 Can Jones
Can Jones Stanislav Ivanov
Stanislav Ivanov Pablo Ferraro1,2
Pablo Ferraro1,2- 1Memorial Healthcare System, Pembroke Pines, FL, United States
- 2Memorial Cancer Institute, Pembroke Pines, FL, United States
- 3Moffitt Malignant Hematology & Cellular Therapy at Memorial Healthcare System, Pembroke Pines, FL, United States
VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) is a novel disorder first described in 2020. Patients are diagnosed by identifying a somatic mutation of the ubiquitin-like modifier-activating enzyme 1 (UBA1) gene. They usually have systemic inflammation and present with a combination of hematologic and rheumatologic abnormalities such as myelodysplastic syndrome and polychondritis. VEXAS syndrome patients are at increased risk of developing hematologic malignancies. We present a case of a 60-year-old male who developed transfusion-dependent macrocytic anemia, was found to have UBA1 mutation in the bone marrow, and was diagnosed with VEXAS syndrome. The patient responded well to steroid treatment and did not require more blood transfusion. The two main goals of treating VEXAS syndrome are eradicating the UBA1 mutated hematopoietic cells and inhibiting the inflammatory process. Early stem cell transplant evaluation is necessary as VEXAS-related complications may limit the efficacy of transplantation. Further research is required to improve the prognosis and quality of life of VEXAS syndrome patients.
Introduction
VEXAS syndrome (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) is a novel condition first described in 2020 (1). Patients with this condition are defined by the presence of a somatic mutation of the ubiquitin-like modifier-activating enzyme 1 (UBA1) gene. The UBA1 gene is located on the X chromosome and encodes the master enzyme of cellular ubiquitylation, a process that modifies proteins and facilitates protein degradation via the proteasome or the autophagy-lysosome systems (2). Loss of function mutation in the UBA1 gene leads to dysfunctional ubiquitination, overactivation of inflammatory processes, late onset of autoimmune/inflammatory syndromes, and hematologic disorders (3).
The clinical presentation involves a combination of hematologic and rheumatologic abnormalities. The most observed conditions include myelodysplastic syndrome (MDS) and relapsing polychondritis (4). VEXAS syndrome patients are at high risk of developing hematologic malignancies, leading to high morbidity and mortality (5).
Here we present a case of a 60-year-old male with a medical history of systemic lupus erythematosus (SLE) who developed transfusion-dependent macrocytic anemia and was later diagnosed with VEXAS syndrome.
Case presentation
A 60-year-old white male patient with a past medical history of systemic lupus erythematosus on hydroxychloroquine was initially admitted in November of 2023 for urinary retention in the context of a urinary tract infection. Admission laboratories were notable for macrocytic anemia with hemoglobin at 5.8 g/dL and mean corpuscular volume (MCV) at 103.3 fL. He received 2 units of packed red blood cells.
A review of his history revealed that the patient was previously diagnosed with macrocytic anemia in 2021 for which he had required frequent blood transfusions due to symptomatic anemia. At the time the patient had undergone extensive evaluation including nutritional deficits, hemolysis as well as occult signs of bleeding. Bone marrow biopsy and aspirate in March of 2021 was unremarkable and his esophagogastroduodenoscopy and colonoscopy in September of 2021 were negative for bleeding. The patient continued to receive supportive treatments and management of his SLE. The patient had a subsequent bone marrow biopsy in April of 2023 secondary to persistent anemia as well as mild neutropenia. The biopsy result noted mildly hypercellular marrow with maturing trilineage hematopoiesis, myeloid predominance, and approximately 2% blasts. Some neutrophilic precursors demonstrated cytoplasmic vacuolization.
During the current hospitalization, the patient was found to have diffuse non-tender erythematous rash involving his abdomen and limbs (Figures 1A, B). Evaluation at the time noted normal vitamin B12/folate levels, negative hemolysis panel, normal thyroid function, viral panel, heavy metal panel, and infectious disease panel. Iron studies were consistent with anemia of chronic disease.
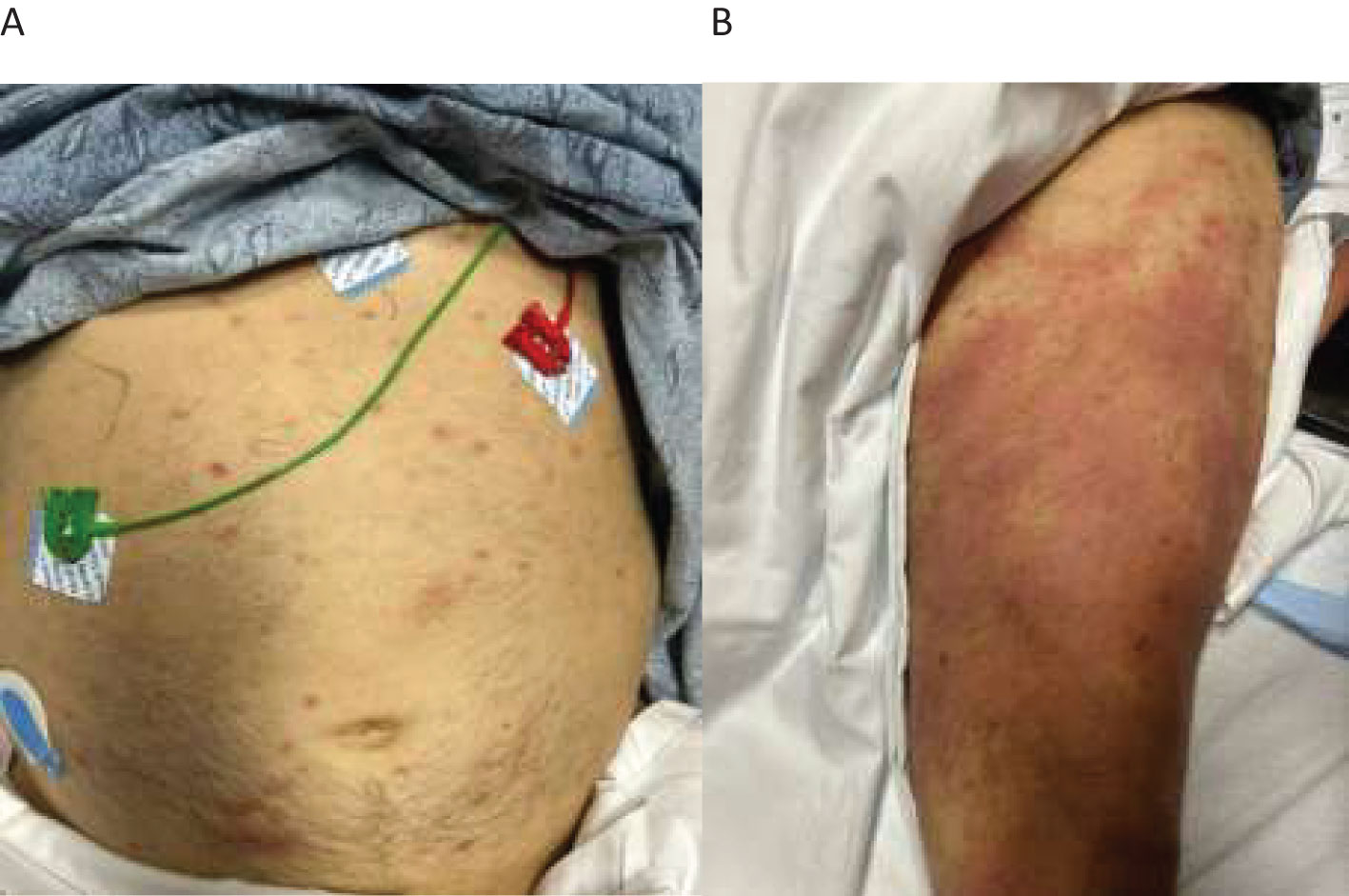
Figure 1. Diffuse non-tender erythematous rash seen on the patient’s abdomen and limbs. (A) scattered erythematous rashes on the abdomen. (B) diffuse large erythematous rashes on the thigh.
The clinical course of the patient was complicated by the incidental discovery of a small pulmonary embolism at the right lower lobe with minimal clot burden. He was started on an anticoagulant. Given late-onset disease, positive bone marrow vacuolization in a male patient, and his history of SLE, VEXAS syndrome was highly suspected. A third bone marrow biopsy was performed, and the patient was started on high-dose steroids to alleviate the inflammation. The results of the marrow biopsy demonstrated normocellular to slightly hypercellular for age marrow with trilineage hematopoiesis with complete maturation. Cytoplasmic vacuolization of some of the myeloid and erythroid precursors was observed (Figures 2A, B). The Next-Generation Sequencing test was positive for UBA1 mutation.
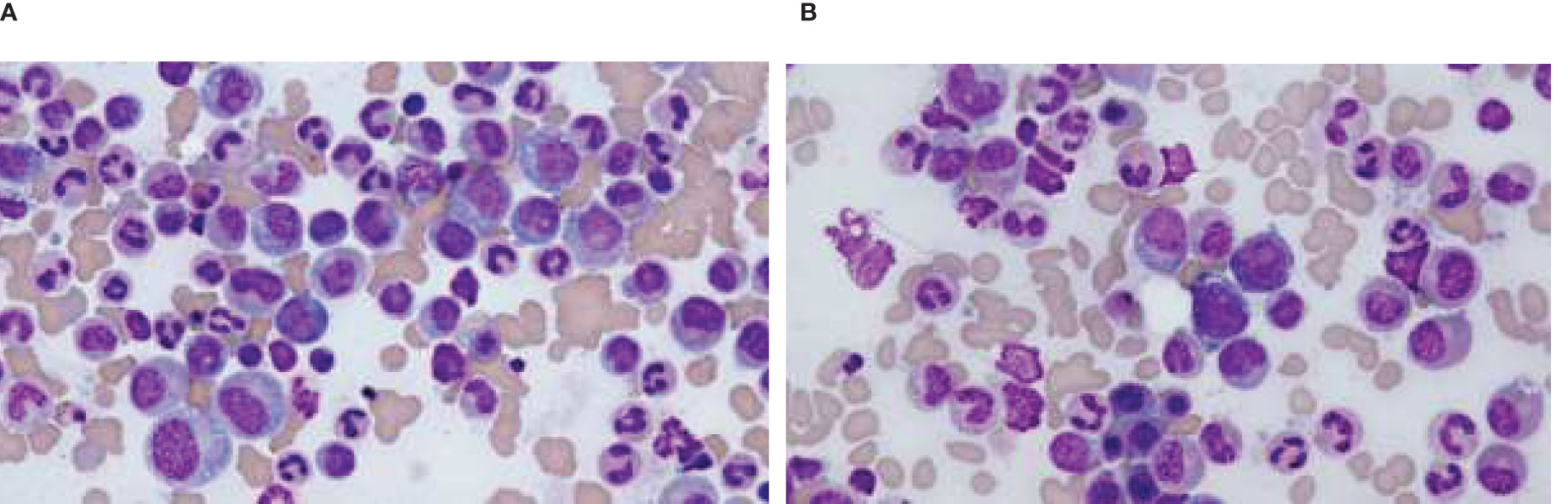
Figure 2. Cytoplasmic vacuoles seen in the myeloid cells in bone marrow smear. (A) Vacuoles seen in the myeloid cells. (B) Vacuoles seen in the myeloid cells.
The patient was formally diagnosed with VEXAS syndrome. His symptoms improved significantly with steroids, and he did not require more blood transfusions. The patient was discharged to home with oral prednisone and followed up with the hematology clinic. The patient's disease course was summarized in Table 1 as below.

Table 1. Detailed disease course of this VEXAS syndrome patient.
Patient perspective
The patient reported feeling better and his energy level improved with the transfusion and steroids treatment. He continues to follow up with hematology and rheumatology in the clinic. He is compliant with lupus treatment. He has not required blood product transfusion since discharge from the hospital.
Timeline of disease course
The patient's disease couse was described in the below table.
Literature review and discussion
Epidemiology
VEXAS syndrome is seen in biological male patients and was first reported in December 2020 with 25 cases by Beck et al. (1). 25 male patients were identified with somatic mutations of methionine-41 in UBA1 by analyzing peripheral-blood exome sequence data. Clinical presentations included inflammatory syndromes and hematologic disorders such as MDS, or multiple myeloma (6).
A further retrospective study evaluating UBA1 variants in exome from an unselected population of 163,096 patients was reported in 2023. 11 patients harbored somatic mutation of UBA1 and most of them were found to have macrocytic anemia, thrombocytopenia. Two of the 11 patients were females without evidence of aneuploidy. All patients were above 50 years old (7). According to this large-scale study, the estimated prevalence is 1 in 14,000 from the entire cohort; 1 in 4,000 for males, 1 in 26,000 for females, and 1 in 8,000 for the combined gender group (8).
Definition
Vacuoles
Vacuoles are physiological intracellular organelles in plants, fungi, and bacteria. The presence of cytoplasmic vacuoles in human cells indicates a pathologic process (5). Almost all VEXAS syndrome patients have vacuolation in the promyelocytes, myelocytes, and erythroid precursors in the bone marrow. Vacuoles can also be found in eosinophils, monocytes, megakaryocytes, and plasma cells, but not in mature lymphocytes likely due to the location of the mutated UBA1 gene (4). Lymphoid cells are found to have increased apoptosis in VEXAS syndrome patients (9). Dense vacuolation in more than 10% neutrophilic precursors is both sensitive and specific to diagnose VEXAS syndrome (10).
Nonetheless, the evaluation of cytoplasmic vacuolation should be taken into careful consideration as it can also be observed in other conditions such as alcohol use, copper deficiency, zinc toxicity, and MDS. Cherniawsky et al. reviewed bone marrow biopsies from 210 patients with identified cytoplasmic vacuolation in myeloid and erythroid precursors but only 6 patients tested positive for UBA1 mutations (2.9%) (11). The most common causes of vacuolation among the group were MDS and acute myeloblastic leukemia (AML). Thorough evaluation and exclusion of these conditions are necessary to pursue a diagnosis of VEXAS syndrome. The mechanism of cell vacuolation in VEXAS syndrome is still under further investigation.
E1 Enzyme
The E1 enzyme coded by the UBA1 gene is detected in myeloid progenitor cells and circulating myeloid cells, but not in mature lymphoid cells. The E1 enzyme is responsible for the initiation of the protein ubiquitination (5). Ubiquitination is a post-translational modification essential to protein turnover and plays an important role in the assembly of multiprotein complexes, intracellular signaling, inflammatory signaling, and DNA repair (5). This process also facilitates protein destruction via the proteasome complex or lysosome and is key in maintaining cellular homeostasis as well as facilitating autophagy when needed (8).
Three enzymes (E1, E2, and E3) are required for the binding of ubiquitin to target proteins. The E1 enzyme initializes the process of ubiquitination by forming a thioester bond with the terminal region of ubiquitin, followed by the transfer of ubiquitin to a sulfide group of the E2 conjugating enzyme. The E3 ligase enzyme then binds the ubiquitin to the target protein to initiate the proteasomal degradation (5). Mutations in UBA1 lead to malfunctioning E1 enzymes resulting in reduced ubiquitylation, decreased protein degradation, and increased inflammation (12).
X-linked
The UBA1 gene is located on the X chromosome (Xp11.3). As such the prevalence of VEXAS syndrome is largely predominant in male patients, particularly those in the latter decades of life. Studies have demonstrated female patients are protected from the manifestation of VEXAS largely due to the presence of additional alleles (5). Rare presentations in female patients can be seen however in patients with acquired X-chromosome monosomy or structural deletion. Presentation is usually a milder form of the disease (13).
Autoinflammatory
A recent study was conducted to address the relationship between UBA1 mutation and inflammation in VEXAS patients using transcriptome sequencing of single bone marrow mononuclear cells, hematopoietic stem cells, and progenitor cells (9). In UBA1 mutated myeloid cells, upregulated inflammatory pathways, active precursor cell cycling, and increased granulocytic differentiation were observed.
The decreased ubiquitination/proteasome pathway activity leads to the accumulation of unfolded proteins. The increased protein response contributes to the elevation of inflammatory cytokines such as interferon-gamma (IFN- γ), and Tumor necrosis factor alpha (TNF)- α, and the activation of multiple inflammatory pathways (14). Meanwhile, neutrophils may also contribute to exacerbating the inflammatory process by promoting the formation of neutrophil extracellular traps. Interestingly, there is an increased apoptosis of lymphoid cells in VEXAS patients (8). Systemic inflammation affects the skin, lungs, vessels, and cartilage. Patients typically present with fever, polychondritis, skin rashes, pulmonary involvement, and vasculitis (4).
Somatic
Somatic mutations accumulate with age. It can be seen in healthy aging people and patients with various diseases. The UBA1 mutation is a somatic mutation that occurs later in life (1).
UBA1 has two protein isoforms: a nuclear form initiated at p. Met1 and a cytoplasmic form initiated at p.Met41. Loss of the cytoplastic isoform is found in most VEXAS patients (2), which causes the disruption of cytoplasmic E1 enzymes and results in systemic inflammation. The timing between the mutation onset and the development of the disease is still under investigation.
Pathogenesis
Almost all VEXAS syndrome patients were found to have missense mutations in codon 41 of UBA1. It results in reduced expression of the cytoplasmic UBA1B isoform along with the formation of the catalytically impaired UBA1C isoform (1). Ubiquitination is an essential post-translational modification, contributing to multiprotein assembly, cell signaling conduction, and inflammatory signaling. Since the ubiquitin-activating enzyme (E1 enzyme) is encoded by the UBA1 gene, this mutation process leads to decreased ubiquitination activity, decreased degradation of unfolded protein, and increased levels of inflammatory proteins. The mutations are restricted to myeloid-lineage cells (15).
Robust activities of neutrophils and monocytes are found in VEXAS syndrome patients. They express proinflammatory cytokines TNF, IL-6, IL-8, and interferon-r, contributing to myeloid inflammation (16). UBA1- mutant neutrophils have a preserved phagocytic capacity, and an enhanced spontaneous neutrophil extracellular trap formation, leading to proinflammatory neutrophil activation. Unfolded protein response with an amplified inflammatory process is also reported (4).
Diagnosis
The key finding in VEXAS syndrome is cytoplasmic vacuoles in myeloid and erythroid precursor cells (17). These can also be seen in alcoholism, MDS, zinc toxicity, and copper deficiency. However, if they are identified in patients with inflammatory conditions or presumed MDS, further workup is warranted to rule out VEXAS syndrome. Other typical marrow findings include hypercellular marrow with granulocytic hyperplasia, minimal dyspoiesis, a normal karyotype, and no increase in blasts in the absence of associated MDS (11).
Clinical presentation
Hematologic manifestations
MDS
MDS is observed in more than half of the VEXAS patients. There are two large retrospective case series that report MDS diagnosis in 31-50% of cases in VEXAS patients (8). This is much higher than the MDS incidence in paroxysmal nocturnal hemoglobinuria over 10 years, which is approximately 2-6% (18). Compared to VEXAS patients, VEXAS-MDS patients have a higher incidence of non-infectious recurrent fever, respiratory symptoms, and gastrointestinal involvement. They also have lower platelet count and higher medullar blasts (4). However, the difference in overall survival rate between VEXAS patients without MDS and VEXAS-MDS patients was insignificant (19).
MDS is caused by clonal alterations of hematopoietic stem cells, causing ineffective hematopoiesis and progressive cytopenias, bone marrow dysplasia, and eventual leukemic transformation (20). The mutational profile of VEXAS-MDS appears to be less complex, classic MDS-associated mutations are not frequently detected in VEXAS patients (2).
Previous studies have indicated that MDS is associated with a systemic inflammatory process (21). Inflammatory status in VEXAS syndrome plays a critical role in the development of MDS because of UBA1 gene mutation. However, UBA1 has not been included in the MDS scoring system (8).
Cytopenia
Macrocytic anemia can be observed in VEXAS syndrome patients with normal B12, folate, and copper levels. Two large case series reported that approximately 32% of VEXAS patients developed transfusion-dependent anemia and had increased the risk of death by 4.5-fold (15). Thrombocytopenia, lymphopenia, and monocytopenia were also reported. Monoclonal gammopathy of unknown significance (MGUS) and multiple myeloma can also be seen in VEXAS syndrome patients (6).
Thrombosis
Thromboembolic events usually occur in the early stage of VEXAS syndrome. The venous thrombosis incidence is higher than arterial thrombosis (8). The overactive immune cells, platelets, and endothelial cells contribute to the dysregulation of hemostasis. Chronic inflammation also plays a role in the development of thrombosis by inappropriate formation of fibrin, reduced synthesis, and increased consumption of the natural anticoagulant (5).
In one large-scale study with 119 VEXAS patients, 49% of patients developed thrombotic events, majorly venous thromboembolism, and approximately two-thirds of them were unprovoked. At a median follow-up time of 4.8 years, the overall survival (OS) of the entire cohort was 88% and there was no significant overall survival difference between VEXAS patients with and without thrombosis (22). Thromboembolic prophylaxis should be administered to VEXAS patients unless there are contraindications.
Other systems
Auricular and nasal chondritis is commonly observed in VEXAS syndrome patients (3). Lung involvement is often seen, associated with pleural effusion. The common gastrointestinal tract symptoms include abdominal pain, diarrhea, and gastrointestinal bleeding. Renal involvement can present with proteinuria, hematuria, and renal insufficiency. Epididymitis is frequently reported, along with orchitis and prostatitis. Lymphadenopathy and splenomegaly are observed (19).
Treatment
The coexistence of hematologic and rheumatologic dysfunction creates a unique challenge in the management of VEXAS syndrome and usually requires a multidisciplinary approach. Two main therapy goals include 1) the targeting and eradication of the UBA1-mutated hematopoietic population, and 2) the inhibition of the inflammatory process. Currently, there are no standard treatment protocols for VEXAS syndrome. The treatment of VEXAS patients is primarily guided by clinician experience and available clinical series due to the lack of data from randomized controlled trials.
Hypomethylating agents
Hypomethylating agents such as azacytidine have shown promising results in managing VEXAS patients. A French cohort study of VEXAS syndrome patients with concomitant MDS showed clinical response after 4 cycles of azacytidine (AZA) treatment in 5/11 (46%) patients (23).
A phase II prospective trial study in 2022 reported that 9/12 (75%) VEXAS patients achieved complete responses or partial responses after 6 cycles of AZA treatment, with improvement in inflammatory manifestations and a decrease in the requirement of steroids (23). However, cytopenia or myelodysplastic features were not improved significantly. Hypomethylating agents may be preferentially considered in patients with concomitant MDS.
Glucocorticoids
The available studies show that high doses of glucocorticoids are effective in ameliorating systemic inflammation, however, attempts to taper steroids have resulted in poor symptom control and even recurrence (4). Prolonged use of high-dose steroids is associated with serious side effects and dependence. Further research needs to be conducted on steroids-sparing agents for the long-term treatment of VEXAS treatment.
Janus kinase inhibitors
JAKi has served a role in treating VEXAX syndrome patients. By suppressing the inflammatory pathway, JAKi decreases the production of inflammatory cytokines such as IL-6, which improves systemic inflammation and related symptoms.
JAKi are effective in relieving inflammatory symptoms, especially skin manifestations. Significant regression of skin lesions has been observed with topical ruxolitinib without increasing the dose of steroids, suggesting the potential effectiveness of JAKi. Heiblig et al. reported complete clinical and biological responses in 7/11 (64%) and 6/11 (54%) patients treated with ruxolitinib for 1 month (21). Subsequently, 87% of patients achieved complete clinical remission after 6 months of treatment with ruxolitinib, with a significant increase in hemoglobin and platelet levels, decreased use of steroids, and even discontinuation of steroids.
These patients were found to have persistent clonal amplification of UBA1 with the treatment, indicating that ruxolitinib aims to alleviate inflammation instead of eradicating UBA1 mutations.
However, side effects of JAKi are common, including severe opportunistic infections, and thromboembolism, which can also occur in the already vulnerable patients with VEXAS syndrome, making the safety of JAK inhibitors challenging to assess (24). Prospective studies are still needed to confirm its efficacy further.
Interleukin 6 inhibitors
Tocilizumab is a monoclonal antibody against the inflammatory cytokine IL-6 receptor. Within short-term follow-up, tocilizumab showed alleviation of inflammatory symptoms and reduced steroid requirement (5), but the long-term efficacy and side effects need to be investigated.
Tocilizumab has been associated with neutropenia, viral infection reactivation (herpes zoster virus), and severe gastrointestinal side effects in VEXAS syndrome patients. Intestinal perforation is one of the severe comorbidities, especially perforation of the jejunum or ileum at the site of diverticulitis (25).
Interleukin 1 inhibitors
The use of IL-1 receptor inhibitors like anakinra has been noted to lead to stabilization of the inflammatory symptoms in some patients for 1-2 years, but it caused severe skin reactions at the site of injection (8).
Hematopoietic stem cell transplantation
For cases with multisystemic involvement, including severe hematological abnormalities, hematopoietic stem cell transplantation (HSCT) may be considered as a curative option. A few studies investigated the outcome of HSCT in VEXAS syndrome patients. The results were mixed.
In 2021, the Mayo Case Series first reported that a VEXAS syndrome patient with coexisting plasma cell myeloma received bortezomib, lenalidomide, and dexamethasone induction therapy, followed by autologous stem cell transplant (26). The patient demonstrated the resolution of arthritis and the discontinuation of steroids and IL-6 inhibitors. A French case series of six VEXAS syndrome patients reported that all patients were in complete remission after HSCT.
In a UK study in 2022, four patients underwent HSCT, and only one patient achieved remission, with one patient developing a significant infection and extensive Graft-Versus-Host Disease and two patients dying of infection (27).
Abhishek et al. published a prospective series in which five patients underwent reduced-intensity conditioning allogeneic stem cell transplant with a median follow-up of 9.6 months (26). Four patients had normalization of bone marrow morphology with eradication of vacuoles and did not require more steroid treatment. Inflammatory markers normalized in at least two patients. UBA1 mutation disappeared in 2 patients. None of those patients experienced the recurrence of inflammatory symptoms or worsened cytopenia.
HSCT evaluation should be conducted in the early stage, as VEXAS-related complications may limit the efficacy of transplantation. It is recommended to control inflammation before HSCT to reduce post-transplant complications (8). Currently, HSCT studies are under conducted and will provide valuable guidance in treating VEXAS syndrome in the future.
Conclusion
Although VEXAS syndrome was identified less than 2 years ago, it has generated a profound medical interest. It is associated with significantly high morbidity and mortality, as well as treatment-related adverse events. VEXAS syndrome patients will benefit from early diagnosis based on multidisciplinary management and regular clinical and molecular follow-up. VEXAS syndrome needs to become a new classification of MDS and an addition to the MDS scoring system. It may be effective to target and eradicate UBA1 mutation gene editing, or HSCT to replace mutated progenitor cells; to block the inflammatory cascade response, or even to consider restoring the function of the UBA1 gene. Further research and clinical studies are necessary to improve the prognosis and quality of life for VEXAS syndrome patients.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
CJ: Writing – original draft, Writing – review & editing. SI: Writing – review & editing. PF: Writing – review & editing. SY: Writing – review & editing. HF: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
VEXAS, Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic; UBA1, ubiquitin-like modifier-activating enzyme 1; MDS, myelodysplastic syndrome; SLE, systemic lupus erythematosus; MCV, mean corpuscular volume; AML, acute myeloblastic leukemia; IFN- γ, interferon-gamma; TNF- α, Tumor necrosis factor-alpha; IL, interleukin; MGUS, Monoclonal gammopathy of unknown significance; OS, overall survival; AZA, azacytidine; JAKi, Janus kinase inhibitors; HSCT, hematopoietic stem cell transplantation.
References
1. Beck DB, Ferrada M, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe Adult-Onset autoinflammatory disease. New Engl J Medicine/the New Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
2. Patel BA, Ferrada M, Grayson PC, Beck DB. VEXAS syndrome: An inflammatory and hematologic disease. Semin Hematol. (2021) 58:201–3. doi: 10.1053/j.seminhematol.2021.10.005
4. Vitale A, Caggiano V, Bimonte A, Caroni F, Tosi GM, Fabbiani A, et al. VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases. Internal Emergency Med. (2023) 18:711–22. doi: 10.1007/s11739-023-03193-z
5. Zhang Y, Dong X, Wang H. VEXAS syndrome—Review. Global Med Genet. (2023) 10:133–43. doi: 10.1055/s-0043-1770958
6. Obiorah IE, Patel B, Groarke EM, Wang W, Trick M, Ombrello AK, et al. Benign and Malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. (2021) 5:3203–15. doi: 10.1182/bloodadvances.2021004976
7. Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA. (2023) 329:318. doi: 10.1001/jama.2022.24836
8. Templé M, Kosmider O. VEXAS Syndrome: a novelty in MDS landscape. Diagnostics. (2022) 12:1590. doi: 10.3390/diagnostics12071590
9. Wu Z, Gao S, Gao Q, Patel BA, Groarke EM, Feng X, et al. Early activation of inflammatory pathways in UBA1-mutated hematopoietic stem and progenitor cells in VEXAS. Cell Rep Med. (2023) 4:101160. doi: 10.1016/j.xcrm.2023.101160
10. Diral E, Campochiaro C, Tomelleri A, Bergonzi G, Pizzano U, Ponzoni M, et al. Case report: Cytopenias in VEXAS syndrome - a WHO 2022 based approach in a single-center cohort. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1354130
11. Cherniawsky H, Friedmann J, Nicolson H, Dehghan N, Stubbins RJ, Foltz L, et al. VEXAS syndrome: A review of bone marrow aspirate and biopsies reporting myeloid and erythroid precursor vacuolation. Eur J Haematol. (2023) 110:633–8. doi: 10.1111/ejh.v110.6
12. Watanabe R, Kiji M, Hashimoto M. Vasculitis associated with VEXAS syndrome: A literature review. Front Med. (2022) 9. doi: 10.3389/fmed.2022.983939
13. Stubbins RJ, McGinnis E, Johal B, Chen LYC, Wilson L, Cardona DO, et al. VEXAS syndrome in a female patient with constitutional 45,X (Turner syndrome). Haematologica. (2021) 107:1011–3. doi: 10.3324/haematol.2021.280238
14. Onuora S. Somatic mutations cause VEXAS syndrome. Nat Rev Rheumatol. (2020) 17:1. doi: 10.1038/s41584-020-00559-x
15. Ferrada M, Savic S, Cardona DO, Collins JC, Alessi H, Gutierrez-Rodrigues F, et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood. (2022) 140:1496–506. doi: 10.1182/blood.2022016985
16. Al-Hakim A, Savic S. An update on VEXAS syndrome. Expert Rev Clin Immunol. (2022) 19:203–15. doi: 10.1080/1744666X.2023.2157262
17. Loeza-Uribe MP, Hinojosa-Azaola A, Sánchez-Hernández BE, Crispín JC, Apodaca-Chávez E, Ferrada M, et al. VEXAS syndrome: Clinical manifestations, diagnosis, and treatment. Reumatología Clínica. (2023) 20(1):47–56. doi: 10.1016/j.reumae.2023.12.004
18. Sun L, Babushok DV. Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood. (2020) 136:36–49. doi: 10.1182/blood.2019000940
19. Georgin-Lavialle S, Terrier B, Guédon A, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients*. Br J Dermatology/British J Dermatol Supplement. (2021) 186:564–74. doi: 10.1111/bjd.20805
20. Zhan D, Park CY. Stem cells in the myelodysplastic syndromes. Front Aging. (2021) 2:719010. doi: 10.3389/fragi.2021.719010
21. Heiblig M, Ferrada M, Koster MJ, Barba T, Gerfaud-Valentin M, Mékinian A, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multicenter study. Blood. (2022) 140:927–31. doi: 10.1182/blood.2022016642
22. Kusne Y, Ghorbanzadeh A, Florea AD, Shalhoub R, Andrade PEA, Nghiem K, et al. Venous and arterial thrombosis in patients with VEXAS syndrome. Blood. (2024) 143(21:2190–220. doi: 10.1182/blood.2023022329
23. Comont T, Heiblig M, Rivière É, Terriou L, Rossignol J, Bouscary D, et al. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. (2021) 196:969–74. doi: 10.1111/bjh.17893
24. Guilpain P. JAK inhibitors in autoinflammatory syndromes? The long road from drug development to daily clinical use. Rheumatology. (2022) 62:1368–9. doi: 10.1093/rheumatology/keac592
25. Van Der Made CI, Potjewijd J, Hoogstins A, Willems HPJ, Kwakernaak AJ, De Sévaux R, et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: A Dutch case series of patients with VEXAS. J Allergy Clin Immunology/Journal Allergy Clin Immunology/the J Allergy Clin Immunol. (2022) 149:432–439.e4. doi: 10.1016/j.jaci.2021.05.014
26. Mangaonkar A, Langer KJ, Lasho TL, Finke C, Litzow MR, Hogan WJ, et al. Reduced intensity conditioning allogeneic hematopoietic stem cell transplantation in VEXAS syndrome: Data from a prospective series of patients. Am J Hematol. (2022) 98:E28–31. doi: 10.1002/ajh.26786
Keywords: VEXAS, myelodysplastic syndrome, UBA1 mutation, inflammation, bone marrow transplant
Citation: Jones C, Ivanov S, Ferraro P, Younes S and Fernandez H (2024) Case report: VEXAS syndrome and literature review. Front. Hematol. 3:1480436. doi: 10.3389/frhem.2024.1480436
Received: 14 August 2024; Accepted: 09 October 2024;
Published: 13 November 2024.
Edited by:
Valentina Giudice, University of Salerno, ItalyReviewed by:
Donglei Zhang, Zhongnan Hospital of Wuhan University, ChinaIoanna Triviai, Max Planck Institute for Molecular Genetics, Germany
Copyright © 2024 Jones, Ivanov, Ferraro, Younes and Fernandez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Can Jones, Y2Fuam9uZXM5MTlAZ21haWwuY29t