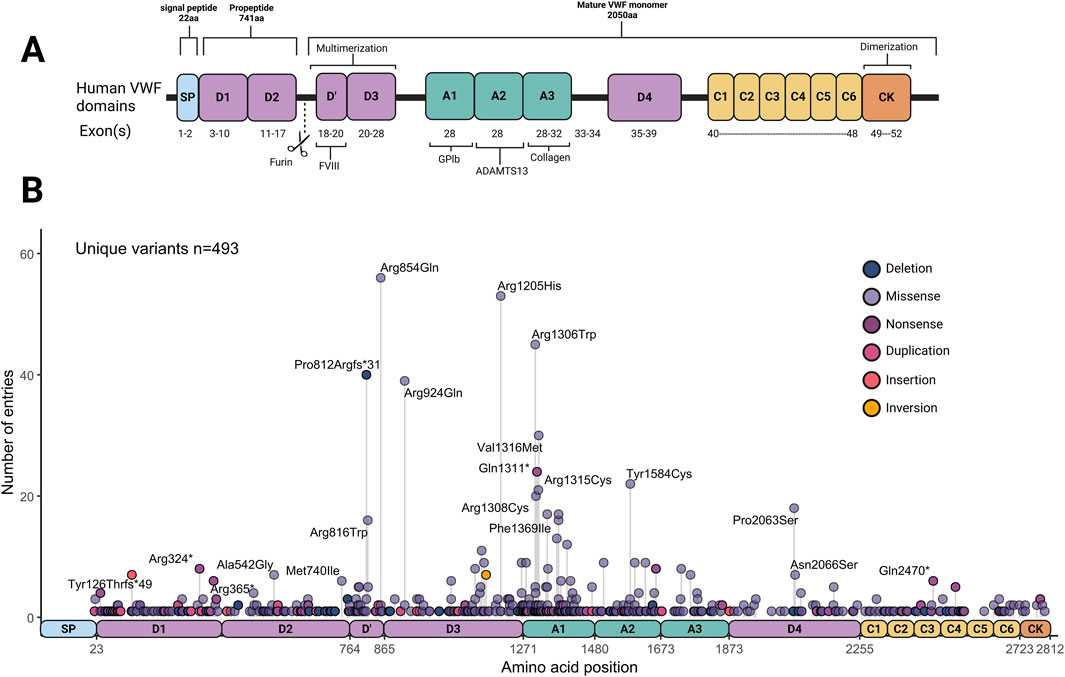
Figure 1. Overview of von Willbrand Factor (VWF) domains, and variant distributions. (A) Von Willebrand Factor domains and binding sites. (B) Distribution and frequency of von Willebrand Factor variants curated in the Leiden Open Variation Database (LOVD) (Accessed and data retrieved on 8-1-2025). Created in BioRender.
Even though in around 30% of patients no pathogenic mutation in VWF could be identified, VWD is generally considered a monogenic disease. Detecting the location of a disease-causing variant within VWF may indicate the impaired domain and binding site of the protein, thus implying the underlying molecular mechanism impairing VWF. However, the majority of variants that are found within VWF are not sufficiently characterized and are thus classified as variants of uncertain significance (VUS). To shed light on this, studies have sequenced large patient populations and performed ex vivo characterization experiments to explore which VWD variants are truly pathogenic (Atiq et al., 2022; Bär et al., 2025; Christopherson et al., 2022; 2024; James et al., 2006; Laan et al., 2024; Tosetto et al., 2020; Vangenechten et al., 2022).
1.2 Inheritance of VWD
VWD is predominantly inherited as an autosomal dominant disease, with the majority of VWF variants being heterozygous missense mutations. This results mostly in VWD type 1 and 2 subtypes, while type 3 and type 2N often derive out of homozygous null mutations or compound heterozygous and are therefore rarer forms of VWD with a typically recessive inheritance pattern. The prevalence for type 3 lays around 1:250,000 to 1:1,000,000 but is challenging to determine because of the increased prevalence in regions where consanguinity is more common (Nichols et al., 2008).
1.3 The star of primary hemostasis: von Willebrand Factor (VWF)
In VWD there can be quantitative or functional deficiency of VWF: a large multimeric protein that is produced by endothelial cells (EC) and megakaryocytes. Long VWF strings are primarily stored in Weibel-Palade bodies (WPB) of ECs and α-granules of platelets, respectively (Lenting et al., 2015). In ECs after synthesis (Figure 2A), VWF is translocated to the endoplasmic reticulum (ER) where the VWF signal peptide is removed, and C-terminal dimerization occurs between VWF monomers via the CK domains (Hordijk et al., 2024). The dimers are subsequently shuttled to the Golgi, where Furin cleaves the VWF propeptide domain (VWFpp) off the dimers, allowing for their multimerization via the N-terminal D′ D3 domains. Importantly, the VWFpp remains non-covalently bound to the VWF multimers which is essential for trafficking and packaging of VWF multimers into WPBs (Haberichter, 2015). These organelles store ultra large (UL) and high molecular weight (HMW) VWF multimers which are secreted into the bloodstream upon endothelial activation. At the same time, low molecular weight (LMW) VWF is constantly secreted by the ECs in a process called constitutive secretion. The third secretion pathway of VWF is the unstimulated fusion of WPBs and the release of its content during basal secretion (Silva et al., 2016). The basally secreted VWF is considered the main contributor to stable VWF levels in plasma. When secreted, the UL and HMW VWF strings undergo cleavage by ADAMTS13, resulting in a distinct multimeric pattern of different sizes (Giblin et al., 2008; Zhang et al., 2009). Biosynthesis of VWF in megakaryocytes also results in the formation of HMW VWF multimers that are eventually stored in tubular sub-compartments of platelet α-granules (Karampini et al., 2020).
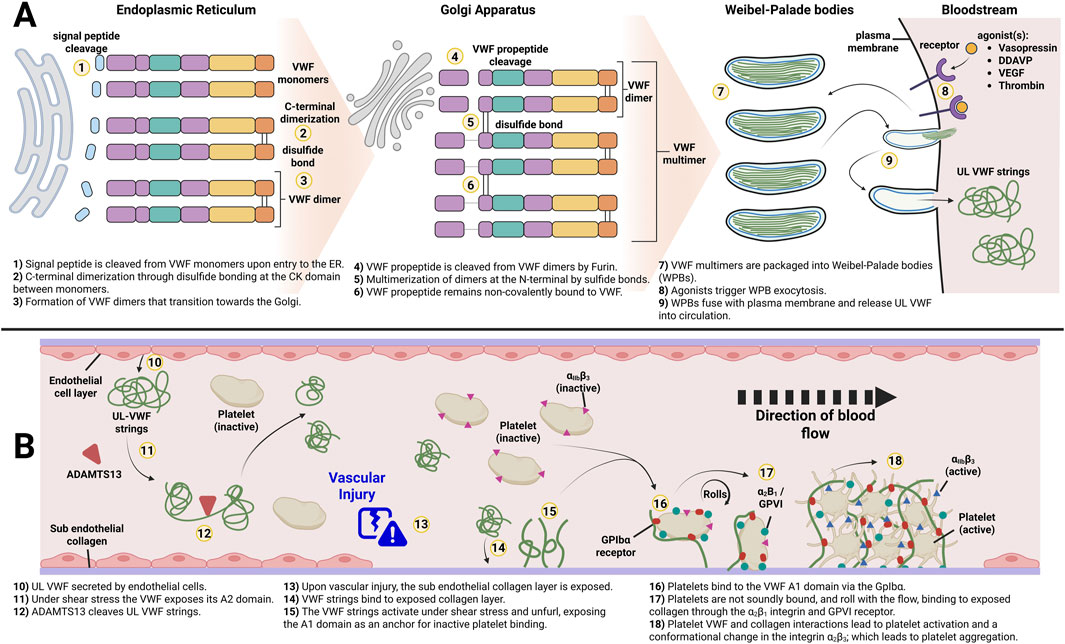
Figure 2. Overview of von Willebrand Factor biosynthesis and its role in primary hemostasis. (A) Key processing steps by endothelial cells of von Willebrand Factor to form Weibel-Palade Bodies. (B) Function of HMW secreted from WPBs. Adapted from Leebeek and Eikenboom, 2016. Created in BioRender.
In plasma, VWF acts as a chaperone for FVIII, protecting it from clearance and proteolytic cleavage (Pipe et al., 2016). At sites of vascular injury, VWF binds to the exposed sub-endothelial collagen layer through its A3 domain, anchoring itself and unfolding to expose its platelet binding sites (Romijn et al., 2001). The unfolded VWF strings provide a platform for platelets to bind to the A1 domain through their glycoprotein Ibα (GPIbα) receptor, initiating the formation of a platelet plug (Lenting et al., 2024; Figure 2B). This completes the process called primary hemostasis (Karampini et al., 2020). Due to the versatile role of VWF and its complex structure including several binding sites, the variety in VWD phenotypes is not surprising. Deficiency of VWF impacts other proteins involved in coagulation and the treatment and prevention of bleeds caused by faulty or missing VWF is challenging.
1.4 Current treatment of VWD
The plethora of pathogenic variants and disease mechanisms complicates treatment of VWD tremendously. In addition, the lack of knowledge about the disease-underlying causes currently prevents personalized treatment in the clinic. Current treatment aims to increase VWF and FVIII plasma concentrations on demand in case of a bleeding event or before and after interventions. A common treatment approach for VWD patients, especially type 1, is to increase circulating VWF and FVIII levels by stimulating release of stored VWF and FVIII from ECs. This is achieved via the administration of desmopressin (DDAVP), a synthetic version of the hormone vasopressin, which interacts with the V2R receptor of ECs to stimulate VWF release (Phua and Erik, 2019). However, not all VWD patients respond to DDAVP and DDAVP administration is contraindicated in patients with type 2B VWD, due to increased risk of thrombocytopenia as their VWF has an increased affinity for platelets (Laan et al., 2025). Also, patients with type 3 VWD do not benefit from DDAVP, since there is no stored VWF that can be released by ECs (Connell et al., 2021). According to the ASH ISTH NHF WFH 2021 VWD guidelines, DDAVP has to be periodically tested in patients for which this could be a viable option, and treatment should be adjusted based on the results of these trials (James et al., 2021).
An alternative for DDAVP for unresponsive patients or in major surgery where DDAVP is not sufficient, is replacement therapy with VWF (FVIII) factor concentrates. Replacement therapy is used in the management of acute bleeds and in severely affected VWD patients to prevent bleeding in the long-term. The latter patients receive factor concentrate 2–3 times per week intravenously (so-called prophylaxis). Plasma-derived VWF concentrates and recombinant VWF (rVWF) are both effective to stop and prevent bleeding (Gill et al., 2015; Leebeek et al., 2022). The antifibrinolytic agent tranexamic acid is mostly used as an adjunctive therapy to DDAVP or VWF concentrates (Laffan et al., 2014).
Taken together, there are effective therapies in place for patients with VWD. However, VWF concentrates need to be administered intravenously by a healthcare professional and therefore require a hospital visit. Especially in severely affected patients, like VWD type 3 patients on long term prophylaxis, regular hospital visits decrease their quality of life. In addition, concentrates are costly and represent a monetary burden for society due to the life-long treatment need. The current treatment is therefore sub-optimal and a durable treatment approach would be ideal to reduce the burden of disease in patients with VWD; allowing them to engage more in their daily activities with reduced concern. Therefore, alternative and innovative therapies are needed to optimize treatment of VWD. In the following paragraphs we will discuss the suitability of different gene therapy approaches for personalized treatment of VWD.
2 Silencing of mutant VWF in heterozygous VWD patients using small interfering RNA (siRNA)
RNA interference (RNAi) is a mechanism whereby mRNA is degraded through targeting by complementary short interfering RNA (siRNA) molecules. In hemophilia patients, siRNA targeting antithrombin has been studied extensively with the aim to restore the hemostatic balance. Patients treated with this siRNA showed a normalization of thrombin generation and reduction of the number of bleedings (Srivastava et al., 2021). A similar approach can be used in heterozygous VWD patients whereby siRNA is targeting the mutated, disease-causing allele. As mentioned above, around 33% of VWF mutations lead to a qualitative impairment, resulting in type 2 VWD (Seidizadeh et al., 2023). In human umbilical vein endothelial cells (HUVECs), siRNA-mediated knock-down of VWF can achieve 90% reduction at the transcript level (Furini et al., 2023; Seidizadeh et al., 2023). De Jong et al. enhanced this approach by encapsulating siRNA targeting mutant VWF mRNA in lipid nanoparticles (LNPs), resulting in allele-selective VWF degradation in a dominant negative type 2A VWD model (Figure 3). Through this approach, they demonstrated ex vivo resolution of VWF ER retention in a VWD type 2A phenotype using patient-derived venous endothelial colony-forming cells (ECFCs) (De Jong et al., 2020). The siRNA was designed to target a non-pathogenic, commonly occurring heterozygous single-nucleotide polymorphism (SNP) that co-segregated with the pathogenic mutation encoding for the dominant negative VWD variant. This concept was then further explored in mice, by crossing two different mouse strains, B6 and 129S, to create a heterozygous B6.129S model. This model mimicked a heterozygous scenario where the siRNA could discriminate between a single benign SNP present in the two VWF alleles (Jongejan et al., 2023; 2024). Following this proof of concept, Linthorst et al. investigated whether this approach could be extended to a heterozygous VWD type 2B mouse model (Linthorst et al., 2024). The resulting selective reduction of mutant VWF protein, the improved multimeric VWF pattern, and the normalized bleeding times in two-thirds of the heterozygous VWD 2B mice highlight the potential of this personalized therapeutic treatment strategy. The targeting of a heterozygous SNP instead of the disease-causing variant also allows for a broader application, independent of the pathogenic VWD mutation. De Jong et al. identified 4 common SNPs with a minor allele frequency of approximately 0.3 in the general Caucasian population. Based on these frequencies, they calculated that a person has a 74% probability of being heterozygous for at least one of these SNPs (De Jong et al., 2020). In another study, WT and a dominant negative VWF variant plasmid that impacts multimerization were co-injected into VWF−/− mice. The VWF variant was then counteracted by a follow-up siRNA injection; demonstrating an improvement in multimerization on the following day (Campioni et al., 2021). This elegant approach may potentially be used for successful treatment of a subset of patients with dominant negative type 2 VWD. However, siRNAs do not provide a permanent solution for patients with VWD but require regular re-administration (Linthorst et al., 2025). How long the siRNAs would be functional in vivo and alleviate the disease phenotype still needs to be evaluated in large animal models and humans.
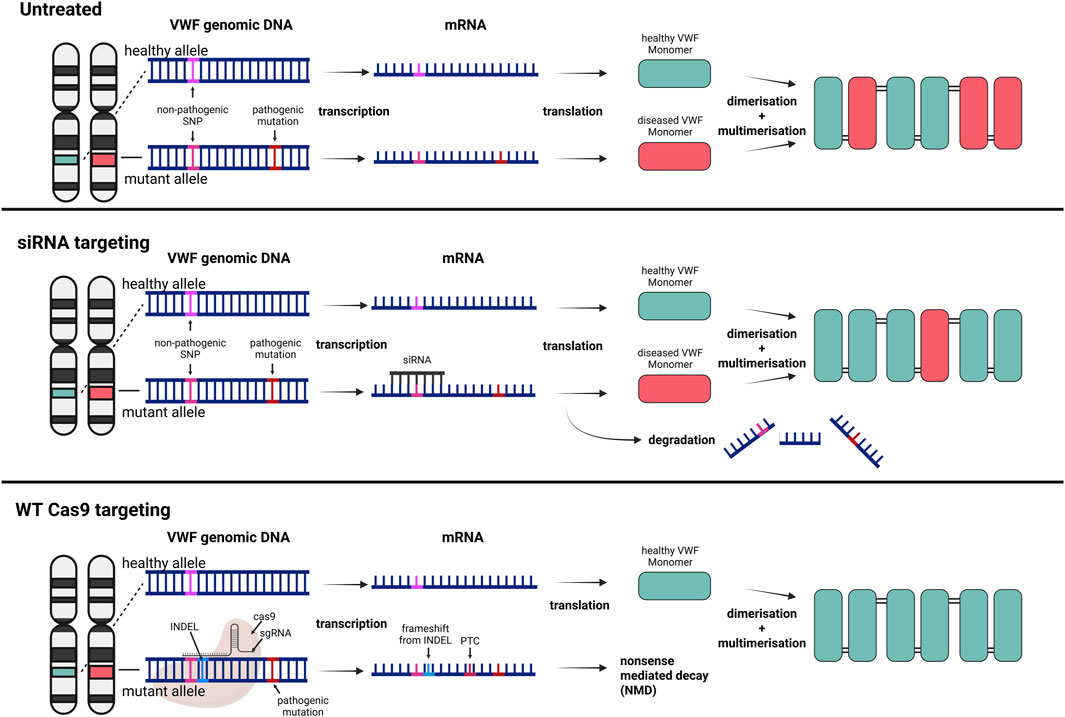
Figure 3. Broad targeting gene therapy strategies for dominant negative VWD variants to increase the ratio of healthy to diseased VWF subunits. Top) Untreated VWD results in VWF multimers consisting of healthy and diseased subunits. Middle) Selective siRNA targeting of a benign SNP on the mRNA originating from the mutant allele results in degradation of the transcript and a higher ratio of healthy to mutant VWF subunits. Bottom) The same targeting approach of a benign SNP, but the Cas9 directly targets the genomic DNA and induces a frameshift and premature termination codon (PTC). The knockout of the mutant allele results in sole expression of healthy VWF monomers. Created in BioRender.
3 CRISPR-cas technologies
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins are adaptive immune systems protecting their prokaryotic host against invading genetic elements. Since 2012, various Cas nucleases have been repurposed as next-generation genetic engineering tools, replacing previous time-consuming and costly technologies such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). CRISPR-Cas systems are categorized into two classes, seven types and several subtypes. Class 2 systems have taken current genome editing applications by storm, due to their simple architecture of a single effector protein (Cas9, Cas12, and Cas13), in contrast to the multi-protein effector complex nature of Class 1 systems. Nowadays, the most widely used Class 2 nucleases are the type II-A Cas9 from Streptococcus pyogenes (SpCas9) and the type V-A Cas12a from Lachnospiraceae bacterium (LbCas12a) or Alicyclobacillus acidoterrestris (AsCas12a). Cas9 and Cas12a nucleases require a small guide RNA (gRNA) molecule to bind to a target DNA sequence (protospacer) flanked by a short, conserved motif (protospacer adjacent motif; PAM) and create a double-strand DNA break (DSDB) (Jinek et al., 2012; Zetsche et al., 2015) Several other naturally occurring Cas9 or Cas12 orthologues and engineered variants thereof have also been harnessed, enriching the current CRISPR-Cas toolbox (Acharya et al., 2019; Agudelo et al., 2020; Chatterjee et al., 2018; Fedorova et al., 2020; Harrington et al., 2017; Hu et al., 2020; Lee et al., 2016; Liu et al., 2019; Müller et al., 2016; Ran et al., 2015; Trasanidou et al., 2023; Wang et al., 2023; Wu T. et al., 2023; Wu et al., 2022). Below is an overview of how different CRISPR-Cas technologies can be employed for treatment and/or correction of VWD, with Figure 4 illustrating their mechanisms.
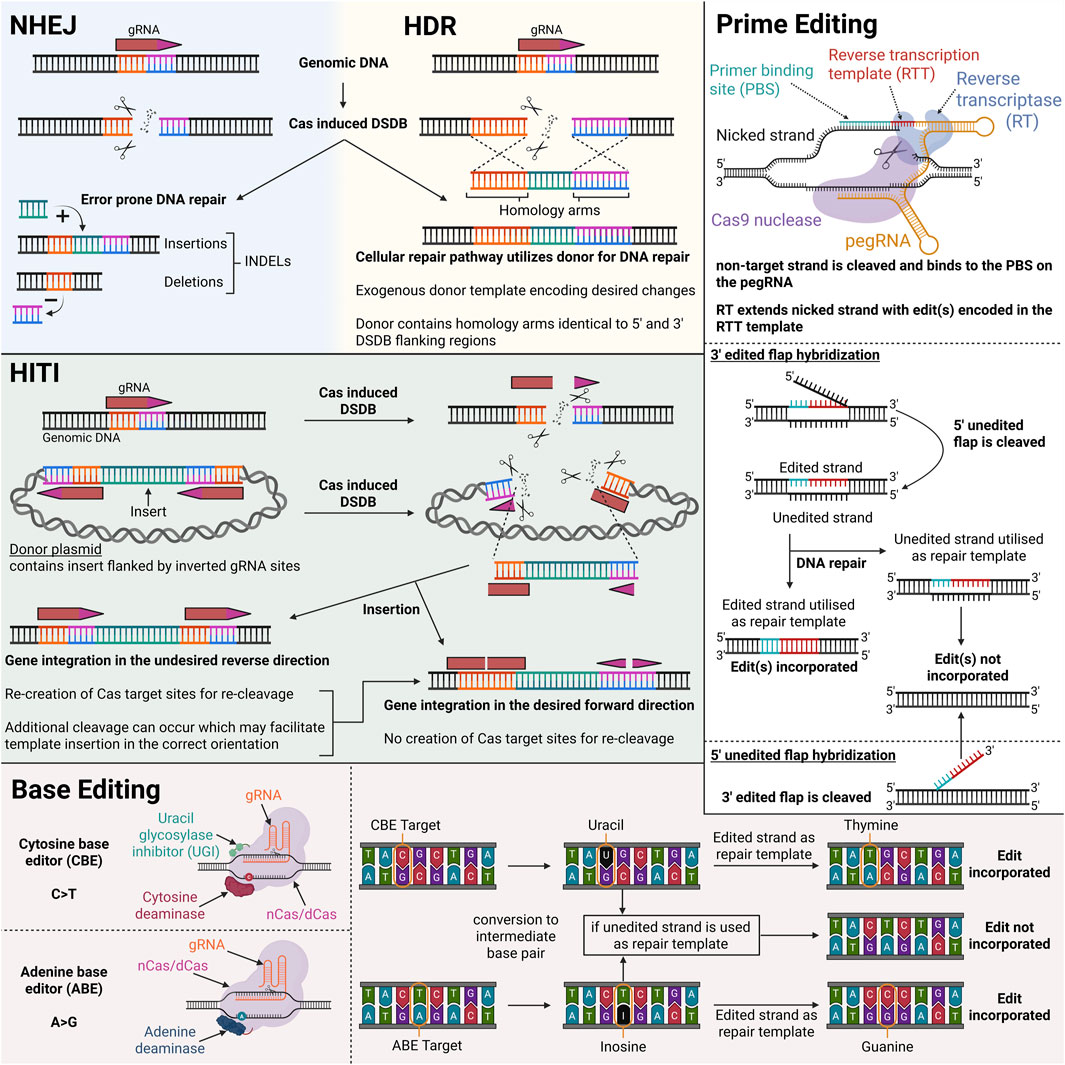
Figure 4. Illustration of different CRISPR-Cas modalities and their mechanisms of genomic DNA modification. Created in BioRender.
3.1 DSDB-based gene editing: non-homologous end joining (NHEJ) and homology directed repair (HDR)
There are two main pathways to repair a DSDB generated by class 2 Cas nucleases: non-homologous end joining (NHEJ) and homology-directed repair (HDR). The NHEJ approach exhibits high efficiency due to its independence from cell cycle and the continuous editing at the recognition site of the gRNA until random insertions and/or deletions (INDELs) are achieved, which usually prevent further Cas:gRNA binding. In contrast, HDR introduces precise genetic modifications and requires a DNA repair template and dividing cells. The presence of an exogenous DNA template tricks the cell to utilize the exogenous template for repair, rather than the sister chromatid during homologous recombination (HR) (Liao et al., 2024). Except from the aforementioned restrictions of HDR, a strong DSDB-induced NHEJ background is also usually observed (Chapman et al., 2012; Cox et al., 2015). Confirmation of CRISPR-induced modifications in the context of VWD can be achieved through both genotypic and phenotypic analyses. Next-generation sequencing enables precise detection of molecular alterations introduced by CRISPR, while phenotypic validation such as improved VWF synthesis observed via ex vivo confocal microscopy of patient-derived ECFCs or changes in VWF plasma levels in vivo could further substantiate successful gene editing.
3.1.1 Non-homologous end joining (NHEJ)
In mammalian cells, the DSDB is mainly repaired via the error prone NHEJ repair pathway; resulting in frameshifts and premature termination codons (PTC) due to small INDELs. This approach has been utilized by Schillemans et al., employing SpCas9 in a lentiviral system in cord-blood (CB) ECFCs to generate clonal VWF knock-out (KO) lines (Schillemans et al., 2019). With this study the effectiveness of Cas9 in ECFCs has been proven and paved the way for further ex vivo patient-derived ECFC studies.
Following the same rationale as allele-selective siRNA targeting, the CRISPR-Cas technology may be exploited for the selective disruption of mutant VWF alleles harboring pathogenic variants (Figure 3). In an ongoing study we observed a phenotypic rescue in type 2A and type 2B VWD patient-derived venous ECFCs by targeting a common heterozygous SNP located on the same allele as the pathogenic variant (Bär et al., 2024). A key consideration of CRISPR-based approaches for selectively silencing pathogenic alleles of VWF lies in the potential for off-target activity. In particular, insufficient discrimination between the mutant and wild-type alleles by the gRNA can lead to unintended silencing of the functional allele. Such off-target effects could exacerbate the clinical phenotype of VWD, rather than ameliorate it, by further reducing the levels of functional VWF. Simultaneously, selective targeting of ECs needs to be ensured to prevent genome-wide off-targeting of the CRISPR construct. As an additional safety measure, an EC specific promoter could be leveraged to ensure limited expression in undesired tissue in vivo. Nevertheless, this targeting strategy is a promising first step towards a permanent rescue of heterozygous VWD types, providing a high allele-selectivity, and limiting disruption of healthy alleles. Whilst the approach of allele-selective targeting, whether with siRNAs or CRISPR-Cas technology, show promising results for alleviating the dominant-negative effect of mutant VWF, the risk of haploinsufficiency remains, which has previously been reported for VWD type 1 (Desch et al., 2013; Sadler et al., 2022). Theoretically, severely affected heterozygous VWD type 2 patients could transition to a mild type 1 VWD following this treatment, shifting from a qualitative to a quantitative deficiency of VWF. Considering the generally lower bleeding phenotype in type 1 VWD patients, this transition would be expected to ease the disease burden, thereby enhancing the quality of life for these patients rather than curing them entirely.
CRISPR-Cas has also been used to facilitate in vivo editing of murine vascular endothelium. Specifically, Cas9 was expressed under the control of the endothelial-specific CDH5 promoter from a plasmid-based expression system. Prior to retro-orbital injection, the plasmid was encapsulated in a PP/PEI (PEG5,000-b-PLGA/ PEI25,000Da) nanoparticle, leading to 40%–45% editing of the targeted Pik3cg in lung ECs of the mice, while no INDELS were detected in lung non-ECs (Zhang et al., 2022). This study shows that a specific and targeted delivery of CRISPR-Cas into ECs is possible, and the selectivity of gene targeting is highly dependent on the gRNA used. A thorough in vitro analysis of efficient and selective gRNAs beforehand is therefore vital and influences the editing percentage drastically.
3.1.2 Homology directed repair (HDR)
While those NHEJ-based approaches are efficient, the NHEJ pathway is an error-prone repair mechanism and the outcome of the editing is not completely predictable. In contrast, HDR provides a mostly error-free repair that alters regions of DNA ranging from a few to thousand base pairs (bps) (Liao et al., 2024; Martin et al., 2019). While HDR provides a more controlled way of editing, efficiency is greatly reduced compared to NHEJ. But the approach can be highly tailored, e.g., by customization of HAs including varying lengths, and in the case of single-stranded DNA (ssDNA) templates, asymmetrical HAs (Di Stazio et al., 2021). The inclusion of truncated gRNA protospacer sequences on the HAs was also shown to aid HDR rates, the rationale being that Cas9 can bind but not cleave the HDR donor; allowing it to traffic the donor into nucleus due to the Cas9 nuclear localization signal(s) (NLS) (Nguyen et al., 2019). There are additional strategies aimed at boosting HDR efficiencies, as greatly summarized by Nambiar et al. (2022). For research purposes, cell synchronization agents and NHEJ inhibitor agents have been applied (Eghbalsaied and Kues, 2023; Selvaraj et al., 2024). Whilst they can improve HDR rates, they are toxic and not viable for therapeutic applications. A recent study on AZD7648, which inhibits NHEJ, resulted in increased translocations and large megabase deletions, including loss of chromosome arms (Cullot et al., 2024). However, chromosome rearrangements also occur spontaneously during HDR due to the involvement of different pathways managing the pairing of homologous sequences or a prematurely terminated HDR repair (Al-Zain and Symington, 2021). A major pitfall of CRISPR-Cas HDR technology is however its restriction to the S and G2 phases, where normally sister chromatids act as repair templates. Due to this, HDR is restricted to dividing cells and might therefore not be the optimal choice to edit the endothelium in vivo. Another limitation of inserting the full-length VWF coding sequence (∼8.4 kb) is the large payload size required, which can reduce the efficiency of in vivo delivery. Targeting specific regions with shorter donor templates would likely be more beneficial in terms of insertion efficiency. In VWD, the exon 4 and 5 deletion has been described in a multitude of VWD patients and would require a considerably shorter template (Bowman et al., 2012; Sutherland et al., 2009). Of course, the trade off from a targeted exon approach such as this sacrifices a broader VWD patient pool that can be treated for a smaller pool with a potential higher correction achieved.
3.1.3 Homology-independent targeted integration (HITI)
Homology-independent targeted integration (HITI), as deployed by Suzuki et al., represents a strategy to increase the efficiency of gene insertions by leveraging the NHEJ pathway for donor DNA integration (Suzuki et al., 2016). This approach aims to overcome the limitations of HDR, which is often inefficient, especially in non-dividing cells. Instead of relying on HA, the donor DNA contains sgRNA binding sites located at one or both sides of the desired insert in an inverted polarity. These sites, when targeted by Cas9, can lead to the linearization of the donor template and its subsequent integration into the host genome at a CRISPR-Cas9 facilitated DSB. The reliance of HITI on NHEJ presents another limitation, as the occurrence of INDELs cannot be ruled out and a small risk of undesired fragments and inverted integration remains. Although HITI demonstrates high efficiency, particularly in non-dividing cells, its general application to VWD - like HDR - requires the delivery and integration of the full VWF coding sequence (CDS). The large size of this insert likely limits the overall efficacy of this approach. Nevertheless, HITI would present an attractive personalized editing strategy for VWD patients harboring exon deletions on VWF, as it has been shown to achieve precise DNA knock--in in various genetic diseases including BCD patient-derived iPSCs and mice, a hemophilia B rat model, and primary CD34+ HSPCs (Bloomer et al., 2020; Chen X. et al., 2022; Meng et al., 2024).
3.2 Base editing technology
DSDBs have been linked to undesired outcomes, such as translocations, complex mixtures of products and inactivation of p53 (Haapaniemi et al., 2018; Ihry et al., 2018; Kosicki et al., 2018). CRISPR-Cas base editing is an attractive gene editing technology due to its capability to modify nucleotides without inducing DSDBs, although unintended DSDBs have been reported in some cases resulting in genotoxic effects (Fiumara et al., 2024; Huang M. et al., 2024). The system is based on a deaminase fused to a (partially) inactive Cas protein. Numerous base editors have been developed to date, presenting variable efficiency, purity, editing window, neighboring nucleotide preference, specificity, targetability and size.
The two main types of base editors are adenine base editors (ABEs) and cytosine base editors (CBEs). ABEs convert adenine to inosine, which is read as guanine during DNA replication or repair. ABEs contain a laboratory-evolved tRNA adenosine deaminase A (TadA) for the conversion (Gaudelli et al., 2017; Shi et al., 2016). A recent ABE variant, ABE9, presents high efficiency, specificity and acts within a window of 1–2 bp (Chen L. et al., 2022). Moreover, Qin et al. developed a range of highly efficient ABE-Ultramax base editors (ABE-Umax) possessing either a wider and flexible editing window or a narrow editing window of 1–2 bp with increased specificity (Qin et al., 2024).
CBEs convert cytosine to uracil, which is read as thymine during DNA replication or repair. To prevent uracil-DNA glycosylase-mediated excision of the uracil, at least one uracil glycosylase inhibitor (UGI) is usually fused to the CBEs. The inclusion of multiple UGIs has been shown to increase efficiency, but also toxicity. Hence, limiting the number of UGIs for in vivo gene editing is critical. The most widely used CBE systems contain the apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC) enzymes, the Petromyzon marinus cytidine deaminase 1 (PmCDA1) and engineered variants thereof. However, the newest CBEs evolved from the TadA from ABEs, decreasing off-targeting probability (Lam et al., 2023).
Except from ABEs (A>G) and CBEs (C>T), additional base editors, such as CGBEs (C>G), AYBEs (A>C or A>T), gTBEs (T>C or T>G), gCBE (C>G), CABEs (C>A) and DBEs (C>T and A>G), have been developed, further expanding the applicability of the base editing technology (Chen L. et al., 2021; Kurt et al., 2020; Lam et al., 2023; Sakata et al., 2020; Tong et al., 2023; 2024). However, when compared to ABEs and CBEs, they currently exhibit lower efficiency and purity of conversion as well as larger window of activity, limiting their use in therapeutic applications. Recent reviews by Xu et al. and Wang et al. provide a very nice summary on the use of different classes of base-editors for precision medicine (Wang D. et al., 2024; Xu F. et al., 2024; Xu W. et al., 2024).
In ECFCs, adenine base editing was successfully applied by Bär et al. as a means of VWD disease modelling (Bär et al., 2025). The pathogenic VWF p.M771V point mutation was installed in healthy CB-ECFCs through lentiviral delivery of the ABE8e-SpG editor. Post enrichment of puromycin selection showed on-target efficiencies of around 70% with minimal undesired bystander editing (<2%) at neighboring base pairs. The introduced patient mutation in healthy ECFCs mimicked the patient-derived ECFC phenotype, making base editing the ideal tool to study patient mutations in the original cell type and under the control of the gene’s natural promoter. Building on this concept, precise correction of pathogenic variants using base editors would be a desirable strategy for curing VWD. However, in addition to the not fully explored side effects in vivo and the specific genomic requirements related to the positioning of the small editing window, the size of the base editors presents a significant limitation, which we will further discuss in the section regarding delivery.
Base editors appear to be the most suitable correction tool for the majority of disease-causing variants in VWF, as the majority of variants are point mutations. An analysis of the LOVD database for splice, missense, and nonsense variants reveals that in principle almost half of these point mutations could theoretically be corrected with ABEs, whereas the theoretical targeting potential for CBEs lays at around 20% (Figure 5). However, the extensive heterogeneity of pathogenic VWD variants makes base editing a largely patient-specific approach, limiting its current viability due to the high costs associated with developing mutation-specific reagents. Whilst in-vivo clinical trials for base editing are underway, further assessment of their suitability for VWD gene therapy is required.
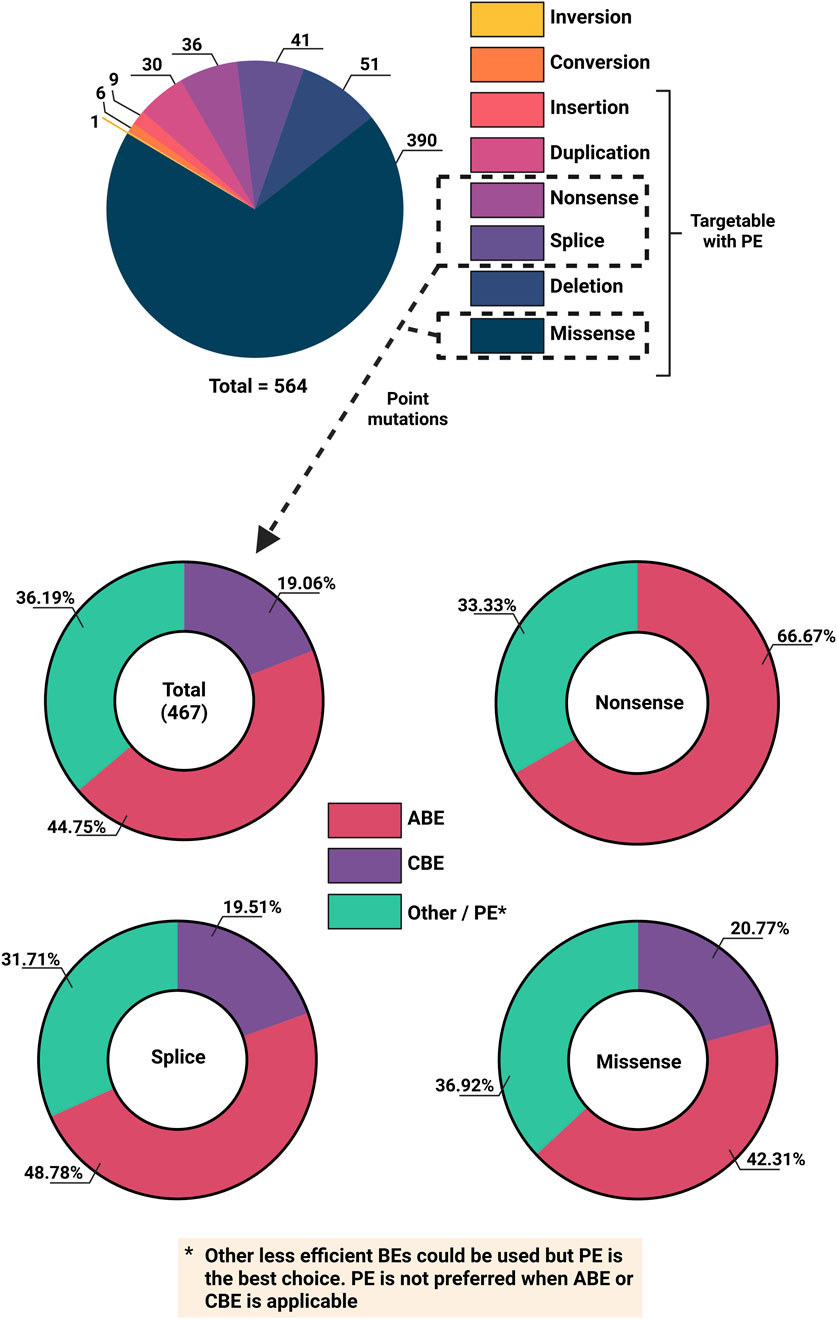
Figure 5. Overview of VWD mutation types and feasibility for base editing from the Leiden Open Variant Database (LOVD). Top) Number of different VWD mutation types. Bottom) Different CRISPR-Cas base editing technologies to target total Splice, Missense and non-Sense variants (and individually).
3.3 Prime editing technology
Similar to base editors, prime editors (PEs) introduce edits on target double-stranded DNA (dsDNA) without DSDBs and donor DNA. Moreover, PEs enable any type of single nucleotide modification including point mutations, small insertions, deletions and replacements, making it a potent editing tool for all the VWF variants that cannot be targeted by BEs (Figure 5). PEs consist of a partially inactive Cas9 or Cas12 protein fused to a reverse transcriptase (usually a mutant variant from Mouse-Moloney Leukemia Virus; MMLV). Instead of the standard spacer-scaffold architecture of the guide RNA, a prime editing guide RNA (pegRNA) consists of the spacer, the scaffold and additional 3′ elements, including the primer binding site (PBS) and the reverse transcription template (RTT) (Anzalone et al., 2019). When PE is guided to the target DNA loci by the spacer, it nicks the DNA, creating a 3′ DNA flap, which then hybridizes to the complementary PBS on the pegRNA. This serves as the initiation point for reverse transcription to extend the sequence using RTT, which encodes desired alterations. This ‘edited’ DNA flap can hybridize to the genome, resulting in a heteroduplex mismatch. Whether the edit is incorporated or removed depends on which strand is used for DNA repair (Anzalone et al., 2019).
Although PEs present limited off-targeting editing due to the triple checkpoint validation, they have several limiting factors, such as degradation of the 3′ end of the pegRNA and DNA mismatch repair (MMR) pathway inhibition. Several strategies have been developed to bypass these issues which have been nicely summarized by Murray et al. (2025):
• Introducing silent mutations around a desired target assists in evading the MMR pathway (Chen P. J. et al., 2021).
• Inclusion of a secondary standard 20bp nicking gRNA to nick the unedited strand, coercing the cell to utilize the edited strand as repair template. This secondary nicking gRNA can be either at the site of editing or at a short distance away (Anzalone et al., 2019).
• Fusion of a MHL1 variant with a dominant negative effect impairing MMR. MLH1 is a DNA mismatch repair protein (Chen P. J. et al., 2021).
• Addition of a 3′ motif with a strong secondary structure on the pegRNA (known as epegRNA) to prevent degradation of the 3′ PBS on the pegRNA (Nelson et al., 2022).
• Fusion of the RNA–binding exonuclease protection factor LA to the PE. LA interacts with polyuridine tracts at the 3′ end of transcripts, protecting them from exonucleases (Yan et al., 2024).
PEs have been used to target the VEGFR-2 gene in human retinal microvascular ECs and in murine vascular ECs. Both cases achieved efficiencies just over 50% utilizing a dual lentiviral approach with a secondary nicking gRNA and MMR inhibitor (Huang X. et al., 2024; Ma et al., 2024). For applications in VWD, just like base editing, prime editing would likely fall under a personalized therapy for patient specific mutations; which currently reduces its applicability.
3.4 Large sequence editing: transposon and integrase-based systems
Despite the high potential of short sequence editing approaches (NHEJ, HDR, BE, PE), the large exon deletions and the high heterogenicity of pathogenic mutations in VWF impose the need for large sequence editors with the ability to efficiently and precisely insert or replace multiple kb in a targeted genomic site, preferably in a DSDB-free and payload size-independent manner.
Transposon systems like the Sleeping Beauty (SB) provide an avenue to deliver large transgenes such as VWF into the cells genome. The SB system has been utilized to transfer full length murine VWF cDNA (8.4 kb) to the liver of VWF−/- mice. Whilst supra-physiological expression was stably maintained for 1.5 years, hepatocytes do not endogenously express VWF. So, the hemostatic efficacy was diminished due to reduced expression of HMW VWF multimers and did not correct the bleeding phenotype in some mice (Portier et al., 2018). Here, both episomal and random integrations into the genome were observed; highlighting the uncontrolled nature of transposons. Alternatively, Cas nucleases have been fused to transposase enzymes, generating homology-independent CRISPR transposon editors with enhanced site-specific integration. We briefly outline some of these technologies in Table 2 with references to further literature about it. Whilst these technologies, including the ones mentioned in Table 2, facilitate delivery of large transgenes such as VWF, their applicability in ECs and in vivo is still unknown. Despite the ability of transposon-based technologies to deliver large transgenes, their in vivo efficacy is currently still limited. Although there is constant progress in the development of these techniques, the on-target insertion efficiencies are overall still low. Therefore, for VWD type 1 and 2 there is an argument that this small percentage will have little impact of the bleeding phenotype and may only be viable for severe VWD type 3. Given the immature state of these techniques, we will not elaborate on them further, but Villiger et al. have discussed alternative CRISPR technologies in detail in their review, which we recommend for further reading (Villiger et al., 2024). Overall, the nascent field of large sequence editing presents several limitations, including poor characterization, low efficiency, complex design, large editor size, co-delivery of donor molecules, and undesired sequence scars. Optimization of these technologies may unlock kilobase-scale gene therapies for VWD.
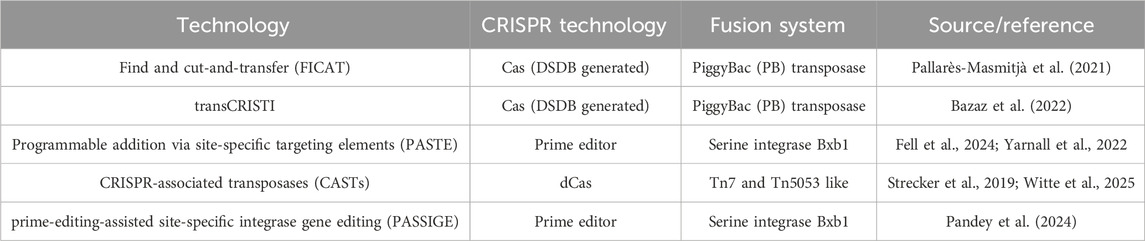
Table 2. Overview of CRISPR-Cas based transposon and integrase technologies to facilitate large insertion templates.
4 In vivo delivery systems
Delivery of gene editing systems is one of the major challenges holding back research and application of gene therapy in vivo. For VWD, multiple studies have been carried out in VWF−/- mice through hydrodynamic injection. As mentioned above, the SB system (SB100x) successfully delivered VWF to the liver for prolonged ectopic expression. Another study by De Meyer et al. also demonstrated that plasmid hydrodynamic injection resulting in ectopic liver VWF expression can be sufficient to restore thrombus formation; highlighting the feasibility of VWF rescue for severe VWD in vivo (Meyer et al., 2008). In this section, we elaborate on different delivery systems including adeno-associated viral vectors (AAV), adenoviral vectors (Ad), retroviral vectors (RV), (integrase-deficient) lentiviral vectors ((ID)LV), lipid nanoparticles (LNPs), and virus-like particles (VLPs); most of which are summarized in Figure 6. Besides these popular delivery systems, other techniques have been explored in the last years for in vivo gene therapy delivery, such as cell-penetrating proteins (CPPs), extracellular vesicles (EVs), and ultrasound-mediated drug delivery (Cecchin et al., 2023; Perolina et al., 2024; Rackear et al., 2024; Yu et al., 2021). While these approaches are not new, they have undergone different modification approaches, tailoring them more towards targeted, and safe delivery. However, they still require more investigation.
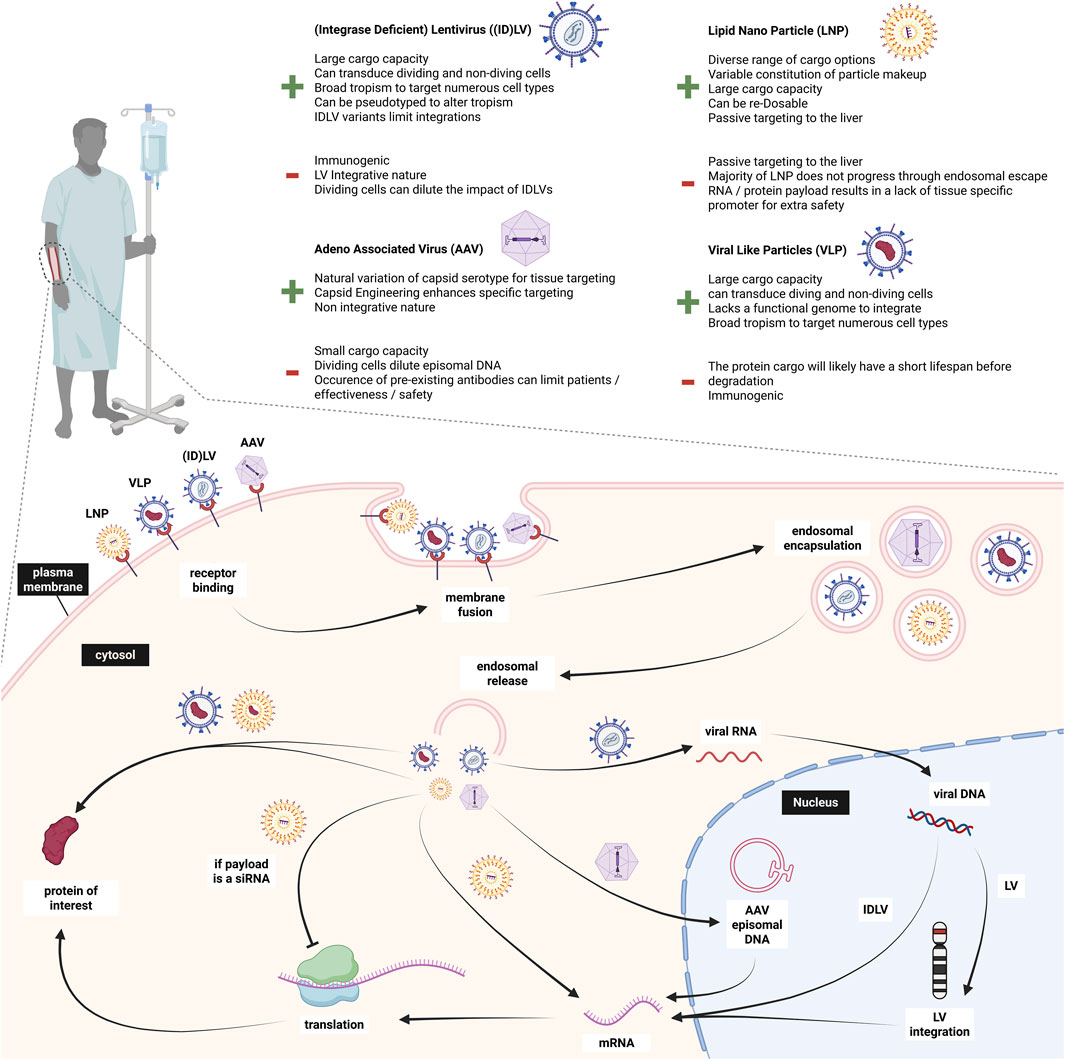
Figure 6. Overview of in vivo delivery approaches to endothelial cells and different payload mechanisms. Created in BioRender.
With the choice of which delivery system to use also comes the decision of which endothelium to target. Since all ECs produce VWF, the preferred target location could be tailored to the delivery approach. Generally, the vascular endothelium lining the blood vessels gets easily exposed by any intravenously administered product and is the main producer of VWF. A significant contributor to VWF levels in blood is the lung endothelium (Kaufmann et al., 2000; Yamamoto et al., 1998). Those ECs could be specifically targeted in view of their large contribution to circulating VWF. A third option of targetable endothelium would be liver sinusoidal endothelial cells (LSEC) (Milani et al., 2024). As any drug, a potential gene therapeutic agent would pass the liver, and the regenerative property of the liver could add another safety measure in case of undesired outcomes (Herman and Santos, 2023). The desired target cell population therefore depends on the chosen delivery strategy and could vary in their safety and efficacy.
4.1 Adeno-associated viral vectors
AAVs consist of a ssDNA genome and an icosahedral protein capsid. They are attractive due to their low immunogenicity and non-integrative nature. Furthermore, the natural occurrence of various serotypes can help elude existing neutralizing antibodies; which are considered a contra-indication for most AAV gene therapies. For bleeding disorders such as hemophilia A and B, AAVs are currently being used with transgenes of factor VIII and factor IX (Blair, 2022; Chowdary et al., 2022; George et al., 2021; Heo, 2023; Ozelo et al., 2022). These studies directed AAVs to the liver to achieve prolonged factor expression. Subsequently, most patients could discontinue prophylactic use of clotting factor concentrate infusions and the number of bleeds were strongly reduced. A review by Zwi-Dantsis et al. greatly summarizes AAVs and their applications in gene therapy (Zwi-Dantsis et al., 2025). Unfortunately, the tropism of AAVs for ECs is rather low, challenging EC transduction (Sreeramoju and Libutti, 2010). This can be enhanced through capsid modification, such as peptide insertion, which can alter specificity and infectivity to specific cells. This was shown by Varadi et al. where capsid engineered AAV2 and 9 containing peptide ligands selected from random peptide libraries were successfully used to enhance transduction of different EC types, such as HUVECS (Varadi et al., 2011). Nevertheless, there are limitations including the fading therapeutic effects over the years, but one of the main limitations of AAVs with respect to VWD is their limited cargo capacity of around 4.5 Kb. Consequently, a single AAV approach is unfeasible for large transgenes such as SpCas9, LbCas12a and VWF, with a CDS length of ∼8.4 Kb.
Dual AAV systems can be applied to circumvent the size issue but add further complications: firstly, they usually require a protein fragment such as a split-intein to combine both terminals together (Wu Y. et al., 2023). This can be less efficient than standard expression. Secondly, a higher dose of AAV titer is required which can cause immunogenic complications during treatment. Finally, target cells need to be transduced by both AAV constructs to be effective. This approach was tested in HUVECs and mice, delivering a human VWF transgene split across dual AAVs utilizing a recombinant region from the AK region of the F1 phage genome (Barbon et al., 2021). The plasma levels were between 1% and 1.5% of WT VWF and determined to be subtherapeutic.
Generally, single AAV outcompetes dual AAV systems, especially when AAV dose or tissue can prevent saturating levels of transduction (Davis et al., 2022). As the field evolves, it is likely that more efficient dual AAV delivery systems will be developed; but as it stands now, it is unlikely that AAVs will be utilized for VWF gene therapy.
4.2 Adenoviral vectors
Ads are commonly studied as vectors for transgene delivery, oncolytic agent, and vaccines (Paul et al., 2003; Peter and Kühnel, 2020; Smaill et al., 2013). In humans, more than 57 serotypes of Ads have been found to form seven Ad groups A-G (Wold and Toth, 2013). While this offers a big variety of Ads, most people are immune to several serotypes due to previous Ad infections. Ad’s genetic material is dsDNA with a length of 25–46 kb. The cargo capacity of Ad5, the most used Ad serotype, exceeds AAVs by far with 8–36 kb compared to AAV’s ∼5 kb (Shiver et al., 2002). Engineered Ad5 has been shown to transduce the vascular endothelium and ECs across multiple organs in mice (Lu et al., 2014). While they have many advantages such as their capacity to infect non-dividing and dividing cells of various tissues, their non-integrative nature and large cargo, the strong host immunogenicity and cellular toxicity of Ad vectors remain a major problem for their use in gene therapy (Wang and Shao, 2023).
4.3 Retroviral vectors
The first human cancer gene therapy approaches were facilitated with retroviruses, which consist of three subcategories: gamma-retroviruses, alpha-retroviruses, and lentiviruses. They all possess the ability to randomly integrate their cargo into the host-cell genome, thereby imposing a higher safety concern when compared to non-integrative gene therapy platforms (Shao et al., 2022). Liu et al. used a retroviral approach to target tumor-associated ECs and developed a successful transduction system of HUVECs and KSY1 ECs (Liu et al., 2000). However, the adverse events of secondary cancer developments, including leukemia, render retroviruses too dangerous to use in the clinic (Li et al., 2002).
As one of the most used retroviruses in research, we only further elaborate on the lentiviral system.
4.4 Lentiviral vectors
LV systems have large size capacities of around 9 Kb. This makes them attractive for delivery of large transgenes and led to the FDA approval of several LV-based gene therapies in the last years (Ali et al., 2019; Jensen et al., 2021; Keam, 2021; Schuessler-Lenz et al., 2019). LVs infect dividing and non-dividing cells–in contrast to gamma-retroviral vectors solely infecting dividing cells–which would make LVs attractive to target ECs, but low transduction efficiency and significant vector-associated cytotoxicity has been reported for ECs (Sreeramoju and Libutti, 2010). In vivo pseudotyping of LVs is pivotal to assure efficient and predominant delivery of agents to desired cells/tissues. The VSV-G pseudotype has successfully delivered Cas9 to CB-ECFCs and canine venous blood-derived ECs ex vivo (Meyer et al., 2006; Schillemans et al., 2019). In a proof of principle study, phenotypic correction of canine VWD ECFCs was obtained by LV-mediated gene delivery (Meyer et al., 2006). The GP64 pseudotyped LV vector has been shown to transduce liver sinusoidal endothelial cells (LSECs) more efficiently than vascular ECs. This was taken further with FVIII delivery to LSECs in FVIII KO mice (Milani et al., 2024). Other in vivo studies showed benefits of HIV-derived LV vectors over AAV vectors due to the decreased prevalence of HIV-directed antibodies in humans, reducing the host immune response (Follenzi et al., 2007). LV delivery was also explored in a canine model to treat hemophilia B with surprisingly positive results and no long-term toxicity in the animal model (Cantore et al., 2015). LV delivery was used to ensure stable gene integration in the liver cells, preventing dilution of the factor IX transgene. Common insertion sites (CIS) have been identified in patients undergoing LV gene therapy for adrenoleukodystrophy (ALD). Biffi et al. demonstrated that these CIS arise from a benign integration bias rather than oncogenic selection (Biffi et al., 2011). However, a recent study using LVs in ALD reported the development of hematological cancers due to clonal vector insertions within oncogenes (Duncan et al., 2024).
To increase the safety profile of LVs, IDLVs have been developed. The vector DNA of IDLV exists as non-replicating episomal DNA which prevents integration (Cortijo-Gutiérrez et al., 2021). Nevertheless, the episomal DNA can be subjected to epigenetic silencing, limiting the efficacy of treatment (Suwanmanee et al., 2014). Furthermore, unintegrated DNA is diluted out through cell division, so safe delivery of transiently acting proteins or protein fusions may be feasible using IDLVs (Wanisch and Yáñez-Muñoz, 2009).
4.5 Lipid nanoparticles (LNPs)
To date, LNPs are the most popular non-viral delivery systems, as they are being developed and optimized since the 1990s. They are FDA-approved (COVID-19 Pfizer/BioNTech and Moderna vaccines) and they exhibit low immunogenicity, transient expression and feasibility for large-scale production (Jung et al., 2022). Generally, LNPs are mainly used to deliver RNAs (siRNA or mRNA) but have also been shown to deliver CRISPR-Cas RNPs (Chen et al., 2024). The natural target site of LNPs are liver hepatocytes due to their neutral charge and the spontaneous binding of apolipoprotein E (ApoE) that functions as an endogenous ligand for hepatocytes. This property has been exploited for the development of LNP-based gene editing approaches for treatment of transthyretin amyloidosis as well as hereditary angioedema (Cohn et al., 2024; Gillmore et al., 2021). Interestingly, depending on their specific composition, some LNPs accumulate in LSECs, which leads to immune responses according to Sato et al. (2017). While this can be circumvented by reengineering the LNP composition, it can also be used to specifically target LSECs by modifying physical size and incorporating active targeting ligands, such as mannose or antibody moieties, to the LNP (Campbell et al., 2018; Kim et al., 2021; Pattipeiluhu et al., 2022). A recent review by Wang et al. provides a comprehensive overview of LNPs focusing on mRNA delivery, therapeutic applications and targeting mechanisms (Wang J. et al., 2024).
4.6 Viral-like particles (VLPs)
VLPs may provide an alternative to traditional LV approaches by combining the benefits of viral and non-viral delivery. VLPs are generated utilizing a fusion of the transgene of interest to the LV matrix gag protein. The most popular VLPs are pseudotyped with VSV-G glycoprotein, HIV-1 envelope glycoprotein and so-called ‘nanoblades’, which are murine leukemia VLPs loaded with Cas9-sgRNA ribonucleoproteins (RNPs) (Mangeot et al., 2019). Essentially a LV capsid is produced encapsulating the protein of interest, but while the envelope and capsids are viral components, the genome is non-viral. The cargo of mRNA, proteins or RNPs increase the safety by staying transiently expressed instead of integrating into the host genome. This improves CRISPR-Cas therapies regarding off-target activity and host immune response against the Cas protein, while still facilitating efficient genome editing (Lyu et al., 2019). The group of David Liu have engineered VLPs specifically to deliver BEs and PEs, which renders them promising delivery tools in vivo (An et al., 2024; Banskota et al., 2022). Furthermore, VLPs are relatively cost-effective and easily produced but the batch variability remains problematic (Jaron et al., 2022). Moreover, the immune response and rapid clearance of VLPs in vivo remains a potential concern. For further reading on this topic, see the reviews from Gupta et al. (2023), Nooraei et al. (2021), and Tariq et al. (2022).
As of now, with fine tuning of capsids, lipid composition, and pseudtyping, an array of delivery vehicles can be directed to the targeting of EC’s in-vivo. Smaller platforms such as AAVs that cannot accommodate the bulky nature of gene correction technologies and VWF currently sit in a suboptimal position; despite their attractive non-integrative nature. This likely leaves non integrative systems than can package larger loads as the foremost options.
5 Is gene therapy the future of VWD treatment?
While the demand for new VWD treatments is clear, the path to achieving them remains uncertain. Exploiting the latest advances in genome editing and gene delivery mentioned in this review, targeting the root cause of VWD through gene therapy now appears increasingly feasible.
Gene therapy can be achieved through either an ex vivo (manipulation of cells outside of the body) or an in vivo (direct manipulation inside the body) approach. As the predominant producers of VWF, ECs would be the optimal candidate for targeted therapy. Furthermore, ECs line the vasculature and therefore would be readily exposed to any agents delivered intravenously. The most common and accessible patient-derived ECs are venous blood-derived ECFCs. These cells are hampered by limited growth potential in vitro, and a vast heterogeneity between donors in terms of growth and isolation success, which could make ex vivo correction and transplantation problematic (Olgasi et al., 2021). Alternatively, human induced pluripotent stem cells (hiPSCs) derived from the patient as an autologous source could give rise to ECFCs through the mesoderm lineage, and multiple studies have successfully produced ECFCs in vitro (Abutaleb and George, 2021; Hamad et al., 2022). However, these models often exhibit a more embryonic state compared to primary ECFCs, and as such often lack WPBs, or produce pseudo-WPBs rather than the clear elongated shape that is expected (de Boer et al., 2024). These phenotypical discrepancies are too severe to use hiPSCs-derived ECs for VWD therapy and their use is even critically discussed for in vitro research. Consequently, in vivo gene therapy is the only feasible option for VWD gene therapy.
The allele-selective KO approach using siRNA that was discussed above seems to be one of the most promising approaches to alleviate VWD in vivo. So far, mouse models show impressive rescue of the bleeding phenotype but the duration of the siRNA approach and the feasibility of repeated treatment still needs further investigation (Jongejan et al., 2023). In vivo delivery of therapeutic siRNA was achieved employing encapsulation into 7C1 oligomeric lipid nanoparticles (composed of PEG, cholesterol and dioleoylphosphatidylethanolamine) specifically containing C14 alkyl PEG moieties and were shown to primarily target ECs (Sago et al., 2018; Yu et al., 2019). The targeting ability of 7C1-containing LNPs paves the way for siRNA- or CRISPR-based allele-selective disruption of mutant VWF alleles in VWD patients carrying dominant negative heterozygous mutations on their VWF. Furthermore, these studies provide a basis for the cutting-edge base and prime editing technologies, especially these composed of compact Cas proteins, that can be tailored to a variety of mutation types.
Proof-of-principle for the feasibility of gene editing employing a lentiviral-based CRISPR-Cas9 ABE in cord blood derived ECs has been shown with the goal of introducing a suspected pathogenic mutation in exon 18 of VWF (Bär et al., 2025). This observation provides an avenue towards in vivo correction of VWD employing CRISPR-Cas9 base and prime editing strategies. First in vitro experiments using a CBE on patient-derived ECFCs to correct the patient mutation p.M771V showed that correction of at least one allele leads to a rescue of the ECFC phenotype. However, in the current state efficiency of this method remains too low (<6%) to translate the genetic correction to a rescue of the bleeding phenotype. In addition, the low proliferative nature of ECs in vivo excludes most of the other genetic tools and enhances the risks associated with stable CRISPR-Cas expression in the cells. Hence, non-viral delivery of base and prime editors using the aforementioned 7C1-containing LNPs would prevent continuous editing and thus limit off-target effects that would most likely result from viral delivery systems.
Another point to consider for VWD gene therapy is the huge variety of pathogenic variants along VWF. Gene correction therapy needs to be personalized, which currently is associated with high costs. However, targeting the endothelium seems to be a durable approach and could potentially alleviate patient burden for several years.
Taking everything into consideration, considerable progress has been made with respect to the development of novel approaches for VWD gene therapy. In vivo delivery of siRNA-loaded LNPs targeting ECs has emerged as a potential treatment strategy for dominant negative VWD type 2. In vitro evidence for base editing approaches in ECFCs has recently been obtained. We anticipate that continuously evolving editing systems will allow for the generation of highly specific on-target editing tools with low off-target effects. The simultaneous evolution of delivery systems that can efficiently target therapeutic cargo to ECs has the potential to launch a new era of gene therapy for patients suffering from VWD.
Author contributions
AB: Data curation, Writing – review and editing, Visualization, Writing – original draft. IB: Writing – original draft, Writing – review and editing. Tv: Visualization, Writing – review and editing. KF: Writing – review and editing. JE: Writing – review and editing. FL: Writing – review and editing. RB: Writing – review and editing. JV: Writing – review and editing. DT: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research received funding from the Netherlands Organization for Scientific Research (NWO), Domain Applied and Engineering Sciences (TTW), ‘Connecting Innovators’ Open Technology Programme, Project#18712.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fgeed.2025.1719330.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abutaleb, N. O., and George, A. T. (2021). Differentiation and characterization of human IPSC-derived vascular endothelial cells under physiological shear stress. Star. Protoc. 2 (2), 100394. doi:10.1016/J.XPRO.2021.100394
Acharya, S., Mishra, A., Paul, D., Ansari, A. H., Azhar, M., Kumar, M., et al. (2019). Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proc. Natl. Acad. Sci. U. S. A. 116 (42), 20959–20968. doi:10.1073/PNAS.1818461116
Agudelo, D., Carter, S., Velimirovic, M., Duringer, A., Rivest, J. F., Levesque, S., et al. (2020). Versatile and robust genome editing with Streptococcus thermophilus CRISPR1-Cas9. Genome Res. 30 (1), 107–117. doi:10.1101/gr.255414.119
Al-Zain, A. M., and Symington, L. S. (2021). The dark side of homology-directed repair. DNA Repair 106, 103181. doi:10.1016/J.DNAREP.2021.103181
Ali, S., Kjeken, R., Niederlaender, C., Markey, G., Saunders, T. S., Opsata, M., et al. (2019). The european medicines agency review of kymriah (tisagenlecleucel) for the treatment of acute lymphoblastic leukemia and diffuse large b-cell lymphoma. Oncol. 25 (2), e321–e327. doi:10.1634/THEONCOLOGIST.2019-0233
An, M., Raguram, A., Du, S. W., Banskota, S., Davis, J. R., Newby, G. A., et al. (2024). Engineered virus-like particles for transient delivery of prime editor ribonucleoprotein complexes in vivo. Nat. Biotechnol. 42 (10), 1526–1537. doi:10.1038/s41587-023-02078-y
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-Replace genome editing without double-strand breaks or donor DNA. Nature 576 (7785), 149–157. doi:10.1038/s41586-019-1711-4
Atiq, F., Boender, J., Van Heerde, W. L., Tellez Garcia, J. M., Schoormans, S. C., Krouwel, S., et al. (2022). Importance of Genotyping in von Willebrand Disease to Elucidate Pathogenic Mechanisms and Variability in Phenotype. HemaSphere 6 (6), E718. doi:10.1097/HS9.0000000000000718
Atiq, F., Blok, R., van Kwawegen, C. B., Doherty, D., Lavin, M., van der Bom, J. G., et al. (2023). Type 1 VWD classification revisited: novel insights from combined analysis of the LoVIC and WiN studies. Blood 143 (14), 1414–1424. doi:10.1182/BLOOD.2023022457
Banskota, S., Raguram, A., Suh, S., Du, S. W., Davis, J. R., Choi, E. H., et al. (2022). Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 185 (2), 250–265.e16. doi:10.1016/j.cell.2021.12.021
Bär, I., Bürgisser, P., van Moort, I., van Kwawegen, C., Voorberg, J., Eikenboom, J., et al. (2024). Oral communication abstracts. Res. Pract. Thrombosis Haemostasis 8, 102503. doi:10.1016/j.rpth.2024.102503
Bär, I., Barraclough, A., Bürgisser, P. E., van Kwawegen, C., Fijnvandraat, K., Eikenboom, J. C. J., et al. (2025). The Severe von Willebrand Disease Variant p.M771V Leads to Impaired Anterograde Trafficking of von Willebrand Factor in Patient-Derived and Base-Edited Endothelial Colony-Forming Cells. J. Thrombosis Haemostasis JTH 23 (2), 466–479. doi:10.1016/J.JTHA.2024.10.023
Barbon, E., Kawecki, C., Marmier, S., Sakkal, A., Collaud, F., Charles, S., et al. (2021). Development of a Dual Hybrid AAV Vector for Endothelial-Targeted Expression of von Willebrand Factor. Gene Ther. 30 (3), 245–254. doi:10.1038/s41434-020-00218-6
Bazaz, R., MahereSeno, M. M. G., and Dehghani, H. (2022). Transposase-CRISPR mediated targeted integration (TransCRISTI) in the human genome. Sci. Rep. 12 (1), 3390. doi:10.1038/S41598-022-07158-8
Biffi, A., Bartholomae, C. C., Cesana, D., Cartier, N., Aubourg, P., Ranzani, M., et al. (2011). Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 117 (20), 5332–5339. doi:10.1182/BLOOD-2010-09-306761
Blair, H. A. (2022). Valoctocogene roxaparvovec: first approval. Drugs 82 (14), 1505–1510. doi:10.1007/s40265-022-01788-y
Bloomer, H., Smith, R. H., Hakami, W., and Larochelle, A. (2020). Genome editing in human hematopoietic stem and progenitor cells via CRISPR-Cas9-Mediated homology-independent targeted integration. Mol. Ther. 29 (4), 1611–1624. doi:10.1016/J.YMTHE.2020.12.010
Bowman, M. L., Li, L., Pluthero, F., Christensen, H., Walker, I., Kahr, W. H., et al. (2012). Comparative cellular studies of the VWF exon 4–5 deletion mutation using patient-derived blood outgrowth endothelial cells (BOEC) and megakaryocytes. Blood 120 (21), 1076. doi:10.1182/BLOOD.V120.21.1076.1076
Campbell, F., Bos, F. L., Sieber, S., Arias-Alpizar, G., Koch, B. E., Huwyler, J., et al. (2018). Directing nanoparticle biodistribution through evasion and exploitation of Stab2-Dependent nanoparticle uptake. ACS Nano 12 (3), 2138–2150. doi:10.1021/acsnano.7b06995
Campioni, M., Legendre, P., Loubiere, C., Lunghi, B., Pinotti, M., Christophe, O. D., et al. (2021). In vivo Modulation of a Dominant-negative Variant in Mouse Models of von Willebrand Disease Type 2A. J. Thrombosis Haemostasis 19 (1), 139–146. doi:10.1111/JTH.15131
Cantore, A., Ranzani, M., Bartholomae, C. C., Volpin, M., Valle, P. D., Sanvito, F., et al. (2015). Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 7 (277), 277ra28. doi:10.1126/scitranslmed.aaa1405
Cecchin, R., Troyer, Z., Witwer, K., and Morris, K. V. (2023). Extracellular vesicles: the next generation in gene therapy delivery. Mol. Ther. J. Am. Soc. Gene Ther. 31 (5), 1225–1230. doi:10.1016/J.YMTHE.2023.01.021
Chapman, J. R., Taylor, M. R. G., and Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47 (4), 497–510. doi:10.1016/J.MOLCEL.2012.07.029
Chatterjee, P., Jakimo, N., and Jacobson, J. M. (2018). Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 4 (10), eaau0766. doi:10.1126/SCIADV.AAU0766
Chen, K., Han, H., Zhao, S., Xu, B., Yin, B., Lawanprasert, A., et al. (2024). Lung and liver editing by lipid nanoparticle delivery of a stable CRISPR–Cas9 ribonucleoprotein. Nat. Biotechnol. 2024, 1–13. doi:10.1038/s41587-024-02437-3
Chen, L., Park, J. E., Paa, P., Rajakumar, P. D., Prekop, H. T., Chew, Y. T., et al. (2021). Programmable C:G to G:C genome editing with CRISPR-Cas9-Directed base excision repair proteins. Nat. Commun. 12 (1), 1384–1387. doi:10.1038/s41467-021-21559-9
Chen, L., Zhang, S., Xue, N., Hong, M., Zhang, X., Zhang, D., et al. (2022). Engineering a precise adenine base editor with minimal bystander editing. Nat. Chem. Biol. 19 (1), 101–110. doi:10.1038/s41589-022-01163-8
Chen, P. J., Hussmann, J. A., Yan, J., Knipping, F., Ravisankar, P., Chen, P. F., et al. (2021). Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184 (22), 5635–5652.e29. doi:10.1016/J.CELL.2021.09.018
Chen, X., Niu, X., Liu, Y., Zheng, R., Yang, L., Lu, J., et al. (2022). Long-term correction of hemophilia B through CRISPR/Cas9 induced homology-independent targeted integration. J. Genet. Genomics 49 (12), 1114–1126. doi:10.1016/J.JGG.2022.06.001
Chowdary, P., Shapiro, S., Makris, M., Evans, G., Boyce, S., Kate, T., et al. (2022). Phase 1–2 trial of AAVS3 gene therapy in patients with hemophilia B. N. Engl. J. Med. 387 (3), 237–247. doi:10.1056/NEJMoa2119913
Christopherson, P. A., Haberichter, S. L., Flood, V. H., Perry, C. L., Sadler, B. E., Bellissimo, D. B., et al. (2022). Molecular pathogenesis and heterogeneity in type 3 VWD families in U.S. zimmerman program. J. Thrombosis Haemostasis 20 (7), 1576–1588. doi:10.1111/jth.15713
Christopherson, P. A., Tijet, N., Haberichter, S. L., Flood, V. H., Ross, J., Notley, C., et al. (2024). The common VWF variant p.Y1584C: detailed pathogenic examination of an enigmatic sequence change. J. Thrombosis Haemostasis 22 (3), 666–675. doi:10.1016/J.JTHA.2023.11.016
Cohn, D. M., Gurugama, P., Magerl, M., Katelaris, C. H., Launay, D., Bouillet, L., et al. (2024). CRISPR-based therapy for hereditary angioedema. N. Engl. J. Med. 392, 458–467. doi:10.1056/NEJMoa2405734
Connell, N. T., Flood, V. H., Brignardello-Petersen, R., Abdul-Kadir, R., Arapshian, A., Couper, S., et al. (2021). ASH ISTH NHF WFH 2021 Guidelines on the Management of von Willebrand Disease. Blood Adv. 5 (1), 301–325. doi:10.1182/BLOODADVANCES.2020003264
Cortijo-Gutiérrez, M., Sánchez-Hernández, S., Tristán-Manzano, M., Maldonado-Pérez, N., Lopez-Onieva, L., Real, P. J., et al. (2021). Improved functionality of integration-deficient lentiviral vectors (IDLVs) by the inclusion of IS2 protein docks. Pharmaceutics 13 (8), 1217. doi:10.3390/pharmaceutics13081217
Cox, D. B. T., Platt, R. J., and Zhang, F. (2015). Therapeutic genome editing: prospects and challenges. Nat. Med. 21 (2), 121–131. doi:10.1038/NM.3793
Cullot, G., Aird, E. J., Schlapansky, M. F., Yeh, C. D., Venn, L. van de, Vykhlyantseva, I., et al. (2024). Genome editing with the HDR-enhancing DNA-PKcs inhibitor AZD7648 causes large-scale genomic alterations. Nat. Biotechnol. 2024, 1–5. doi:10.1038/s41587-024-02488-6
Daniel, M. Y., Ternisien, C., Castet, S., Falaise, C., D’Oiron, R., Volot, F., et al. (2024). Type 2N von Willebrand Disease: Genotype Drives Different Bleeding Phenotypes and Treatment Needs. J. Thrombosis Haemostasis 22 (10), 2702–2712. doi:10.1016/J.JTHA.2024.06.020
Davis, J. R., Wang, X., Witte, I. P., Huang, T. P., Levy, J. M., Raguram, A., et al. (2022). Efficient in vivo base editing via single adeno-Associated viruses with size-optimized genomes encoding compact adenine base editors. Nat. Biomed. Eng. 6 (11), 1272–1283. doi:10.1038/s41551-022-00911-4
de Boer, S., Laan, S., Dirven, R., and Eikenboom, J. (2024). Approaches to induce the maturation process of human induced pluripotent stem cell derived-endothelial cells to generate a robust model. PLOS ONE 19 (2), e0297465. doi:10.1371/JOURNAL.PONE.0297465
de Jong, A., and Eikenboom, J. (2017). Von Willebrand Disease Mutation Spectrum and Associated Mutation Mechanisms. Thrombosis Res. 159, 65–75. doi:10.1016/J.THROMRES.2017.09.025
De Jong, A., Dirven, R. J., Boender, J., Atiq, F., Yahya Anvar, S., Leebeek, F. W. G., et al. (2020). Ex Vivo Improvement of a von Willebrand Disease Type 2A Phenotype Using an Allele-Specific Small-Interfering RNA. Thrombosis Haemostasis 120 (11), 1569–1579. doi:10.1055/S-0040-1715442
Desch, K. C., Ozel, A. B., Siemieniak, D., Kalish, Y., Shavit, J. A., Thornburg, C. D., et al. (2013). Linkage Analysis Identifies a Locus for Plasma von Willebrand Factor Undetected by Genome-Wide Association. Proc. Natl. Acad. Sci. U. S. A. 110 (2), 588–593. doi:10.1073/pnas.1219885110
Di Stazio, M., Foschi, N., Athanasakis, E., Gasparini, P., and Pio d’Adamo, A. (2021). Systematic analysis of factors that improve Homologous direct repair (HDR) efficiency in CRISPR/Cas9 technique. PLOS ONE 16 (3), e0247603. doi:10.1371/JOURNAL.PONE.0247603
Duncan, C. N., Bledsoe, J. R., Grzywacz, B., Beckman, A., Bonner, M., Eichler, F. S., et al. (2024). Hematologic cancer after gene therapy for cerebral adrenoleukodystrophy. N. Engl. J. Med. 391 (14), 1287–1301. doi:10.1056/NEJMoa2405541
Eghbalsaied, S., and Kues, W. A. (2023). CRISPR/Cas9-Mediated targeted Knock-in of large constructs using nocodazole and RNase HII. Sci. Rep. 2023 13 (1), 2690–15. doi:10.1038/s41598-023-29789-1
Eikenboom, J. C. J. (2001). Congenital von Willebrand Disease Type 3: clinical Manifestations, Pathophysiology and Molecular Biology. Best Pract. and Res. Clin. Haematol. 14 (2), 365–379. doi:10.1053/BEHA.2001.0139
Fedorova, I., Vasileva, A., Selkova, P., Abramova, M., Arseniev, A., Pobegalov, G., et al. (2020). PpCas9 from pasteurella pneumotropica - a compact type II-C Cas9 ortholog active in human cells. Nucleic Acids Res. 48 (21), 12297–12309. doi:10.1093/NAR/GKAA998
Fell, C. W., Schmitt-Ulms, C., Tagliaferri, D. V., Gootenberg, J. S., and Abudayyeh, O. O. (2024). Precise kilobase-scale genomic insertions in mammalian cells using PASTE. Nat. Protoc. 20, 1546–1583. doi:10.1038/s41596-024-01090-z
Fiumara, M., Ferrari, S., Omer-Javed, A., Beretta, S., Albano, L., Canarutto, D., et al. (2024). Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nat. Biotechnol. 42 (6), 877–891. doi:10.1038/s41587-023-01915-4
Follenzi, A., Santambrogio, L., and Annoni, A. (2007). Immune responses to lentiviral vectors. Curr. Gene Ther. 7 (5), 306–315. doi:10.2174/156652307782151515
Furini, G., De Carli, A., Fonnesu, R., Spezia, P. G., Scebba, F., Pistello, M., et al. (2023). Gene Silencing of Endothelial von Willebrand Factor Reduces the Susceptibility of Human Endothelial Cells to SARS-CoV-2 Infection. FEBS J. 290 (17), 4300–4315. doi:10.1111/FEBS.16808
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2017). Programmable base editing of A˙T to G˙C in genomic DNA without DNA cleavage. Nature 551 (7681), 464–471. doi:10.1038/nature24644
George, L. A., Monahan, P. E., Elaine Eyster, M., Sullivan, S. K., Ragni, M. V., Croteau, S. E., et al. (2021). Multiyear factor VIII expression after AAV gene transfer for hemophilia A. N. Engl. J. Med. 385 (21), 1961–1973. doi:10.1056/NEJMoa2104205
Giblin, J. P., Hewlett, L. J., and Hannah, M. J. (2008). Basal Secretion of von Willebrand Factor from Human Endothelial Cells. Blood 112 (4), 957–964. doi:10.1182/BLOOD-2007-12-130740
Gill, J. C., Castaman, G., Windyga, J., Kouides, P., Ragni, M., Leebeek, F. W. G., et al. (2015). Hemostatic Efficacy, Safety, and Pharmacokinetics of a Recombinant von Willebrand Factor in Severe von Willebrand Disease. Blood 126 (17), 2038–2046. doi:10.1182/BLOOD-2015-02-629873
Gillmore, J. D., Gane, J. T., Kao, J., Fontana, M., Maitland, M. L., Jessica, S., et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385 (6), 493–502. doi:10.1056/nejmoa2107454
gnomAD (2025). gnomAD.boradinstitte. Available online at: https://gnomad.broadinstitute.org/ (Accessed April 1, 2025).
Gupta, R., Arora, K., Roy, S. S., Joseph, A., Rastogi, R., Arora, N. M., et al. (2023). Platforms, advances, and technical challenges in virus-like particles-based vaccines. Front. Immunol. 14, 1123805. doi:10.3389/FIMMU.2023.1123805
Haapaniemi, E., Botla, S., Persson, J., Schmierer, B., and Taipale, J. (2018). CRISPR–Cas9 genome editing induces a P53-Mediated DNA damage response. Nat. Med. 24 (7), 927–930. doi:10.1038/s41591-018-0049-z
Haberichter, S. L. (2015). Von Willebrand Factor Propeptide: biology and Clinical Utility. Blood 126 (15), 1753–1761. doi:10.1182/BLOOD-2015-04-512731
Hamad, S., Derichsweiler, D., Gaspar, J. A., Brockmeier, K., Hescheler, J., Sachinidis, A., et al. (2022). High-efficient serum-free differentiation of endothelial cells from human IPS cells. Stem Cell Res. Ther. 13 (1), 251–16. doi:10.1186/s13287-022-02924-x
Harrington, L. B., Paez-Espino, D., Staahl, B. T., Chen, J. S., Ma, E., Kyrpides, N. C., et al. (2017). A thermostable Cas9 with increased lifetime in human plasma. Nat. Commun. 8 (1), 1424–1428. doi:10.1038/s41467-017-01408-4
Heo, Y.-A. (2023). Etranacogene dezaparvovec: first approval. Drugs 83 (4), 347–352. doi:10.1007/s40265-023-01845-0
Herman, T. F., and Santos, C. (2023). “First-pass effect,” in XPharm: the comprehensive pharmacology reference, 1–2. doi:10.1016/B978-008055232-3.60022-4
HGMD (2025). HGMD® home page. Available online at: https://www.hgmd.cf.ac.uk/ac/index.php (Accessed April 1, 2025).
Hordijk, S., Carter, T., and Bierings, R. (2024). A New Look at an Old Body: molecular Determinants of Weibel-Palade Body Composition and von Willebrand Factor Exocytosis. J. Thrombosis Haemostasis JTH 22 (5), 1290–1303. doi:10.1016/J.JTHA.2024.01.015
Hu, Z., Wang, S., Zhang, C., Gao, N., Li, M., Wang, D., et al. (2020). A compact Cas9 ortholog from Staphylococcus auricularis (SauriCas9) expands the DNA targeting scope. PLoS Biol. 18 (3), e3000686. doi:10.1371/JOURNAL.PBIO.3000686
Huang, M. E., Qin, Y., Shang, Y., Qian, H., Zhan, C., Lian, C., et al. (2024). C-to-G editing generates double-strand breaks causing deletion, transversion and translocation. Nat. Cell Biol. 26 (2), 294–304. doi:10.1038/s41556-023-01342-2
Huang, X., Wu, W., Qi, H., Yan, X., Dong, L., Yang, Y., et al. (2024). Exploitation of enhanced prime editing for blocking aberrant angiogenesis. J. Adv. Res. 72, 121–133. doi:10.1016/J.JARE.2024.07.006
Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D., Theriault, K., et al. (2018). P53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 24 (7), 939–946. doi:10.1038/s41591-018-0050-6
Jacobi, P. M., Gill, J. C., Flood, V. H., Jakab, D. A., Friedman, K. D., and Haberichter, S. L. (2012). Intersection of mechanisms of type 2A VWD through defects in VWF multimerization, secretion, ADAMTS-13 susceptibility, and regulated storage. Blood 119 (19), 4543–4553. doi:10.1182/BLOOD-2011-06-360875
James, P. D., Paterson, A. D., Notley, C., Cameron, C., Hegadorn, C., Tinlin, S., et al. (2006). Genetic Linkage and Association Analysis in Type 1 von Willebrand Disease: results from the Canadian Type 1 VWD Study. J. Thrombosis Haemostasis JTH 4 (4), 783–792. doi:10.1111/J.1538-7836.2006.01860.X
James, P. D., Connell, N. T., Ameer, B., Di Paola, J., Eikenboom, J., Giraud, N., et al. (2021). ASH ISTH NHF WFH 2021 Guidelines on the Diagnosis of von Willebrand Disease. Blood Adv. 5 (1), 280–300. doi:10.1182/BLOODADVANCES.2020003265
Jaron, M., Lehky, M., Zarà, M., Zaydowicz, C. N., Lak, A., Ballmann, R., et al. (2022). Baculovirus-free SARS-CoV-2 virus-like particle production in insect cells for rapid neutralization assessment. Viruses 14 (10), 2087. doi:10.3390/v14102087
Jensen, T. L., Gøtzsche, C. R., and Woldbye, D. P. D. (2021). Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front. Mol. Neurosci. 14, 695937. doi:10.3389/FNMOL.2021.695937
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable Dual-RNA-Guided DNA endonuclease in adaptive bacterial immunity. Science 337 (6096), 816–821. doi:10.1126/science.1225829
Jongejan, Y. K., Elisa, S. E., Dirven, R. J., Paunovska, K., Linthorst, N. A., de Jong, A., et al. (2023). Small Interfering RNA–Mediated Allele-Selective Silencing of von Willebrand Factor in vitro and in vivo. Blood Adv. 7 (20), 6108–6119. doi:10.1182/BLOODADVANCES.2023010643
Jongejan, Y. K., Linthorst, N. A., Schrader Echeverri, E., Laan, S. N. J., Dirven, R. J., Dahlman, J. E., et al. (2024). Impact of Allele-Selective Silencing of von Willebrand Factor in Mice Based on a Single Nucleotide Allelic Difference in von Willebrand Factor. Thrombosis Res. 236, 201–208. doi:10.1016/J.THROMRES.2024.03.002
Jung, H.N., Lee, S. Y., Lee, S., Youn, H., and Jun Im, H. (2022). Lipid nanoparticles for delivery of RNA therapeutics: current status and the role of in vivo imaging. Theranostics 12 (17), 7509–7531. doi:10.7150/THNO.77259
Karampini, E., Bierings, R., and Jan, V. (2020). Orchestration of primary hemostasis by platelet and endothelial lysosome-related organelles. Arteriosclerosis, Thrombosis, Vasc. Biol. 40 (6), 1441–1453. doi:10.1161/ATVBAHA.120.314245
Kaufmann, J. E., Oksche, A., Wollheim, C. B., Günther, G., Rosenthal, W., and Vischer, U. M. (2000). Vasopressin-Induced von Willebrand Factor Secretion from Endothelial Cells Involves V2 Receptors and CAMP. J. Clin. Investigation 106 (1), 107–116. doi:10.1172/JCI9516
Keam, S. J. (2021). Elivaldogene autotemcel: first approval. Mol. Diagnosis and Ther. 25 (6), 803–809. doi:10.1007/S40291-021-00555-1
Kim, M., Jeong, M., Hur, S., Cho, Y., Park, J., Jung, H., et al. (2021). Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 7 (9), eabf4398. doi:10.1126/SCIADV.ABF4398
Kosicki, M., Tomberg, K., and Bradley, A. (2018). Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36 (8), 765–771. doi:10.1038/nbt.4192
Kurt, I. C., Zhou, R., Iyer, S., Garcia, S. P., Miller, B. R., Langner, L. M., et al. (2020). CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 39 (1), 41–46. doi:10.1038/s41587-020-0609-x
Laan, S. N. J., Lenderink, B. G., Eikenboom, J. C. J., and Bierings, R.SYMPHONY consortium (2024). Endothelial Colony-Forming Cells in the Spotlight: insights into the Pathophysiology of von Willebrand Disease and Rare Bleeding Disorders. J. Thrombosis Haemostasis JTH 22 (12), 3355–3365. doi:10.1016/J.JTHA.2024.08.011
Laan, S., Castillo Alferez, J. D., Cannegieter, S., Fijnvandraat, K., Kruip, M., le Cessie, S., et al. (2025). DDAVP response and its determinants in bleeding disorders: a systematic review and meta-analysis. Blood 145, 1814–1825. doi:10.1182/BLOOD.2024026804
Laffan, M. A., Lester, W., O’Donnell, J. S., Will, A., Campbell Tait, R., Goodeve, A., et al. (2014). The Diagnosis and Management of von Willebrand Disease: a United Kingdom Haemophilia Centre Doctors Organization Guideline Approved by the British Committee for Standards in Haematology. Br. J. Haematol. 167 (4), 453–465. doi:10.1111/bjh.13064
Lam, D. K., Feliciano, P. R., Arif, A., Bohnuud, T., Fernandez, T. P., Gehrke, J. M., et al. (2023). Improved cytosine base editors generated from TadA variants. Nat. Biotechnol. 41 (5), 686–697. doi:10.1038/s41587-022-01611-9
Lee, C. M., Cradick, T. J., and Bao, G. (2016). The Neisseria meningitidis CRISPR-Cas9 System enables specific genome editing in mammalian cells. Mol. Ther. J. Am. Soc. Gene Ther. 24 (3), 645–654. doi:10.1038/MT.2016.8
Leebeek, F. W. G., and Eikenboom, J. C. J. (2016). Von Willebrand’s Disease. N. Engl. J. Med. 375 (21), 2067–2080. doi:10.1056/NEJMRA1601561
Leebeek, F. W. G., Peyvandi, F., Escobar, M., Tiede, A., Castaman, G., Wang, M., et al. (2022). Recombinant von Willebrand Factor Prophylaxis in Patients with Severe von Willebrand Disease: phase 3 Study Results. Blood 140 (2), 89–98. doi:10.1182/BLOOD.2021014810
Lenting, P. J., Christophe, O. D., and Denis, C. V. (2015). Von Willebrand Factor Biosynthesis, Secretion, and Clearance: connecting the Far Ends. Blood 125 (13), 2019–2028. doi:10.1182/BLOOD-2014-06-528406
Lenting, P. J., Denis, C. V., and Christophe, O. D. (2024). How Unique Structural Adaptations Support and Coordinate the Complex Function of von Willebrand Factor. Blood 144 (21), 2174–2184. doi:10.1182/BLOOD.2023023277
Li, Z., Düllmann, J., Schiedlmeier, B., Schmidt, M., Von Kalle, C., Meyer, J., et al. (2002). Murine leukemia induced by retroviral gene marking. Science 296 (5567), 497. doi:10.1126/science.1068893
Liao, H., Wu, J., VanDusen, N. J., Li, Y., and Zheng, Y. (2024). CRISPR-Cas9-Mediated homology-directed repair for precise gene editing. Mol. Ther. Nucleic Acids 35 (4), 102344. doi:10.1016/j.omtn.2024.102344
Linthorst, N. A., Jongejan, Y. K., Dirven, R. J., Laan, S. N. J., Bierings, R., Casari, C., et al. (2024). Amelioration of a von Willebrand Disease Type 2B Phenotype in vivo upon Treatment with Allele-Selective SiRNAs. Blood Adv. 9 (2), 310–320. doi:10.1182/BLOODADVANCES.2024014601
Linthorst, N. A., van Vlijmen, B. J. M., and Eikenboom, J. C. J. (2025). The Future of SiRNA-Mediated Approaches to Treat von Willebrand Disease. Expert Rev. Hematol. 18 (2), 109–122. doi:10.1080/17474086.2025.2459259
Liu, L., Liu, L., Anderson, W. F., Beart, R. W., Gordon, E. M., and Hall, F. L. (2000). Incorporation of tumor vasculature targeting motifs into moloney murine leukemia Virus env escort proteins enhances retrovirus binding and transduction of human endothelial cells. J. Virology 74 (11), 5320–5328. doi:10.1128/jvi.74.11.5320-5328.2000
Liu, J. J., Orlova, N., Oakes, B. L., Ma, E., Spinner, H. B., Baney, K. L. M., et al. (2019). CasX enzymes comprise a distinct family of RNA-Guided genome editors. Nat. 2019 566 (7743), 218–223. doi:10.1038/s41586-019-0908-x
Lu, Z. H., Kaliberov, S., Zhang, J., Muz, B., Azab, A. K., Sohn, R. E., et al. (2014). The myeloid-binding peptide adenoviral vector enables multi-organ vascular endothelial gene targeting. Lab. Investig. 94 (8), 881–892. doi:10.1038/labinvest.2014.78
Lyu, P., Javidi-Parsijani, P., Atala, A., and Lu, B. (2019). Delivering Cas9/SgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ‘Hit-and-Run’ genome editing. Nucleic Acids Res. 47 (17), e99. doi:10.1093/NAR/GKZ605
Ma, G., Qi, H., Deng, H., Dong, L., Zhang, Q., Ma, J., et al. (2024). Prime editing of vascular endothelial growth factor receptor 2 attenuates angiogenesis in vitro. arXiv 7 (4), 188–196. doi:10.1089/CRISPR.2024.0019
Maas, D. P. M. S. M., Atiq, F., Blijlevens, N. M. A., Brons, P. P. T., Krouwel, S., Laros-van Gorkom, B. A. P., et al. (2021). Von Willebrand Disease Type 2M: correlation between Genotype and Phenotype. J. Thrombosis Haemostasis 20 (2), 316–327. doi:10.1111/JTH.15586
Mangeot, P. E., Risson, V., Fusil, F., Marnef, A., Laurent, E., Blin, J., et al. (2019). Genome editing in primary cells and in vivo using viral-derived nanoblades loaded with Cas9-SgRNA ribonucleoproteins. Nat. Commun. 10 (1), 45–15. doi:10.1038/s41467-018-07845-z
Martin, R. M., Ikeda, K., Kyle Cromer, M., Uchida, N., Nishimura, T., Romano, R., et al. (2019). Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-Cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell 24 (5), 821–828. doi:10.1016/j.stem.2019.04.001
Meng, X., Jia, R., Zhao, X., Zhang, F., Chen, S., Yu, S., et al. (2024). In vivo genome editing via CRISPR/Cas9-Mediated homology-independent targeted integration for bietti crystalline corneoretinal dystrophy treatment. Nat. Commun. 15 (1), 3773. doi:10.1038/S41467-024-48092-9
Meyer, D., Vanhoorelbeke, K., Chuah, M. K., Pareyn, I., Gillijns, V., Hebbel, R. P., et al. (2006). Phenotypic Correction of von Willebrand Disease Type 3 Blood-Derived Endothelial Cells with Lentiviral Vectors Expressing von Willebrand Factor. Blood 107 (12), 4728–4736. doi:10.1182/BLOOD-2005-09-3605
Meyer, D., Vandeputte, N., Pareyn, I., Petrus, I., Lenting, P. J., Chuah, M. K. L., et al. (2008). Restoration of Plasma von Willebrand Factor Deficiency Is Sufficient to Correct Thrombus Formation after Gene Therapy for Severe von Willebrand Disease. Arteriosclerosis, Thrombosis, Vasc. Biol. 28 (9), 1621–1626. doi:10.1161/ATVBAHA.108.168369
Milani, M., Canepari, C., Assanelli, S., Merlin, S., Borroni, E., Starinieri, F., et al. (2024). GP64-Pseudotyped lentiviral vectors target liver endothelial cells and correct hemophilia A mice. EMBO Mol. Med. 16 (6), 1427–1450. doi:10.1038/S44321-024-00072-8
Müller, M., Lee, C. M., Gasiunas, G., Davis, T. H., Cradick, T. J., Siksnys, V., et al. (2016). Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing of the human genome. Mol. Ther. J. Am. Soc. Gene Ther. 24 (3), 636–644. doi:10.1038/MT.2015.218
Murray, J. B., Harrison, P. T., and Scholefield, J. (2025). Prime editing: therapeutic advances and mechanistic insights. Gene Ther. 32 (2), 83–92. doi:10.1038/s41434-024-00499-1
Nambiar, T. S., Baudrier, L., Billon, P., and Ciccia, A. (2022). CRISPR-based genome editing through the lens of DNA repair. Mol. Cell 82 (2), 348–388. doi:10.1016/J.MOLCEL.2021.12.026
NCBI (2025). ClinVar. Available online at: https://www.ncbi.nlm.nih.gov/clinvar/ (Accessed April 1, 2025).
Nelson, J. W., Randolph, P. B., Shen, S. P., Everette, K. A., Chen, P. J., Anzalone, A. V., et al. (2022). Engineered PegRNAs improve prime editing efficiency. Nat. Biotechnol. 40 (3), 402–410. doi:10.1038/S41587-021-01039-7
Nguyen, D. N., Roth, T. L., Li, P. J., Chen, P. A., Apathy, R., Mamedov, M. R., et al. (2019). Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 38 (1), 44–49. doi:10.1038/s41587-019-0325-6
Nichols, W. L., Hultin, M. B., James, A. H., Manco-Johnson, M. J., Montgomery, R. R., Ortel, T. L., et al. (2008). Von Willebrand Disease (VWD): evidence-based Diagnosis and Management Guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel Report (USA). Haemoph. Official J. World Fed. Hemophilia 14 (2), 171–232. doi:10.1111/J.1365-2516.2007.01643.X
Nooraei, S., Bahrulolum, H., Sadat Hoseini, Z., Katalani, C., Hajizade, A., Easton, A. J., et al. (2021). Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnology 19 (1), 59–27. doi:10.1186/S12951-021-00806-7
Olgasi, C., Borsotti, C., Merlin, S., Bergmann, T., Bittorf, P., Adewoye, A. B., et al. (2021). Efficient and safe correction of hemophilia A by lentiviral vector-transduced BOECs in an implantable device. Mol. Ther. - Methods and Clin. Dev. 23, 551–566. doi:10.1016/J.OMTM.2021.10.015
Orphanet (2025a). Von Willebrand disease type 1. Available online at: https://www.orpha.net/en/disease/detail/166078?name=Von%20willebrand%20disease&mode=name (Accessed July 10, 2025).
Orphanet (2025b). Von Willebrand disease type 2. Available online at: https://www.orpha.net/en/disease/detail/166081?name=Von%20willebrand%20disease&mode=name (Accessed July 10, 2025).
Othman, M., and Favaloro, E. J. (2021). 2B von Willebrand Disease Diagnosis: considerations Reflecting on 2021 Multisociety Guidelines. Res. Pract. Thrombosis Haemostasis 5 (8), e12635. doi:10.1002/RTH2.12635
Ozelo, M. C., Mahlangu, J., Pasi, K. J., Giermasz, A., Leavitt, A. D., Laffan, M., et al. (2022). Valoctocogene roxaparvovec gene therapy for hemophilia A. N. Engl. J. Med. 386 (11), 1013–1025. doi:10.1056/NEJMoa2113708
Pallarès-Masmitjà, M., Ivančić, D., Mir-Pedrol, J., Jaraba-Wallace, J., Tagliani, T., Oliva, B., et al. (2021). Find and cut-and-transfer (FiCAT) Mammalian genome engineering. Nat. Commun. 12 (1), 1–9. doi:10.1038/s41467-021-27183-x
Pandey, S., Gao, X. D., Krasnow, N. A., McElroy, A., Allen Tao, Y., Duby, J. E., et al. (2024). Efficient site-specific integration of large genes in Mammalian cells via continuously evolved recombinases and prime editing. Nat. Biomed. Eng. 9 (1), 22–39. doi:10.1038/S41551-024-01227-1
Pattipeiluhu, R., Arias-Alpizar, G., Basha, G., Chan, K. Y. T., Bussmann, J., Sharp, T. H., et al. (2022). Anionic lipid nanoparticles preferentially deliver MRNA to the hepatic reticuloendothelial system. Adv. Mater. Deerf. Beach, Fla. 34 (16), e2201095. doi:10.1002/ADMA.202201095
Paul, R., Haydon, R. C., Cheng, H., Ishikawa, A., Nenadovich, N., Jiang, W., et al. (2003). Potential use of Sox9 gene therapy for intervertebral degenerative disc disease. Spine 28 (8), 755–763. doi:10.1097/01.BRS.0000058946.64222.92
Perolina, E., Meissner, S., Raos, B., Harland, B., Thakur, S., and Svirskis, D. (2024). Translating ultrasound-mediated drug delivery technologies for CNS applications. Adv. Drug Deliv. Rev. 208, 115274. doi:10.1016/J.ADDR.2024.115274
Peter, M., and Kühnel, F. (2020). Oncolytic adenovirus in cancer immunotherapy. Cancers 12 (11), 3354. doi:10.3390/CANCERS12113354
Phua, C. W., and Erik, B. (2019). A personalized approach to the management of VWD. Transfus. Apher. Sci. 58 (5), 590–595. doi:10.1016/J.TRANSCI.2019.08.009
Pipe, S. W., Montgomery, R. R., Pratt, K. P., Lenting, P. J., and Lillicrap, D. (2016). Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A. Blood 128 (16), 2007–2016. doi:10.1182/BLOOD-2016-04-713289
Portier, I., Vanhoorelbeke, K., Verhenne, S., Pareyn, I., Vandeputte, N., Deckmyn, H., et al. (2018). High and Long-term von Willebrand Factor Expression after Sleeping Beauty Transposon-mediated Gene Therapy in a Mouse Model of Severe von Willebrand Disease. J. Thrombosis Haemostasis 16 (3), 592–604. doi:10.1111/JTH.13938
Qin, W., Fang, L., Lin, S. J., Petree, C., Huang, K., Zhang, Y., et al. (2024). ABE-ultramax for high-efficiency biallelic adenine base editing in zebrafish. Nat. Commun. 15 (1), 1–14. doi:10.1038/s41467-024-49943-1
Rackear, M., Elias, Q., Ianniello, Z., Colón-Ríos, D. A., Krysztofiak, A., Rashed, A., et al. (2024). Next-generation cell-penetrating antibodies for tumor targeting and RAD51 inhibition. Oncotarget 15, 699–713. doi:10.18632/ONCOTARGET.28651
Ran, F. A., Cong, L., Yan, W. X., Scott, D. A., Gootenberg, J. S., Kriz, A. J., et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520 (7546), 186–191. doi:10.1038/NATURE14299
Romijn, R. A. P., Bouma, B., Wuyster, W., Gros, P., Kroon, J., Sixma, J. J., et al. (2001). Identification of the Collagen-Binding Site of the von Willebrand Factor A3-Domain. J. Biol. Chem. 276 (13), 9985–9991. doi:10.1074/jbc.M006548200
Sadler, B., Castaman, G., and O’Donnell, J. S. (2022). Von Willebrand Disease and von Willebrand Factor. Haemoph. Official J. World Fed. Hemophilia 28 (Suppl. 4), 11–17. doi:10.1111/HAE.14547
Sago, C. D., Lokugamage, M. P., Paunovska, K., Vanover, D. A., Monaco, C. M., Shah, N. N., et al. (2018). High-throughput in vivo screen of functional MRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. U. S. A. 115 (42), E9944–52. doi:10.1073/pnas.1811276115
Sakata, R. C., Ishiguro, S., Mori, H., Tanaka, M., Tatsuno, K., Ueda, H., et al. (2020). Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol. 38 (7), 865–869. doi:10.1038/s41587-020-0509-0
Sato, Y., Matsui, H., Yamamoto, N., Sato, R., Munakata, T., Kohara, M., et al. (2017). Highly specific delivery of SiRNA to hepatocytes circumvents endothelial cell-mediated lipid nanoparticle-associated toxicity leading to the safe and efficacious decrease in the hepatitis B virus. J. Control. Release 266, 216–225. doi:10.1016/J.JCONREL.2017.09.044
Schillemans, M., Kat, M., Westeneng, J., Gangaev, A., Hofman, M., Nota, B., et al. (2019). Alternative Trafficking of Weibel-Palade Body Proteins in CRISPR/Cas9-engineered von Willebrand Factor–Deficient Blood Outgrowth Endothelial Cells. Res. Pract. Thrombosis Haemostasis 3 (4), 718–732. doi:10.1002/RTH2.12242
Schuessler-Lenz, M., Enzmann, H., and Vamvakas, S. (2019). Regulators’ advice can make a difference: european medicines agency approval of zynteglo for beta thalassemia. Clin. Pharmacol. Ther. 107 (3), 492–494. doi:10.1002/CPT.1639
Seidizadeh, O., Cairo, A., Baronciani, L., Valenti, L., and Peyvandi, F. (2023). Population-Based Prevalence and Mutational Landscape of von Willebrand Disease Using Large-Scale Genetic Databases. Npj Genomic Med. 8 (1), 31–10. doi:10.1038/s41525-023-00375-8
Selvaraj, S., Feist, W. N., Viel, S., Vaidyanathan, S., Dudek, A. M., Gastou, M., et al. (2024). High-efficiency transgene integration by homology-directed repair in human primary cells using DNA-PKcs inhibition. Nat. Biotechnol. 42 (5), 731–744. doi:10.1038/S41587-023-01888-4
Shao, L., Shi, R., Zhao, Y., Liu, H., Lu, A., Ma, J., et al. (2022). Genome-wide profiling of retroviral DNA integration and its effect on clinical pre-infusion CAR T-Cell products. J. Transl. Med. 20 (1), 514–515. doi:10.1186/s12967-022-03729-5
Shi, K., Carpenter, M. A., Banerjee, S., Shaban, N. M., Kurahashi, K., Salamango, D. J., et al. (2016). Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. and Mol. Biol. 24 (2), 131–139. doi:10.1038/nsmb.3344
Shiver, J. W., Tong, M.F., Chen, L., Casimiro, D. R., Davies, M. E., Evans, R. K., et al. (2002). Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nat. 2002 415 (6869), 331–335. doi:10.1038/415331a
Silva, D., Lopes, M., and Cutler, D. F. (2016). Von Willebrand Factor Multimerization and the Polarity of Secretory Pathways in Endothelial Cells. Blood 128 (2), 277–285. doi:10.1182/blood-2015-10-677054
Smaill, F., Jeyanathan, M., Smieja, M., Medina, M.F., Thanthrige-Don, N., Zganiacz, A., et al. (2013). A human type 5 adenovirus-based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci. Transl. Med. 5 (205), 205ra134. doi:10.1126/SCITRANSLMED.3006843
Soucie, M., Miller, C. H., Byams, V. R., Payne, A. B., Abe, K., Sidonio, R. F., et al. (2021). Occurrence Rates of von Willebrand Disease among People Receiving Care in Specialized Treatment Centres in the United States. Haemoph. 27 (3), 445–453. doi:10.1111/HAE.14263
Sreeramoju, P., and Libutti, S. K. (2010). Strategies for targeting tumors and tumor vasculature for cancer therapy. Adv. Genet. 69 (C), 135–152. doi:10.1016/S0065-2660(10)69015-3
Srivastava, A., Rangarajan, S., Kavakli, K., Klamroth, R., Kenet, G., Khoo, L., et al. (2021). Fitusiran, an investigational SiRNA therapeutic targeting antithrombin for the treatment of hemophilia: first results from a phase 3 study to evaluate efficacy and safety in people with hemophilia a or B without inhibitors (ATLAS-A/B). Blood 138 (Suppl. 2), LBA-3. doi:10.1182/BLOOD-2021-155018
Stonebraker, J. S., Iorio, A., Lavin, M., Rezende, S. M., Srivastava, A., Pierce, G. F., et al. (2023). Reported Prevalence of von Willebrand Disease Worldwide in Relation to Income Classification. Haemophilia 29 (4), 975–986. doi:10.1111/HAE.14810
Strecker, J., Ladha, A., Gardner, Z., Schmid-Burgk, J. L., Makarova, K. S., Koonin, E. V., et al. (2019). RNA-guided DNA insertion with CRISPR-associated transposases. Sci. (New York, N.Y.) 365 (6448), 48–53. doi:10.1126/SCIENCE.AAX9181
Sutherland, M. S., Keeney, S., Bolton-maggs, P. H. B., Hay, C. R. M., Will, A., and Cumming, A. M. (2009). The Mutation Spectrum Associated with Type 3 von Willebrand Disease in a Cohort of Patients from the North West of England. Haemophilia 15 (5), 1048–1057. doi:10.1111/J.1365-2516.2009.02059.X
Suwanmanee, T., Hu, G., Tong, G., Bartholomae, C. C., Kutschera, I., Von Kalle, C., et al. (2014). Integration-Deficient lentiviral vectors expressing codon-optimized R338L human FIX restore normal hemostasis in hemophilia B mice. Mol. Ther. 22 (3), 567–574. doi:10.1038/MT.2013.188
Suzuki, K., Tsunekawa, Y., Hernandez-Benitez, R., Wu, J., Zhu, J., Kim, E. J., et al. (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540 (7631), 144–149. doi:10.1038/nature20565
Tariq, H., Batool, S., Asif, S., Ali, M., and Abbasi, B. H. (2022). Virus-Like particles: revolutionary platforms for developing vaccines against emerging infectious diseases. Front. Microbiol. 12, 790121. doi:10.3389/fmicb.2021.790121
Tong, H., Wang, X., Liu, Y., Liu, N., Li, Y., Luo, J., et al. (2023). Programmable A-to-Y base editing by fusing an adenine base editor with an N-Methylpurine DNA glycosylase. Nat. Biotechnol. 41 (8), 1080–1084. doi:10.1038/s41587-022-01595-6
Tong, H., Wang, H., Wang, X., Liu, N., Li, G., Wu, D., et al. (2024). Development of deaminase-free T-to-S base editor and C-to-G base editor by engineered human uracil DNA glycosylase. Nat. Commun. 15 (1), 4897–12. doi:10.1038/s41467-024-49343-5
Tosetto, A., Badiee, Z., Baghaipour, M. R., Baronciani, L., Battle, J., Berntorp, E., et al. (2020). Bleeding Symptoms in Patients Diagnosed as Type 3 von Willebrand Disease: results from 3WINTERS-IPS, an International and Collaborative Cross-sectional Study. J. Thrombosis Haemostasis 18 (9), 2145–2154. doi:10.1111/JTH.14886
Trasanidou, D., Barendse, P., Bouzetos, E., De Haan, L., Bouwmeester, H., Staals, R. H. J., et al. (2023). Efficient genome and base editing in human cells using ThermoCas9. CRISPR J. 6 (3), 278–288. doi:10.1089/CRISPR.2023.0005
UCSC (2025). UCSC genome browser home. Available online at: https://genome.ucsc.edu/ (Accessed April 1, 2025).
van Kwawegen, C. B., and Leebeek, F. W. G. (2024). Prophylaxis in von Willebrand Disease with von Willebrand Factor Concentrate and Nonfactor Therapies. Res. Pract. Thrombosis Haemostasis 8 (8), 102599. doi:10.1016/J.RPTH.2024.102599
Vangenechten, I., Smejkal, P., Zavrelova, J., Zapletal, O., Wild, A., Berneman, Z., et al. (2022). Analysis of von Willebrand Disease in the ‘Heart of Europe. TH Open Companion J. Thrombosis Haemostasis 6 (4), e335–e346. doi:10.1055/S-0042-1757635
Varadi, K., Michelfelder, S., Korff, T., Hecker, M., Trepel, M., Katus, H. A., et al. (2011). Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene Ther. 19 (8), 800–809. doi:10.1038/gt.2011.143
Villiger, L., Joung, J., Koblan, L., Weissman, J., Abudayyeh, O. O., and Gootenberg, J. S. (2024). CRISPR technologies for genome, epigenome and transcriptome editing. Nat. Rev. Mol. Cell Biol. 25 (6), 464–487. doi:10.1038/s41580-023-00697-6
Wang, Y., and Shao, W. (2023). Innate immune response to viral vectors in gene therapy. Viruses 15 (9), 1801. doi:10.3390/V15091801
Wang, D., Zhang, Y. Z., Zhang, J., and Zhao, J. J. (2024). Advances in base editing: a focus on base transversions. Mutat. Res. - Rev. Mutat. Res. 794, 108515. doi:10.1016/J.MRREV.2024.108515
Wang, Y., Qi, T., Liu, J., Yang, Y., Wang, Z., Wang, Y., et al. (2023). A highly specific CRISPR-Cas12j nuclease enables allele-specific genome editing. Sci. Adv. 9 (6), eabo6405. doi:10.1126/sciadv.abo6405
Wang, J., Ding, Y., Chong, K., Cui, M., Cao, Z., Tang, C., et al. (2024). Recent advances in lipid nanoparticles and their safety concerns for MRNA delivery. Vaccines 12 (10), 1148. doi:10.3390/VACCINES12101148
Wanisch, K., and Yáñez-Muñoz, R. J. (2009). Integration-Deficient lentiviral vectors: a slow coming of age. Mol. Ther. J. Am. Soc. Gene Ther. 17 (8), 1316–1332. doi:10.1038/MT.2009.122
Weyand, A. C., and Veronica, H. F. (2021). Von Willebrand Disease: current Status of Diagnosis and Management. Hematology/Oncology Clin. N. Am. 35 (6), 1085–1101. doi:10.1016/J.HOC.2021.07.004
Witte, I. P., Lampe, G. D., Eitzinger, S., Miller, S. M., Berríos, K. N., McElroy, A. N., et al. (2025). Programmable gene insertion in human cells with a laboratory-evolved CRISPR-associated transposase. Science 388 (6748), eadt5199. doi:10.1126/SCIENCE.ADT5199
Wold, W. S. M., and Toth, K. (2013). Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 13 (6), 421–433. doi:10.2174/1566523213666131125095046
Wu, W. Y., Mohanraju, P., Liao, C., Adiego-Pérez, B., Creutzburg, S. C. A., Makarova, K. S., et al. (2022). The miniature CRISPR-Cas12m effector binds DNA to block transcription. Mol. Cell 82 (23), 4487–4502.e7. doi:10.1016/J.MOLCEL.2022.11.003
Wu, T., Liu, C., Zou, S., Lyu, R., Yang, B., Yan, H., et al. (2023). An engineered hypercompact CRISPR-Cas12f system with boosted gene-editing activity. Nat. Chem. Biol. 19 (11), 1384–1393. doi:10.1038/s41589-023-01380-9
Wu, Y., Wan, X., Zhao, D., Chen, X., Wang, Y., Tang, X., et al. (2023). AAV-mediated base-editing therapy ameliorates the disease phenotypes in a mouse model of retinitis pigmentosa. Nat. Commun. 14 (1), 4923–12. doi:10.1038/s41467-023-40655-6
Xu, F., Zheng, C., Xu, W., Zhang, S., Liu, S., Chen, X., et al. (2024). Breaking genetic shackles: the advance of base editing in genetic disorder treatment. Front. Pharmacol. 15, 1364135. doi:10.3389/fphar.2024.1364135
Xu, W., Zhang, S., Qin, H., and Yao, K. (2024). From bench to bedside: cutting-edge applications of base editing and prime editing in precision medicine. J. Transl. Med. 22 (1), 1133–1143. doi:10.1186/S12967-024-05957-3
Yamamoto, K., De Waard, V., Fearns, C., and Loskutoff, D. J. (1998). Tissue Distribution and Regulation of Murine von Willebrand Factor Gene Expression in vivo. Blood 92 (8), 2791–2801. doi:10.1182/BLOOD.V92.8.2791
Yan, J., Oyler-Castrillo, P., Ravisankar, P., Ward, C. C., Levesque, S., Jing, Y., et al. (2024). Improving prime editing with an endogenous small RNA-binding protein. Nature 628 (8008), 639–647. doi:10.1038/s41586-024-07259-6
Yarnall, M. T. N., Ioannidi, E. I., Schmitt-Ulms, C., Krajeski, R. N., Lim, J., Villiger, L., et al. (2022). Drag-and-Drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat. Biotechnol. 41 (4), 500–512. doi:10.1038/s41587-022-01527-4
Yu, Q., Tai, Y. Y., Tang, Y., Zhao, J., Negi, V., Culley, M. K., et al. (2019). BOLA (bolA family member 3) deficiency controls endothelial metabolism and Glycine homeostasis in pulmonary hypertension. Circulation 139 (19), 2238–2255. doi:10.1161/CIRCULATIONAHA.118.035889
Yu, S., Yang, H., Li, T., Pan, H., Ren, S., Luo, G., et al. (2021). Efficient intracellular delivery of proteins by a multifunctional chimaeric peptide in vitro and in vivo. Nat. Commun. 12 (1), 5131–13. doi:10.1038/s41467-021-25448-z
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-Guided endonuclease of a class 2 CRISPR-cas system. Cell 163 (3), 759–771. doi:10.1016/J.CELL.2015.09.038
Zhang, X., Halvorsen, K., Zhang, C. Z., Wong, W. P., and Springer, T. A. (2009). Mechanoenzymatic Cleavage of the Ultralarge Vascular Protein von Willebrand Factor. Science 324 (5932), 1330–1334. doi:10.1126/science.1170905
Zhang, X., Jin, H., Huang, X., Chaurasiya, B., Dong, D., Shanley, T. P., et al. (2022). Robust genome editing in adult vascular endothelium by nanoparticle delivery of CRISPR-Cas9 plasmid DNA. Cell Rep. 38 (1), 110196. doi:10.1016/J.CELREP.2021.110196
Zwi-Dantsis, L., Mohamed, S., Massaro, G., and Moeendarbary, E. (2025). Adeno-associated virus vectors: principles, practices, and prospects in gene therapy. Viruses 17 (2), 239. doi:10.3390/V17020239
Glossary
(ID)LV (integrase-deficient) lentiviral vector
AAV Adeno-associated viral vector
ABE/CBE Adenine base editor/cytosine base editor
Ad Adenoviral vector
Cas CRISPR-associated
CAST CRISPR-associated transposase
CB Cord blood
CDS Coding DNA sequence
CIS Common integration site
CPP Cell-penetrating protein
CRISPR Clustered regularly interspaced short palindromic repeats
DDAVP Desmopressin
DSDB Double strand DNA break
EC Endothelial cell
ECFC Endothelial colony forming cell
ER Endoplasmic reticulum
EV Extracellular vesicle
FiCAT Find and cut and transfer
HA Homology arm
HDR Homology-directed repair
hiPSC Human induced pluripotent stem cell
HITI Homology-independent targeted integration
HMW High molecular weight
INDELs Insertions and/or deletions
LMW Low molecular weight
LNP Lipid nanoparticle
NHEJ Non-homologous end joining
PAM Protospacer adjacent motif
PASTE Programmable addition via site-specific targeting elements
PE Prime editor
RV Retroviral vector
TALEN Transcription activator-like effector nucleases
TransCRISTI Transposase-CRISPR mediated targeted integration
UL Ultra large
VLPs Viral-like particle
VUS Variants of uncertain significance
VWD Von Willebrand disease
VWF Von Willebrand factor
VWFpp Von Willebrand factor propeptide
WPB Weibel Palade Bodie
ZFN Zinc-finger nuclease
Keywords: gene therapy, VWD, endothelial cells, delivery, CRISPR-Cas, in vivo delivery
Citation: Barraclough A, Bär I, van Duijl T, Fijnvandraat K, Eikenboom JCJ, Leebeek FWG, Bierings R, Voorberg J and Trasanidou D (2025) Rewriting the script: gene therapy and genome editing for von Willebrand Disease. Front. Genome Ed. 7:1620438. doi: 10.3389/fgeed.2025.1620438
Received: 29 June 2025; Accepted: 25 July 2025;
Published: 22 September 2025; Corrected: 07 November 2025.
Edited by:
Ruchita Selot, Narayana Nethralaya Eye Hospital, IndiaReviewed by:
Antonio Casini, Alia Therapeutics, ItalyElena Barbon, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), Italy
Asim Mushtaq, Zhejiang University, China
Copyright © 2025 Barraclough, Bär, van Duijl, Fijnvandraat, Eikenboom, Leebeek, Bierings, Voorberg and Trasanidou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Voorberg, ai52b29yYmVyZ0BzYW5xdWluLm5s
†These authors have contributed equally to this work