 Michael Hebeisen1
Michael Hebeisen1 Mathilde Allard1†
Mathilde Allard1† Philippe O. Gannon1†
Philippe O. Gannon1† Julien Schmidt2,3
Julien Schmidt2,3 Daniel E. Speiser1,2
Daniel E. Speiser1,2 Nathalie Rufer1,2*
Nathalie Rufer1,2*
- 1Department of Oncology, Lausanne University Hospital Center (CHUV), University of Lausanne, Epalinges, Switzerland
- 2Ludwig Center for Cancer Research, University of Lausanne, Epalinges, Switzerland
- 3TCMetrix Sàrl, Epalinges, Switzerland
Cytotoxic T cells recognize, via their T cell receptors (TCRs), small antigenic peptides presented by the major histocompatibility complex (pMHC) on the surface of professional antigen-presenting cells and infected or malignant cells. The efficiency of T cell triggering critically depends on TCR binding to cognate pMHC, i.e., the TCR–pMHC structural avidity. The binding and kinetic attributes of this interaction are key parameters for protective T cell-mediated immunity, with stronger TCR–pMHC interactions conferring superior T cell activation and responsiveness than weaker ones. However, high-avidity TCRs are not always available, particularly among self/tumor antigen-specific T cells, most of which are eliminated by central and peripheral deletion mechanisms. Consequently, systematic assessment of T cell avidity can greatly help distinguishing protective from non-protective T cells. Here, we review novel strategies to assess TCR–pMHC interaction kinetics, enabling the identification of the functionally most-relevant T cells. We also discuss the significance of these technologies in determining which cells within a naturally occurring polyclonal tumor-specific T cell response would offer the best clinical benefit for use in adoptive therapies, with or without T cell engineering.
What Defines a Protective CD8 T Cell Response?
Antibody- and T Cell-Based Immunotherapies
During the last few years, immunotherapy has brought significant progress to clinical oncology. Major breakthroughs were made for melanoma patients (1–4), and progress becomes also evident for patients with frequent diseases, such as lung and kidney cancer (5). Specifically, immunotherapy aims at mobilizing the body’s immune cells to fight against cancer in highly specific ways. Several strategies have been developed over the last two decades to exploit the therapeutic potential of T cells (6). Administration of high-dose IL-2 (7) and tumor-associated (TA)-specific monoclonal antibodies (mAbs) (8) has initially provided long-term clinical benefits, albeit only for relatively few patients. More recently, mAbs that target immune checkpoints have shown remarkable results. In 2010, the successful outcomes of randomized phase III clinical trials with the anti-CTLA-4-specific mAb Ipilimumab offered strong clinical evidence that in humans, as in experimental animal models, the host’s immune system can control tumor growth (9). So far, several antibody-based drugs (anti-CTLA-4 mAb Ipilimumab-Yervoy, anti-PD-1 mAb Nivolumab-Opdivo, and anti-PD-1 mAb Pembrolizumab-Keytruda) have been approved for the treatment of melanoma, and the first FDA approval for carcinoma took place earlier this year, with the introduction of Opdivo for routine therapy of patients with non-small-cell lung cancer. Importantly, the rapidly increasing use of these antibodies represents a major breakthrough in the treatment of cancer patients (1, 2, 4, 9–11).
Already prior to immune checkpoint inhibitors, several lines of evidence suggested that antitumor immune responses might correlate with clinical outcome in patients with cancers. Among them is the very frequently observed correlation between the presence of tumor-infiltrating CD8 T cells and the improved clinical outcome for patients with solid tumors (12–14). The notion that antitumor T cells play a major role in controlling tumor growth was also largely demonstrated in clinical trials with adoptive transfer of autologous tumor-infiltrating T lymphocytes (TILs) (15). Even though technically and clinically challenging, the results are promising in terms of objective clinical responses and durability of responses (3, 16, 17). Moreover, genetic modification of T cells before adoptive cell transfer, such as inserting T cell receptors (TCRs) (18) and chimeric antigen receptors (19), was shown to further increase the clinical efficacy.
Despite that both antibody- and T cell-based immunotherapies can improve clinical outcome in cancer patients, multiple challenges still lay before us to improve the efficacy of cancer immunotherapies in the clinic. Indeed, many patients continue to experience disease relapse and/or progressive disease despite receiving these novel immune-based treatments. The potential reasons for this cancer “resistance” and “evasion” are based on cancer cell-internal and -external mechanisms, i.e., the two backbones of malignant diseases. For the former, cancer cells develop intrinsic therapy resistance. As for the latter, cancer cells corrupt the surrounding tissue microenvironment to support their own growth and suppress the anticancer immune responses. In short, the tumor cells and their microenvironment become a “wound that never heals” (20).
CD8 T Cells Play a Central Role in Tumor-Specific Immune Responses
The recent immunotherapy successes in clinical oncology were build on the profound experience acquired over the years in hemato-oncology, as well as an increase in understanding of the roles of T cells in generating a potent and sustained antitumor immune response. After allogeneic hematopoietic stem cell transplantation, the graft-versus-leukemia effects assure long-term remission of patients with hematological malignancies. Importantly, T cells play a central role in graft-versus-leukemia by controlling tumor growth, progression, and recurrence. Similarly, T cells are also essential players in generating a protective and durable immune response against solid tumors. T cells can act against both cancer cell-internal and -external resistance mechanisms. First, they can specifically target and directly destroy cancer cells. Second, they can revert a tumor-promoting microenvironment into a tumor-hostile one, by changing the patient’s tumor biology toward a “healing wound.” Moreover, as therapeutic successes depend on broad and long-term protection, T cells are important players since, in contrary to pharmaceutical drugs alone, they can generate a therapeutic memory. Therefore, by furthering the development of novel T cell-based therapies against cancer, therapeutic pressure could be applied simultaneously against both malignancy backbones, and tumor escape would be minimized.
For successful immune defense, activated antigen-specific T cells must reach high frequencies, differentiate into numerous and powerful effector and memory cells, and exert multiple functions. To achieve this, T cells must first be primed following the specific recognition by the TCRs of antigenic peptides bound to self-major histocompatibility complex (MHC) molecules (referred to as pMHC thereafter) at the surface of antigen-presenting cells (APCs). Second, upon differentiation and expansion, T cells must migrate and localize to the tumor bed. An essential point is that the TCR–pMHC interaction should be sufficiently strong to enable the efficient recognition of tumor antigens (which are naturally presented at low levels) and to trigger potent tumor-specific T cell effector functions. Finally, robust memory T cells must be established, assuring long-term immune responses for durable disease control.
Since T cells play a major role in immune protection against cancer, it is important to determine which T cell properties are essential to achieve clinical benefit. Several “correlates of protection” have been identified; TCR–pMHC binding affinity/avidity, T cell frequency, poly-functionality, poly-clonality, poly-restriction (i.e., T cells specific for multiple antigens that are presented by different HLA alleles), migration to the tumor, and survival/persistence (21) (Figure 1). The assessment of these criteria can greatly help distinguishing between powerful and ineffective antitumor T cell responses and thus provides essential information on the quality of a patient’s immune response. In this review, we will specifically focus on T cell avidity, both in terms of TCR–pMHC binding properties and functional capacities.
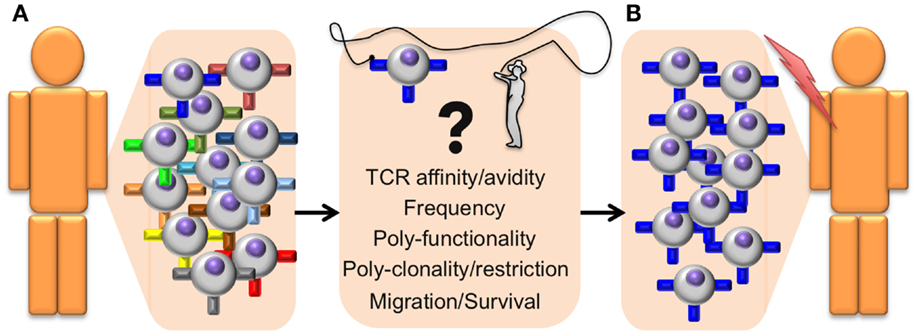
Figure 1. Identifying antitumor T cells of high avidity and high function for adoptive cell transfer immunotherapy. General outline presenting a step-by-step optimized protocol for the identification and adoptive transfer of the most potent tumor-specific CD8 T cells in cancer patients. (A) The selection of therapeutic autologous CD8 T cells (isolated from the tumor or from PBMC) is based on the following correlates of protection parameters that include structural TCR–pMHC affinity/avidity, T cell frequency, polyfunctionality (with differentiation and effector properties), poly-clonality, and poly-restriction to multiple antigens presented by different HLA alleles, cell migration capacity to the tumor site, as well as memory/survival properties with long-term persistence. (B) Selected T cells with optimal combination of those correlates will be isolated and expanded ex vivo, before being re-infused back in the patient. These selected tumor-specific CD8 T cell subpopulations should be highly effective at targeting and eliminating tumors in vivo and achieve enhanced and durable clinical benefits.
Functional Avidity of CD8 T Cells
The functional avidity is a biological measure that describes how well a T cell responds in vitro to a given peptide concentration. It is determined by the in vitro quantification of T cell functions, such as cytotoxic activity, IFN-γ production, and proliferation. Pioneering the field more than 20 years ago, we demonstrated that low T cell avidity is sufficient for in vitro proliferation or cytotoxicity to peptide-coated target cells but not for in vivo protection (22), a finding that was subsequently confirmed and extended by others (23). Meanwhile, there is a general consensus that CD8 T cell responses with increasing functional avidity are better in controlling virus infections (24, 25). There exists a close relationship between T cell functional avidity and target cell recognition, as shown in several antigenic systems (23, 24, 26–29).
In 1998, an elegant study revealed important insights into the roles of essential parameters for in vivo protection from lymphocytic choriomeningitis virus (LCMV) in mice (30). Besides the functional avidity of T cells, the authors analyzed the density of peptide antigen on infected cells, the binding strength of peptide to MHC, the magnitude of T cell responses, and the broadness of the TCR repertoire. They found that the T cells specific for the NP396 peptide provided the highest protection, based on their highest functional avidity and strongest binding of NP396 to MHC. Interestingly, the least protective were the T cells specific for GP33, despite their highest magnitude and TCR diversity, and a GP33 peptide density on infected cells about sixfold higher than of NP396. Finally, T cells specific for GP276 showed an intermediate potency for protection, based on intermediate functional avidity, but lowest TCR diversity, to the peptide with the lowest density (30). Also for T cell responses against tumors, results obtained from both mouse and human models suggest that T cells of high functional avidities (31) and strong peptide binding to MHC (32) are required for efficient protection.
Together, functional avidity stands out as a highly important correlate of protection (Figure 1). Nevertheless, functional avidity has yet to be evaluated much more systematically in the development and routine application of immunotherapy. Limitations of laboratory techniques are the major reasons why this is infrequently done. In general, T cell assessment is mostly limited to assays measuring antigen specificity, target cell killing and cytokine production (e.g., IFNγ) to fixed stimulation doses (17, 33–36). Importantly, these functional assays do not directly measure the TCR–pMHC affinity or avidity (Figure 1), despite representing a major determinant of T cell responsiveness and possibly a more relevant metric of the T cell response.
What Defines an Optimal Self/Tumor Antigen-Specific CD8 T Cell Response?
TCR–pMHC Affinity, Avidity, and Structural Avidity
T cell receptor–pMHC binding and kinetic interactions can be measured in terms of affinity or avidity. The TCR–pMHC binding affinity refers to the physical strength by which a single TCR binds to a single pMHC complex (37) and is inversely proportional to the dissociation equilibrium constant KD. Under equilibrium conditions, KD is defined as the ratio of the dissociation rate and association rate (koff/kon), which are typically measured by surface plasmon resonance (SPR) (Figure 2). The rate at which the TCR dissociates from the pMHC complex, referred to as t1/2 (or half-life), represents another important parameter, and is related to the dissociation rate constant koff by the equation t1/2 = ln 2/koff (38). Conversely, the TCR avidity describes, in the cellular context, the association of multiple TCRs with their respective pMHC complexes (39). TCR–pMHC avidity depends on the TCR affinity and incorporates the potential effects of coreceptors (e.g., CD8), TCR density, and T cell functional (activation) status (40). Using the novel NTA–His tag-containing multimer technology (also termed NTAmers), we recently quantified monomeric TCR–pMHC dissociation rates (koff or t1/2) of living tumor antigen-specific CD8 T cells (41, 42). Since NTAmers (TCMetrix, Epalinges, Switzerland) allow the quantitative assessment of TCR–pMHC binding interactions directly at the monomeric level and include the binding of the CD8 coreceptor, we now refer to this type of measurement as the structural TCR–pMHC avidity (42) (Figure 2). Finally, the overall CD8 T cell response is defined as the functional avidity and depends on the productive TCR–pMHC interactions, integrating the binding of multiple TCR–pMHC complexes and coreceptors together with the strength of cell–cell interactions (39).
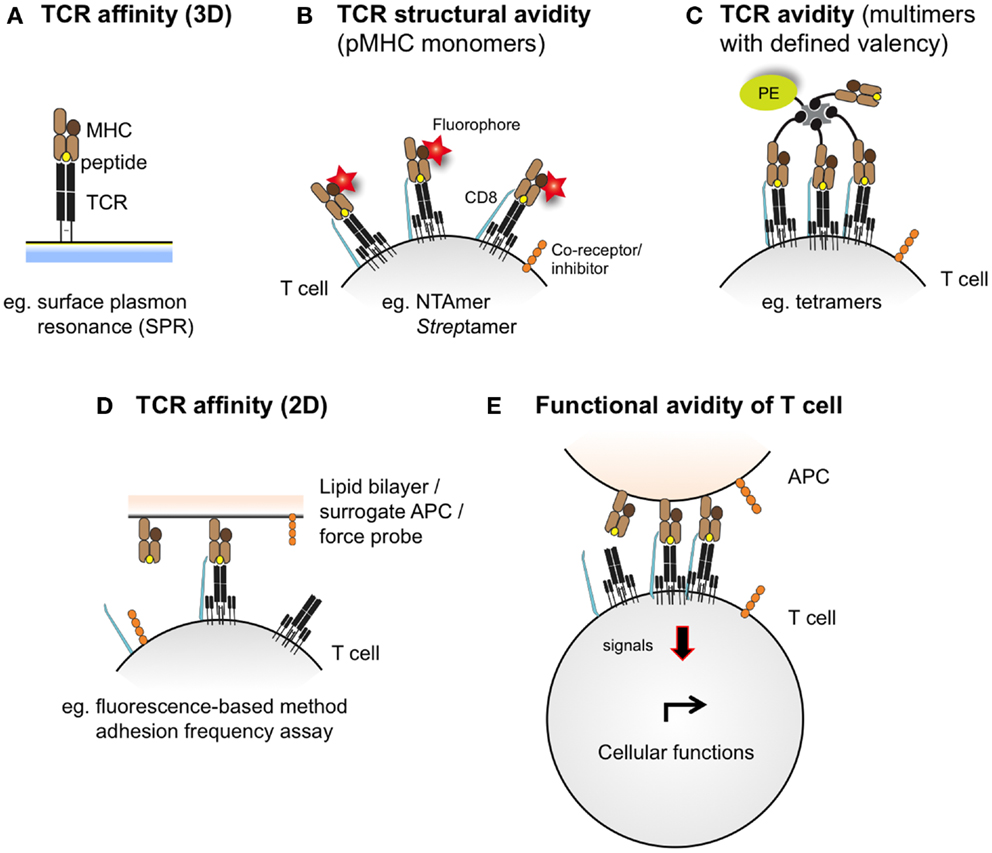
Figure 2. Schematic representations integrating the different assessment levels of TCR–pMHC binding interactions. TCR–pMHC affinity (A) refers to the binding strength of one TCR to one pMHC complex and is typically assessed by SPR (also defined as 3D interaction). At the cellular level (e.g., living antigen-specific CD8 T cells), the TCR–pMHC structural avidity (B) refers to the strength of interaction between monovalent TCR–pMHC complexes, as measured by reversible multimers (e.g., NTAmers, Streptamers). Importantly, monomeric binding measurements contrast to the multimeric TCR–pMHC binding avidity (C), which integrates the binding strength of multiple TCRs and pMHC complexes and is conventionally assessed by fluorescent pMHC multimers of known valency (e.g., tetramers). Recently, 2D-kinetic measurements (D) enable the assessment of TCR–pMHC binding affinity directly at the interface between a living T cell and a juxtaposed surface (e.g., a supported planar lipid bilayer or a surrogate APC) using fluorescent-based or micropipette adhesion frequency assays. T cell functional avidity (E) refers to the productive TCR–pMHC triggering integrating multiple TCR–pMHC binding interactions and represents the relative efficiency of T cell functionality as assessed in the presence of titrated peptide concentrations in various biological read-outs (e.g., target cell killing, cytokine production and proliferation potential).
Antiself/Tumor-Specific CD8 T Cell Responses Are Mediated by TCRs of Low Affinity/Avidity
The TCR–pMHC affinity and avidity vary dramatically between self and non-self-antigens (Figure 3). TCRs that interact with non-self peptides are frequently found among naive T cells and cover the whole physiological affinity range (29), with a preferential distribution of KD found between 25 and 1 μM (43). In fact, it is now commonly accepted that immune responses to pathogens are dominated by cytotoxic T cells that express high-affinity TCRs (44, 45). By contrast, mature CD8 T cells specific for self/tumor antigens express TCRs of weak TCR–pMHC affinity (43), whereas high-affinity cells are very rare due to mechanisms of central and peripheral tolerance (46) (Figure 3). The KD values of these interactions are typically in the range of 200–10 μM (with the mean around 100 μM) (43). Indeed, most TA antigens, such as cancer testis antigens (e.g., NY-ESO-1 and MAGEs) and differentiation antigens (e.g., Melan-A/MART-1, gp100, and tyrosinase) (47, 48), are expressed in the thymus (49). Consequently, thymocytes with high TCR–pMHC affinity/avidity for these antigens are negatively selected. Self/tumor-reactive T cells can further be eliminated in the periphery through mechanisms of peripheral tolerance (50). Nonetheless, these mechanisms spare cytotoxic T cells that can react to self/tumor antigens with relative low TCR–pMHC affinity/avidity (51–53) but which might be too low to mediate an effective antitumor immunity (Figure 3). Therefore, increasing the TCR–pMHC affinity and/or avidity of tumor-specific T cells is of particular interest for immunotherapy based on adoptive T cell transfer.
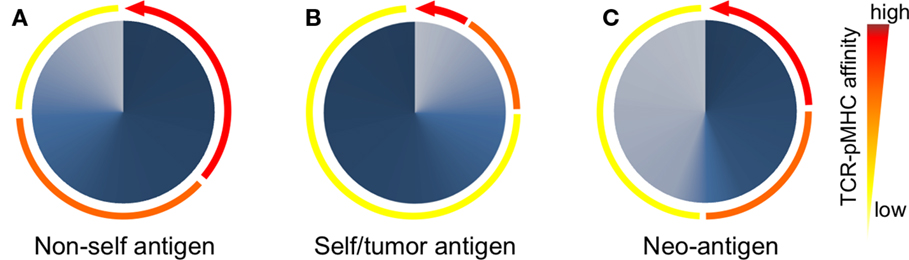
Figure 3. Peripheral T cell repertoires available to respond to non/self- and self-antigens are shaped according to the TCR–pMHC affinities of individual T cells. (A) After thymic selection, CD8 T cells specific for non-self (foreign) antigens express TCRs that span the entire physiological range from low (100 μM) to high (1 μM) affinity (depicted as colored arcs). In these non-self-specific repertoires, a large proportion (depicted as dark blue gradients) of T cells bear TCRs of intermediate to high-affinity TCRs (orange-red arcs). (B) Due to self-tolerance mechanisms, most but not all self/tumor antigen-specific T cells of high-affinity TCRs are deleted (red arcs). Consequently, T cell repertoires specific for self/tumor antigens are mainly composed (dark blue gradients) of low affinities (yellow arc). (C) T cells recognizing neoantigens are not deleted by self-tolerance mechanisms, since tumor-specific mutations generating neoantigens are “non-self like” epitopes. Thus, the repertoire of neoantigen-specific T cells is composed of increased proportions (dark blue gradients) of tumor-specific and high affinities TCRs (red arc).
Improving TCR–pMHC Affinity/Avidity Against Cancer Cells
Strategies developed to improve TCR–pMHC affinity involve the modification of TCR sequences by inserting point mutations within the complementary-determining regions (CDRs) of the variable domains of TCR αβ chains (54), and followed by the screening of yeast or phage variant libraries (55, 56). Such approaches result in the generation of TCR variants with supraphysiological binding strengths for peptide-MHC ligands up to the picomolar affinity range (55–57). However, as yeast and phage display technologies rely on large libraries and are based on random mutagenesis, the generation of such TCR mutants may be associated with loss of target cell specificity. In fact, T cells engineered with TCRs of very high affinities (KD < 1 nM) become crossreactive (or alloreactive) to other pMHC complexes (58–60). To overcome this problem, TCR–pMHC affinity can be optimized in a highly controlled manner by structure-based methods (61–63). These approaches consist of in silico analyses made on available crystallographic structures of TCR–pMHC complexes and aim at identifying the key residues critically involved in the TCR–pMHC interactions. Such residues can subsequently be replaced with other structurally compatible ones, resulting in either increased or decreased TCR–pMHC affinity (64). The structure-based design of TCRs allows an increase of the TCR–pMHC affinity, while preserving antigen specificity and avoiding broad crossreactivity to other pMHCs (62, 65).
Through stepwise rational design of TCR–pMHC affinity (63, 64), we created a unique panel of human CD8 TCRs specific for the cancer testis antigen NY-ESO-1157–165 presented by HLA-A*0201 (defined as A2 thereafter) (66). This affinity-optimization process resulted from the introduction and combination of point mutations either within the CDR2α and CDR2β regions, and/or within the CDR3β region (63, 64). The TCR–pMHC binding and kinetic parameters (KD, koff, and kon) derived from SPR confirmed the in silico predicted changes in TCR affinity for A2/NY-ESO-1157–165, with KD values ranging from 100 μM to 15 nM (65). By characterizing the functional potential of T cells expressing this panel of affinity-optimized TCRs, we found that T cells expressing TCRs with affinities lying in the upper physiological range (KD from 5 to 1 μM) displayed greater biological responses (e.g., cell activation, signaling, proliferation, cytokine/chemokine secretion, and target cell killing) than T cells expressing intermediate/wild-type TCRs (KD at 21.4 μM) or very low affinity (KD > 100 μM) (65–67).
Strikingly, further increase within the supraphysiological TCR affinity range (KD < 1 μM) led to drastic functional decline, with impairment in global gene expression and surface expression of activatory/costimulatory receptors (65, 67). Yet, such engineered T cells retained a high degree of antigen specificity toward the cognate pMHC (65). Importantly, T cell effectiveness was limited by at least two mechanisms. We observed preferential PD-1 expression on T cells expressing very high TCR affinities, as well as a full functional recovery upon PD-1 ligand blockade. In contrast, the upregulation of SHP-1 and SHP-2 phosphatases was broad, with the gradual enhanced expression in engineered T cells along the TCR affinity gradient (67). Together, these observations revealed that maximal biological activity, for the panel of human A2/NY-ESO-1157–165-specific engineered CD8 T cells, occurred within a well-defined affinity window with KD ranging from 5 to 1 μM, above which T cell effectiveness was limited by TCR-mediated regulatory mechanisms (67).
TCR–pMHC Affinity Threshold for Maximal Antitumoral CD8 T Cell Responses
Our findings nicely fit with other studies performed both in mice and human models, as well as in pathogen- and self/tumor-specific T cell responses strongly supporting the notion that T cell activation, signaling and subsequent function are limited to a given TCR–pMHC affinity window. It is known that a minimal threshold of TCR–pMHC binding is required for CD4 and CD8 T cell activation (68–72). Moreover, under physiological conditions, i.e., within the natural TCR affinity range (KD from 200 to 1 μM) and under low peptide concentrations, numerous studies now provide strong evidence that enhanced TCR–pMHC affinity (KD) or off-rate (koff) correlates with improved T cell responsiveness (59, 60, 62, 65, 66, 73–83). However, the correlation between TCR–pMHC affinity and T cell function is not linear as there is a decrease in functionality for TCR–pMHC interactions taking place beyond the natural TCR affinity range (KD < 1 μM) (65, 67, 81, 84, 85) or with prolonged half-lives (75, 77, 78, 80, 86).
Kalergis and colleagues were the first to propose that T cell activation might occur within an optimal range of half-life for TCR–pMHC interactions (78). Using altered peptide ligands derived from the ovalbumin OVA257–264 peptide, they showed that the duration of TCR–pMHC interactions (t1/2) regulated effector function and tumor-killing capacities of OT-1-specific CD8 T cells (80). Specifically, intermediate TCR–pMHC half-lives induced the strong expression of cytotoxic effector molecules, cytokine secretion, and consequently the efficient in vivo tumor clearance mediated by cytotoxic T lymphocytes (CTLs) (80). In line with these observations, Corse et al. studied the effect of TCR–pMHC interaction parameters on in vivo CD4 T cell activation, effector and memory responses upon immunization with lipopolysaccharide and moth cytochrome c (MCC) peptide, or related ligands exhibiting a range of TCR–pMHC half-lives (77). They found that the in vivo response to a superagonist ligand for the MCC-reactive TCR was associated with attenuated intracellular signaling, proliferation and effector functions over time following immunization. These data pointed to an upper limit of T cell ligand potency in vivo, with optimal T cell responses occurring to TCR–pMHC interactions of intermediate half-lives (77). Similar conclusions were reached in another study using vaccination with a panel of AH1 peptide ligands of increasing affinities specific for the immunodominant H-2Ld-restricted antigen from the colon tumor CT26 (84). Data from this report revealed that although TCR–pMHC affinity correlated well with the functional activity of the T cell clone in vitro, only vaccination with peptide ligands of intermediate affinities elicited functional T cell responses and provided best tumor growth control in BALB/c mice. Lastly, Kranz and colleagues explored the functional impact of increased TCR–pMHC affinity using the well-known 2C TCR-based mouse model (54, 55, 87). Excessive enhancement of TCR affinity through yeast display resulted in crossreactivity with other cognate pMHCs (58). Furthermore, they reported rapid in vivo disappearance of specific CD8 T cells expressing the m33 TCR of nanomolar affinity through mechanisms of peripheral deletion in both the TIL population and lymphoid organs, suggesting that TCR affinity controlled the survival and tumor infiltration of the transferred T cells (88, 89).
Clinical Trials with TCR Affinity-Improved T Cells Against Tumor Antigens
The importance of TCR–pMHC binding parameters in the context of human antitumoral T cell mediated responses was originally demonstrated in clinical trials whereby melanoma patients received autologous peripheral blood mononuclear cells transduced with specific TCRs against the differentiation tumor antigen Melan-AMART-126–35 (90). Compared to the native low affinity TCR isolated from the patient’s TIL, referred as DMF4 [KD 29 μM (91)], with moderate ability to recognize the tumor antigen, Johnson and coworkers (92) showed a trend of improved clinical efficacy for the DMF5 TCR of higher affinity [KD 5.6 μM; (91)]. Whereas objective responses were seen in 30% of patients who received the DMF5 TCR, several patients experienced in addition the destruction of normal melanocytes in the skin, ear, and eye, which led to uveitis and hearing loss (92). Similar studies based on the A2/NY-ESO-1157–165 tumor antigen model showed that specific CD8 T cells engineered to express a TCR (defined as 1G4) with affinity lying just beyond the natural affinity range (at KD 730 nM) were those that displayed maximal functionality with the lowest crossreactivity in vitro (59). Importantly, clinical trials conducted with this affinity-improved TCR in adoptive cell transfer of autologous engineered T cells in patients with metastatic melanoma and sarcoma led to objective clinical responses without major adverse events (93). Recently, Tan et al. (81) examined the impact of TCR affinity on the functional properties of human transduced CD8 T cells of three different TA peptide antigens (hTERT540–548, NY-ESO-1157–165, and MageA3161–169) across a wide range of binding parameters. These authors found that the TCR affinity controls T cell antigen sensitivity and poly-functionality, further supporting the presence of an optimal range for CD8 T cell functional improvement but which varies with antigen specificity. Collectively, our data and those by others performed in the human tumor setting recapitulate the findings from mouse models, showing that maximal T cell functional responses occurred at intermediate TCR–pMHC binding strengths (Tables 1 and 2).
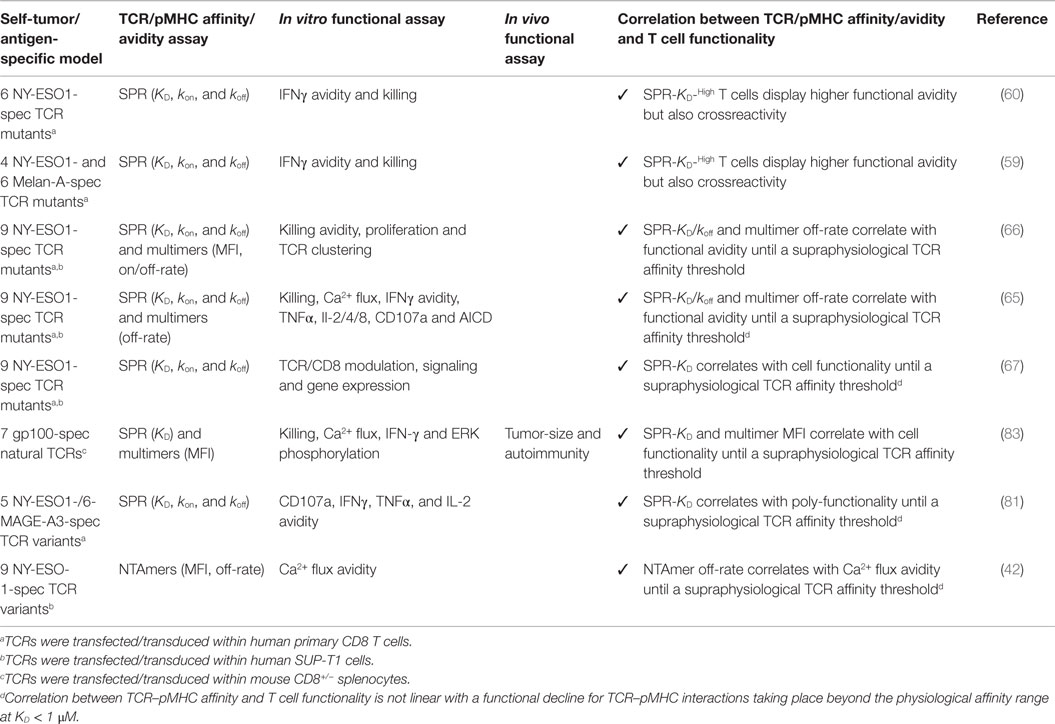
Table 1. Engineered tumor-specific CD8 T cells with affinity-optimized TCR panels.
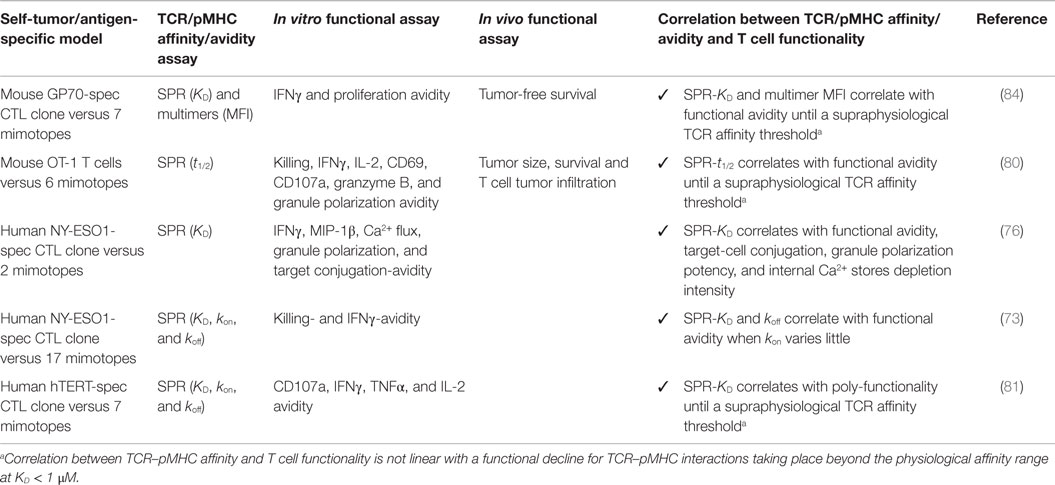
Table 2. Tumor-specific CD8 T cell clones identified by the altered ligand peptide approach.
Increasing Antitumor TCR–pMHC Affinity/Avidity is Associated with Autoreactivity
T cell immunotherapy against cancer should ideally require T cells expressing affinity-improved TCRs, which efficiently control tumor growth without inducing on-target autoimmune reactivity against normal tissues expressing the same self-antigen. Yet, several reports have shown that in contrast to T cells of low avidity, high-avidity tumor-specific T cell responses were often associated with autoimmunity (92, 94–96). Recently, using seven human gp100209–217-specific TCRs isolated from melanoma patients and covering the physiological affinity range (1–100 μM), Zhong et al. (83) carefully evaluated the TCR affinity threshold defining the optimal balance between effective antitumoral activity and autoimmunity in vivo. Their results revealed that T cell antitumor activity and autoimmunity were closely coupled, whereby increasing TCR affinity/avidity correlated with improved tumor regression but was also associated with severe ocular autoimmunity in adoptively transduced A2-Kb mice. Together, these observations suggest that a relatively low-affinity threshold may be required for the immune system to avoid self-damage (KD around 10 μM for A2/gp100209–217) (83). Intriguingly, another recent study reported that two TCR variants of increased binding interactions for the WT1 self/tumor antigen compared to the wild-type TCR were safe and did not mediate autoimmune tissue infiltration or damage when transduced into peripheral CD8 T cells and transferred in vivo (97). These findings are supported by a recent clinical trial showing that the usage of T cells transduced with a TCR of increased affinity for the cancer testis antigen A2/NY-ESO-1157–165 [1G4 α95:LY, KD at 730 nM; (59)] did not lead to autoimmunity in melanoma and sarcoma patients (93). These apparently contradicting results could be explained by the differences in distribution and expression levels of self/tumor antigen expression in normal tissues and in tumors. For instance, NY-ESO-1 is expressed in very restricted germinal tissues (e.g., in testis cells) and WT1 antigen is expressed at low levels in normal self-tissues. This contrasts with other self/tumor antigens, such as Melan-A, gp100, and tyrosinase, that are widely found in melanocytes of the skin, eye, and ear, and whose expression has been shown to lead to melanoma/melanocyte-associated autoimmunity (92, 96, 98). Consequently, the choice of antigen specificity for adoptive cell transfer of affinity-improved T cells is of crucial importance.
Neoantigens, which result from gene mutations or aberrant expression in tumor cells and whose expression is uniquely found in tumor tissues, may represent ideal and safe targets for T cell therapy (99). Recently, using whole-exome sequencing combined with MHC-binding algorithms, Robbins et al. (100) identified mutated antigens expressed on autologous tumor cells that were recognized by three TIL lines from three melanoma patients. Importantly, these patients demonstrated regression of bulky metastatic lesions after adoptive transfer of autologous TILs, suggesting that neoantigens were able to generate strong immune responses in those patients. Tumor-derived neoantigens can trigger potent T cell immunity (101), probably because they are perceived as foreign by the immune system, allowing neoantigen-specific T cells to escape negative selection and express high-affinity/avidity TCRs (102). Thus, similar to non-self specificities, T cells recognizing neoantigen should theoretically express TCR–pMHC affinities spanning the entire physiological range with possibly a large proportion of high-affinity TCRs (Figure 3).
Off-Target Toxicities in Clinical Trials Using TCR Affinity-Improved T Cells
Another important parameter to be considered when using TCR affinity-improved T cells for adoptive-based therapies is that such cells also bear the risk of increased crossreactivity to structurally related self-peptides, resulting in off-target toxicities (103). Recently, two patients treated with TCRs engineered for enhanced affinity toward the cancer testis HLA-A1/MAGE-A3 tumor antigen developed off-target recognition of a similar but not identical peptide from the cardiac muscle-specific protein Titin (104), resulting in cardiogenic shock and death within a few days of T cell infusion (105). Another clinical trial based on the infusion of autologous anti-HLA-A2/MAGE-A3 TCR-engineered T cells further revealed unpredictable adverse effects with a possible crossreactivity to the MAGE-A12 self-antigen expressed in rare neurons, and leading to neurologic toxicities and death in two patients (106). These results highlight not only the functional potency of affinity-improved T cells toward tumor target cells but also the urgent need for improved preclinical systems to carefully assess on-target and off-target reactivity (e.g., in silico proteome screens and in vitro peptide specificity assays), to ensure the safety of self/tumor-specific TCR-engineered T cells in future clinical trials (99, 103, 107).
Collectively, we and others (108, 109) propose that the rational design of improved self/tumor-specific TCRs for adoptive T cell therapy may not need to be optimized beyond the natural TCR affinity range (KD < 1 μM) to achieve optimal T cell function and avoiding possibly unpredictable risk of crossreactivity. Indeed, antigen-specific T cells may only naturally function within a well-defined narrow range of affinities under most conditions to ensure optimal responses against foreign pathogens and minimal responses against autoantigens (109). In that regard, new technological strategies are currently required allowing identifying and selecting for those naturally occurring but rare self/tumor antigen-specific T lymphocytes of the highest TCR affinity/avidity and functional capacities within the physiological TCR affinity range (Figure 3), as described below.
How Can we Identify Self-Tumor/Antigen-Specific CD8 T Cells of High TCR–pMHC Structural Avidity?
Molecular and Cellular TCR–pMHC Binding Measurements
Early TCR–pMHC binding analyses were typically performed by SPR, which allows for the simultaneous detection of molecular kinetics (kon and koff) and affinity (equilibrium dissociation constant KD) of TCR–pMHC interactions in a single assay where one of the two molecules is attached to a sensor chip and the other one is flowing in soluble form (also defined as 3D interactions) (Figure 2) (110). Extensive studies revealed that natural human TCR–pMHC interactions were of relative weak affinities, with KD ranging from 500 to 1 μM, rapid off-rate and slow on-rates (46, 111, 112). However, an inherent caveat of SPR analysis is that it requires the laborious and expensive production of soluble TCRs and ignores the contribution of the binding of the CD8 coreceptor and/or other molecules present in the vicinity of the TCR to the overall TCR–pMHC avidity.
Recently, novel technologies have emerged that enable the deduction of kon and koff kinetics directly at the interface between a living T cell and a surrogate APC, or between a T cell and a supported planar lipid bilayer (also defined as 2D interaction) (Figure 2) (113–115). Such 2D-kinetic analyses were shown to correlate with T cell activation, proliferation and cytokine secretion (79), and calcium signaling (116) in both CD8 and CD4 subsets (117). These studies also highlighted the cooperative role of CD8 coreceptor binding to the preexisting TCR–pMHC complex in a two-stage Lck-dependent manner (118, 119). Unexpectedly, 2D binding parameters are poorly correlating with 3D kinetic measurements, when compared side-by-side [reviewed in Ref. (120)]. Recent models of induced rebinding for TCR triggering, taking into account the TCR clustering effect and conformational changes occurring after initial pMHC encounter, now offer a reconciliation to these initial contradictory reports (121). Although 2D approaches allow for the measurements of TCR–pMHC binding parameters in a more physiological way than 3D SPR technology, both approaches should therefore be viewed as complementary. Importantly, 2D analyses require specialized equipment and time, precluding for the rapid and high-throughput screen of living antigen-specific T cells that could be useful for adoptive cell immunotherapy, and currently limiting their application to fundamental research (122, 123).
TCR–pMHC Binding and Kinetic Measurements by Multimeric pMHC Molecules
To better understand the biophysical parameters regulating T cell activation, numerous studies of TCR–pMHC binding parameters were conducted using soluble pMHC of well-defined valencies directly on living cells (122, 124–126). Despite these advances in TCR staining, attempts to use soluble pMHC multimers to precisely determine the TCR affinity/avidity provided ambiguous results. While some reports showed a relationship between antitumor functional responses and the staining brightness (MFI) of multimeric pMHC attachment to the cell surface TCRs (Figure 2) (27, 83, 127, 128), others demonstrated a clear lack of correlation (129–131) (Table 3). Thus, the level of TCR–pMHC staining intensity (MFI) does not consistently correlate to the TCR–pMHC affinity/avidity, nor to the underlying T cell responsiveness.
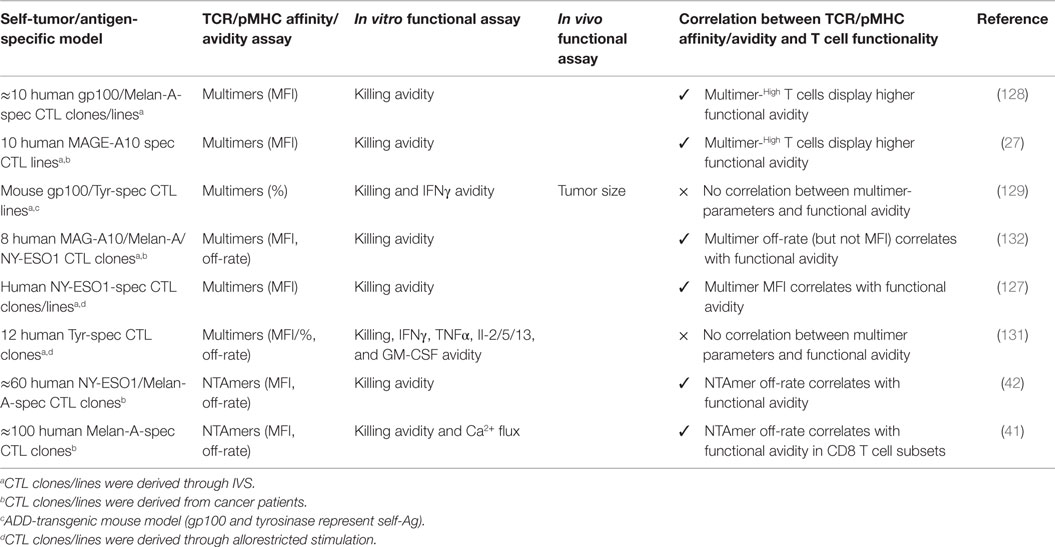
Table 3. Characterization of natural tumor-specific CD8 T cell clones and lines.
Due to these discrepancies, multimeric association and dissociation kinetic rates from the T cell surface were evaluated as potential read-outs for determining TCR–pMHC affinity/avidity and correlates of T cell function. But again, several studies showed correlations between multimeric off-rates and antitumor T cell functions (62, 66, 132), whereas others failed to demonstrate any in vitro functional or in vivo protective association (82, 129, 131, 133) (Table 3). Major reasons for these inconsistencies include the kinetic bias generated by the multivalent nature of the pMHC complexes when compared to monomeric-based molecules, the impact of uncontrolled rebinding during dissociation assays (134), as well as by the induction of signaling events through TCR–pMHC multimerization that can trigger cell death (122, 134).
Improved Detection and Isolation of Antigen-Specific CD8 T Cells by Reversible Multimers
Over the last decade, a major technological improvement was achieved with the development of reversible multimers (i.e., reagents in which pMHC monomers can be disrupted from the multimeric scaffold upon addition of a stimulus). By comparison with conventional multimers, reversible multimer staining allows for the isolation of practically “untouched” T cells, without inducing their TCR-mediated activation, phenotypic change, or activation-induced cell death (122). Knabel and colleagues (135) developed the first class of reversible multimers called Streptamers (IBA, Goettingen, Germany). These molecules are made of fluorescent StrepTactin, a derivative of streptavidin, coupled to several pMHC monomers carrying a streptag, a linear peptide optimized to bind to StrepTactin (135). The molecule d-biotin binds StepTactin with higher affinity than streptag and is able to compete for the same binding site, disrupting the multimeric complex. Consequently, the addition of free d-biotin releases the fluorescent StrepTactin, breaking the multimers into non-activating pMHC monomers at the cell surface. Since MHC monomers do not stably bind to TCRs, they rapidly dissociate, allowing for the identification and isolation of antigen-specific CD8 T cells, while preserving their functional status, in contrast to conventional multimers (136).
Based on the same principle, pMHC monomers containing a desthiobiotin (DTB), a derivative of biotin that binds streptavidin with lower affinity, were multimerized using fluorescent streptavidin and used for sorting untouched antigen-specific CD8 T cells (137). Sorting and cloning of Melan-A-specific CD8 T cells using DTB-based multimers yielded over two times more clones than when using irreversible multimers, mainly because of avoidance of multimer induced apoptosis. Despite these technological advances, a major drawback of these reagents remained the lag time in the switch from multimeric to monomeric form and their weak molecular stability associated with their multimeric form, especially at high temperatures (122). More recently, Schmidt and colleagues (138) reported the third generation of reversible multimers, called NTAmers (TCMetrix), and made of His-tagged pMHC and fluorescent streptavidin carrying an engineered nitrilotriacetic acid (NTA) linker. NTAmer complexes are highly stable even at elevated temperature and have the advantage of decaying very rapidly into monomers, within 2–3 s upon addition of imidazole at doses that are non-toxic for T cells. Moreover, the benefit of using reversible NTAmers for sorting antigen-specific T cells without activation-induced cell death was further confirmed (138).
Monomeric TCR–pMHC Off-Rate Measurements Specific for Non-Self Viral Antigens
Major efforts have been made to develop technologies enabling the measurements of monomeric TCR–pMHC binding strength and kinetics directly at the surface of live T cells. Indeed, rapid interrogation of these parameters may help discriminating CD8 T cells into high or low structural TCR–pMHC avidities (Figure 2) and thus permit the selection of T cells with optimal functional avidity (e.g., high T cell function and low autoreactivity). Thanks to the fluorescent labeling of individual pMHC monomers contained in the reversible multimeric complexes, it became possible to monitor and quantify monomeric dissociation of pMHC from the TCRs directly on living T cells. In 2013, Nauerth et al. (139) reported a real-time microscopic-based method using reversible two-color Streptamers (IBA, Goettingen, Germany) to determine monomeric dissociation kinetics of non-self virus-specific TCRs on human and mouse T cells. The monomeric dissociation koff rates were found to correlate with in vitro functional avidity as well as with in vivo protection capacity. CMV-specific CD8 T cells expressing TCRs with slow dissociation rates (long half-lives) were functionally superior to those with rapid dissociation rates (short half-lives) (139). The authors concluded that monomeric pMHC–TCR dissociation kinetics represent a valuable parameter to identify the best T cells for adoptive cell transfer, and to evaluate the quality of existing or induced immune T cell responses (140). However, the Achilles heel of the Streptamer technology resides in the scaffold of the molecule itself. The significant lag time (about 60 s) required for the Streptamer to decay into monomeric pMHC molecules after addition of free d-biotin as well as the photobleaching effect associated with the microscopic assay prevent for the precise determination of rapid TCR–pMHC off-rates, which are typically found within the self/tumor-specific CD8 T cell repertoires of lower TCR affinities. In turn, a great advantage is realized by the use of real-time fluorescence microscopy to enable simultaneous measurement of koff on several CD8 T cells, typically as many as displayed by the field of view, and is as such not limited to clonal T cell populations.
Monomeric TCR–pMHC Off-Rates Measurements Specific for Self/Tumor Antigens by NTAmers
We recently used a two-color version of the reversible NTAmer molecule, in which pMHC monomers are made with Cy5-labeled β2m and complexed with PE-streptavidin (122) (Figure 4). The rapid decay (2–3 s) of the NTAmer complex into its pMHC monomeric constituents made it possible to precisely analyze by flow cytometry the dissociation kinetics of a wide spectrum of TCR–pMHC affinities, with a special emphasis for self/tumor-specific CD8 T cells (Figure 4). The reliability and accuracy of the NTAmer approach were validated by finding strong correlations between NTAmer-based monomeric dissociation rates and those obtained by SPR measurements (42), when applied on our previously described panel of TCR-engineered A2/NY-ESO-1157–165-specific T cells (65, 67). Using the NTAmer technology, we also successfully measured the monomeric TCR–pMHC dissociation rates of a large series of natural A2/NY-ESO-1157–165- and
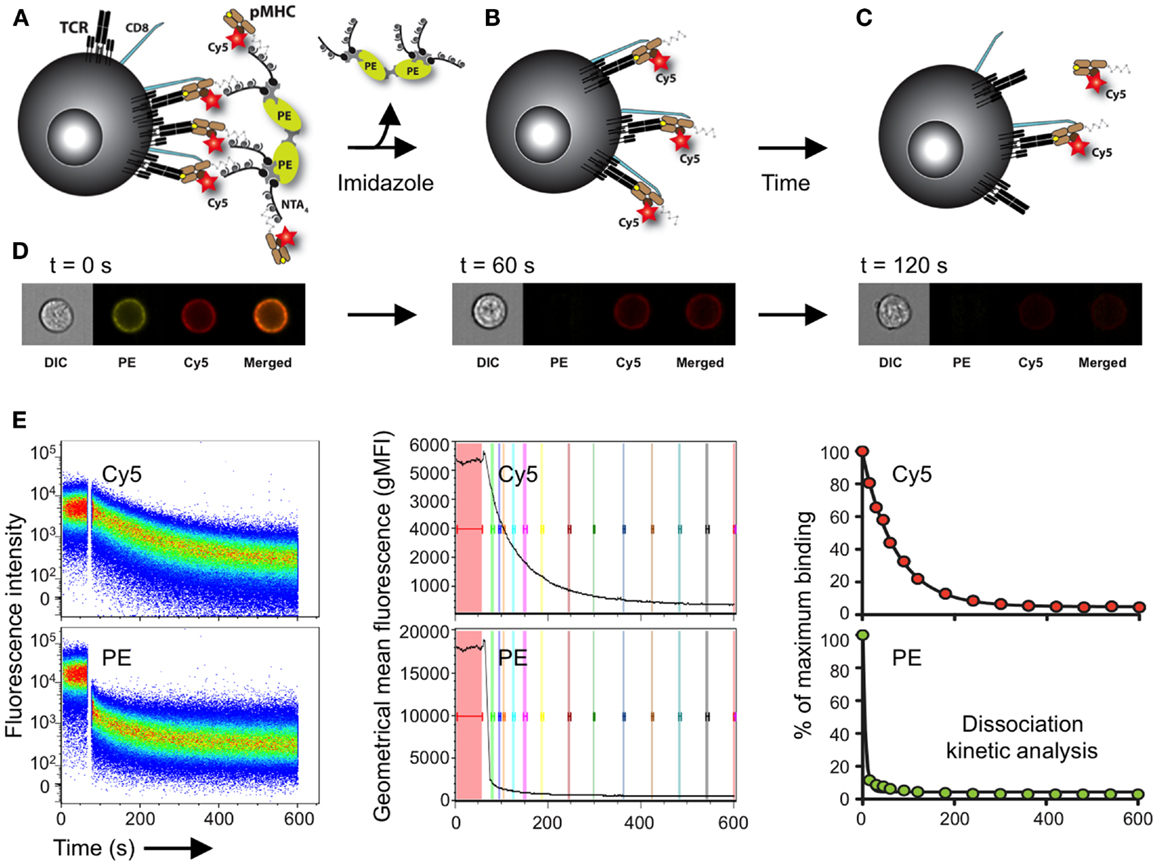
Figure 4. Schematic representation of the NTAmer-based monomeric dissociation assay. (A) CD8 T cells are stained at 4°C with multimeric NTAmers composed of streptavidin-PE (green)-NTA4 (gray) and peptide–MHC (brown) monomers containing His6-tag and Cy5-labeled β2m (red). (B) NTAmers are highly stable but upon addition of imidazole (100 mM), they rapidly decay in Cy5-labeled pMHC monomers and streptavidin-PE-NTA4 scaffolds. (C) Monomers subsequently dissociate from cell-associated TCRs (black) and CD8 (blue) according to the intrinsic TCR/CD8–pMHC dissociation rate (koff). (D) Representative DIC (differential interference contrast), PE, Cy5 and PE/Cy5 composite images acquired at the indicated time with a high-resolution microscopy flow cytometer (Amnis ImageStreamX Mark II) and illustrating the different stages (A–C) of the NTAmer dissociation assay. (E) Representative example of monomeric dissociation off-rates from a tumor-specific CD8 T cell clone following flow cytometry measurements by NTAmers. Imidazole is added after one minute baseline recording (left, white gap) and dissociation curves are followed over time within the Cy5 (monomers) and PE (NTA scaffold) channels. The kinetic module of FlowJo9 is used for geometric mean fluorescent intensity (gMFI) curve analysis (middle), while kinetic dissociation rates and half-lives are calculated with Prism (Graph Pad software Inc.). Adapted from Schmidt et al. (138) and Hebeisen et al. (42).
In summary, the NTAmer offers the real-time quantification of dissociation kinetics on a wide range of TCR–pMHC affinities directly at the surface of living, primary CD8 T cells, thus providing rapid, easy, and direct measurements of the monomeric TCR–pMHC dissociation rates within large numbers of tumor-specific CD8 T cell clones (41, 42) (Figure 4). Moreover, it is now possible to adequately evaluate the ex vivo impact of TCR–pMHC affinity/avidity on the functionality and differentiation of antitumor T cell responses in well-defined clinical settings. Since the NTAmer approach requires the cloning and expansion of antigen-specific CD8 T cells before measuring their TCR–pMHC dissociation off-rates, current efforts are devoted in translating the NTAmer technology to the single cell level.
Impact of the CD8 Coreceptor on TCR–pMHC Affinity/Avidity
If accumulating evidence pinpoints the pivotal role of TCR–pMHC affinity/avidity in T cell activation capacity, several other molecular and cellular parameters were also shown to greatly impact on T cell responsiveness upon TCR triggering. As such, it is largely accepted that the αβ heterodimeric CD8 coreceptor enhances CD8 T cell activation via two main effects: (i) by recruiting p56lck to the TCR/CD3 complex upon TCR engagement promoting cell signaling and (ii) by stabilizing the TCR–pMHC complexes through its weak but direct interaction with the α3 domain of pMHC class I molecule (40, 46, 118, 119, 141–143).
The biophysical contribution of CD8 attachment during TCR–pMHC triggering has been difficult to assess. Since the SPR technology does not allow the measurement of the dynamic effect of CD8 binding during TCR–pMHC interaction, its impact on TCR–pMHC kinetics were estimated at the surface of T cells with CD8-binding proficient or deficient pMHC tetramers (133, 143). Using a CD8 binding-deficient NTAmer variant, we recently performed precise measurements of the impact of CD8 attachment during monomeric TCR–pMHC binding dissociation assays (42). CD8 coattachment was found to strengthen the TCR–pMHC binding interaction by a factor of 3–4 times, as anticipated by previous tetramer dissociation assays (133, 143). Interestingly, the CD8 stabilization factor on TCR–pMHC dissociation was independent of the TCR–pMHC affinity, in contrast with the CD8 dependence for T cell activation, which can be directly linked to TCR affinity (65, 87, 133), and allows tuning the sensitivity and specificity of T cell responses (144).
Finally, the expression levels of various other receptors/molecules, as for instance costimulatory and inhibitory receptors, cytokine receptors, or adhesion molecules, can also be important for the modulation of T cell activation through the fine tuning of either TCR-dependent signaling pathways, T cell activation thresholds and/or T cell–APC adhesion-strength. Interestingly, while the expression level of these molecules can affect both the intensity and duration of T cell responses upon activation, we recently showed that some (e.g., PD-1 inhibitory receptor and SHP-1/SHP-2 phosphatases) also correlated with the TCR–pMHC affinity/avidity (67). Altogether, these additional receptors/molecules are part of a tunable system that enables T cells to adapt their reactivity to different stimulatory conditions. We have just begun to understand how those are achieved (108).
Does the TCR–pMHC Structural Avidity Predict CD8 T Cell Functional Efficiency?
Relationship Between TCR–pMHC Binding Parameters and CD8 T Cell Responsiveness
Up to date, a debate remains regarding which parameter(s) of the TCR–pMHC interaction (e.g., koff and KD) could better predict T cell activation and function. While several reports showed that koff was the most significant factor contributing to T cell activation (69, 78, 145–149), others reported that KD was the preeminent correlate of T cell responsiveness (58, 74, 82, 84). Recent data suggested that these observations were in fact not in conflict which each other, but rather the manifestation of the association rate parameter (kon) (73, 150). Indeed, for TCRs with faster association rates to the pMHC (and thus high probability for TCR–pMHC rebinding), the affinity KD is the better predictor of T cell activation potency. Conversely, for slower association rates (and low rebinding probability), the off-rate remains the better correlate (73, 150). Nonetheless, in most cases the KD value is mainly driven by the koff.
Regardless of the type of parameter(s) used to quantify the TCR–pMHC binding interaction, data emanating from engineered TCR-variant panels (Table 1) or altered peptide ligand models (Table 2) have provided strong evidence, in the context of self/tumor-specific immune responses, that the functionality of CD8 T cells can be tailored by the TCR–pMHC affinity/avidity. As such, we and others demonstrate that within the range of physiological interactions (KD 100–1 μM), the TCR–pMHC affinity (as determined by SPR) strongly correlates with various T cell functional read-outs (59, 60, 65–67, 73, 76, 80, 81, 83, 84). These include T cell potency for target cell conjugation, phosphorylation of downstream molecules of the TCR-signaling complex, intracellular Ca2+ mobilization, lytic-granule polarization, target cell killing, cytokine production, cell proliferation, polyfunctionality, in vivo tumor infiltration, and protection/survival.
Identifying Rare, High-Avidity Self/Tumor-Specific CD8 T Cells in Melanoma Patients Using the Novel NTAmer Technology
It is important to mention that most studies described above are based on artificial models (e.g., using affinity-optimized TCR variants) and therefore rely on a wide TCR–pMHC affinity spectrum (from KD 100 μM to 1 pM) (Table 1). Since large-scale assessment of endogenous TCR–pMHC affinity/avidity repertoires has remained, until recently, technically challenging, only limited information is available on the overall impact and clinical relevance of TCR–pMHC affinity/avidity in the context of natural self/tumor antigen-specific CD8 T cell repertoires (Table 3). Specifically, the questions whether T cells of high TCR–pMHC affinity/avidity can be found in the endogenous tumor-specific repertoire of cancer patients and whether the TCR–pMHC structural avidity represents an important factor contributing to a robust antitumor T cell stimulation and activity remain open. To address them, novel tools had to be developed allowing the quantification of the endogenous TCR–pMHC affinity/avidity in relation to the tumor-specific T cell functions.
Recently, our group demonstrated that the NTAmer technology could be used to precisely assess the TCR–pMHC structural avidity on living TCR-engineered and natural self/tumor- specific CD8 T cells (42) (Figure 4). Using large panels of Melan-AMART-126–35- and NYESO-1157–165-specific CTL clones isolated from vaccinated melanoma patients, we showed that the NTAmer-derived off-rates strongly correlated with the killing avidity of naturally occurring antitumor T cells (41, 42). Importantly, using this novel technology, we could quantify the potency of an immunotherapy intervention in melanoma patients. Indeed, we confirmed that the type of peptide used for vaccination of cancer patients profoundly influenced the TCR–pMHC structural avidity of tumor-specific T cells, which in turn correlated with T cell functions (41). Patients vaccinated repetitively with the natural Melan-AMART-126–35 decapeptide generated tumor-specific CD8 T cells with increased TCR–pMHC structural avidities as compared to vaccinations with the analog Melan-AMART-126–35 A27L peptide, even if the latter binds more strongly and stably to MHC as compared to the natural peptide. Analog peptides with enhanced MHC binding are commonly used for cancer vaccines. However, vaccination with enhanced MHC binding has likely similar consequences as vaccination with high peptide doses, since both result in the activation and selection of qualitatively inferior T cells, likely due to their lower functional avidity (23, 151). Indeed, we found that the overall functional properties of the tumor-specific CD8 T cells correlated with the biased T cell repertoire selection of vaccination with natural versus analog peptide (41, 152). Thus, vaccination with low peptide doses or peptides with weak/natural MHC binding favors an enrichment of T cell clonotypes with higher functional competence.
Consequently, the assessment of the TCR–pMHC structural avidity on living cells by NTAmers enabled to address which therapeutic vaccine protocol triggered the most potent, high-avidity tumor-specific T cell responses within comparative experimental cohorts, providing precious insights into the choice of peptide to be employed for future cancer vaccines. Furthermore, we were able to evaluate the impact of TCR–pMHC affinity/avidity on T cell differentiation. Our data reveal that, compared to a high-affinity mimotope vaccine, the use of the natural Melan-AMART-126–35 peptide could impact both the functionality and the preferential differentiation of T cells bearing high structural avidity TCRs (41). Hence, we report the feasibility and usefulness of TCR–pMHC structural avidity assessment by NTAmers of naturally occurring polyclonal T cell responses, allowing the identification and selection of rare high-avidity cytotoxic T cells from patients for cancer therapy.
A New Look at Old Questions
Several other hypotheses can now be re-evaluated to further dissect the impact of TCR–pMHC affinity/avidity on T cell (poly)functionality, memory formation, survival and persistence. In terms of functionality, additional studies need to precisely appraise the impact of TCR–pMHC affinity/avidity on the ensuing intracellular signals and effector activities, this in terms of both quantitative (ligand potency versus maximal activity) and qualitative (poly-functionality) aspects. Our current data indicate that the TCR–pMHC structural avidity correlates better with ligand potency (EC50) than with maximal activity using various functional readouts upon stimulation with titrated amounts of peptide [(41) own unpublished observations]. Of note, distinct T cell functions are triggered with different activation thresholds (Ca2+ flux < killing < cytokine-release < proliferation) (153–159), and could therefore be differentially affected by the TCR–pMHC affinity/avidity. Besides, more detailed studies are required to fully characterize the impact of TCR–pMHC affinity/avidity on the ability of CD8 T cells to develop distinct cytokine profiles (e.g., TH1 versus TH2) (131) or polyfunctional responses, a well-established and important indicator of the ability of CD8 T cells to control viral infections, as suggested in various models (160).
Another important question is whether there is a distinct regulation of the repertoire selection, differentiation and persistence of high- versus low-TCR–pMHC affinity/avidity antitumor CD8 T cells in vivo. For instance, numerous studies conducted in mouse models (70, 161–165) or ex vivo analysis of human T cell responses (166, 167) targeting pathogen-derived antigens demonstrate that the secondary repertoire is selectively enriched in high-affinity/avidity TCR-expressing T cells compared to the primary one. Such process of narrowing of the memory repertoire largely results from the loss of low-affinity/avidity TCR-bearing T cells during antigen-driven clonal expansion. Indeed, due to interclonal competition and restricted access to pathogen epitopes, low-avidity T cells are less likely to be primed and rapidly expanded than high-avidity ones (168–170). However, high-affinity/avidity immunodominant clonotypes may also be preferentially driven toward functional impairment, when compared to low-affinity/avidity ones, as a physiological consequence of T cell activation (171, 172).
T cell receptor–pMHC affinity/avidity could also represent a critical determinant for T cell susceptibility to tumor homing, as suggested in transgenic mouse models of adoptive transfer of high versus low-avidity tumor-specific CD8 T cells (173–177). When transferred into tumor-bearing mice, high-avidity CTLs were shown to be more potent than low avidity CTLs to infiltrate tumors (174, 175, 177), which could be partly attributed to the expression of integrins and lectins, such as CD62L and CD11a, on high avidity CTLs (174), and the recognition of tumor antigens (32), emphasizing the role of antigen as homing molecule. High-avidity tumor-infiltrating T lymphocytes (TILs) expressed higher levels of molecules linked with their killing potential (e.g., granzyme B and perforin), associated with reduced levels of inhibitory proteins (e.g., LAG-3, PD-1, and NKG2A), than low avidity TILs (174). In contrast, low-avidity T cells were shown to upregulate members of the apoptosis pathway (e.g., Bim, FasL, and CD24) promoting their own cell death but also that of other tumor-specific T cells in the tumor microenvironment (173, 174). However, high-avidity CTLs were also more prone to tolerization mechanisms, and were preferentially tolerized by dendritic cells or regulatory T cells in the tumor microenvironment, while low-avidity CTLs retained their effector functions (175–177).
Conclusive Remarks
Immunotherapy of cancer has made significant progress with the recent introduction of new therapeutic reagents, such as antibodies specific for CTLA, PD-1, and PDL-1, so-called immune “checkpoints.” Yet, we still need robust techniques allowing the rapid identification and isolation of CD8 T cells of optimal avidity and functions against tumors. Ideally, these technologies should enable the efficient screening of live CD8 T cells derived from a tumor sample or the peripheral blood of cancer patients at the single cell level. In that regard, binding strength analysis with novel fluorescent pMHC systems (e.g., NTAmers and Streptamers), combined with single cell microscopic or LCD camera screening may enable to retrieve cells with the desired dissociation rates, which could then be used for T cell amplification and/or TCR cloning. Among these selected clones, the ones that express TCRs of highest TCR–pMHC structural avidity could then be tested for other parameters that may define optimal antitumoral activity in vivo, including their differentiation status (e.g., naive, effector, and memory), surface activatory/inhibitory receptor profile (e.g., CD69, PD-1, and CTLA4), effector properties (e.g., target cell killing, cytokine and chemokine secretion, and poly-functionality), as well as their proliferation potential. The combination of these parameters should allow the rational selection for tumor-specific, high-avidity cytotoxic T cells that have maximal capacity to control tumor growth and eliminate tumor cells in vivo (Figure 1).
Furthermore, by developing NTAmers or related technologies for the quantification of TCR–pMHC affinity/avidity within polyclonal populations, it may become possible to characterize the impact of immune-based therapies (adoptive cell transfer and checkpoint blockade), as well as chemotherapy and radiotherapy, on the survival and persistence of anticancer T cells. The addition of the TCR–pMHC affinity/avidity read-out may offer a new “biometric,” by which the quality of the T cell response can be directly evaluated and graded in order to better characterize their impact on the efficacy of cancer-based therapies.
Finally, neoantigen-specific T cells are expected to express TCRs of higher affinity/avidity and to be safe to use to treat cancer patients (99), as T cells with such TCRs are not clonally deleted, are truly tumor specific and thus are unlikely to attack healthy cells (102). In the near future, more neoantigens will be identified (102), allowing for the isolation of neoantigen-specific T cells of optimal avidities using reversible multimer-based technologies. Neoantigen-specific TCR cloning may also give rise to libraries of potent transgenic T cells that can be used against common neoantigens shared by various cancers (e.g., BCR-ABL).
Understanding the correlates of immune protection and developing technologies and algorithms allowing selecting for the best (i.e., high avidity and high function) tumor-specific CD8 T cells will support the progress of T cell-based therapies against cancer.
Author Contributions
MH, MA, PG, JS, DS, and NR wrote the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to P. Baumgaertner, M.-N. Duong, P. Guillaume, I. Luescher, O. Michielin, and P. Romero for their contributions and helpful discussions. The authors apologize to all colleagues whose work could not be referred in this review.
Funding
MH is supported by the Department of Oncology and the University of Lausanne, MA is supported by the ISREC Foundation, and PG was supported by the Canadian Institute of Health Research.
References
1. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
2. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364:2517–26. doi:10.1056/NEJMoa1104621
3. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi:10.1158/1078-0432.CCR-11-0116
4. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
5. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372:2018–28. doi:10.1056/NEJMoa1501824
6. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science (2013) 342:1432–3. doi:10.1126/science
7. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol (1999) 17:2105–16.
8. Campoli M, Ferris R, Ferrone S, Wang X. Immunotherapy of malignant disease with tumor antigen-specific monoclonal antibodies. Clin Cancer Res (2010) 16:11–20. doi:10.1158/1078-0432.CCR-09-2345
9. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
10. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med (2015) 372:2006–17. doi:10.1056/NEJMoa1414428
11. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372:2521–32. doi:10.1056/NEJMoa1503093
12. Fridman WH, Galon J, Pages F, Tartour E, Sautes-Fridman C, Kroemer G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res (2011) 71:5601–5. doi:10.1158/0008-5472.CAN-11-1316
13. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature (2011) 480:480–9. doi:10.1038/nature10673
14. Pages F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol (2009) 27:5944–51. doi:10.1200/JCO.2008.19.6147
15. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12:269–81. doi:10.1038/nri3191
16. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res (2013) 19:4792–800. doi:10.1158/1078-0432.CCR-13-0380
17. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res (2012) 18:6758–70. doi:10.1158/1078-0432.CCR-12-1177
18. Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer – what clinicians need to know. Nat Rev Clin Oncol (2011) 8:577–85. doi:10.1038/nrclinonc.2011.116
19. Gill S, Porter DL. CAR-modified anti-CD19 T cells for the treatment of B-cell malignancies: rules of the road. Expert Opin Biol Ther (2014) 14:37–49. doi:10.1517/14712598.2014.860442
20. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med (1986) 315:1650–9. doi:10.1056/NEJM198612253152606
21. Verdeil G, Fuertes Marraco SA, Murray T, Speiser DE. From T cell “exhaustion” to anti-cancer immunity. Biochim Biophys Acta (2015). doi:10.1016/j.bbcan.2015.06.007
22. Speiser DE, Kyburz D, Stubi U, Hengartner H, Zinkernagel RM. Discrepancy between in vitro measurable and in vivo virus neutralizing cytotoxic T cell reactivities. Low T cell receptor specificity and avidity sufficient for in vitro proliferation or cytotoxicity to peptide-coated target cells but not for in vivo protection. J Immunol (1992) 149:972–80.
23. Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci U S A (1996) 93:4102–7. doi:10.1073/pnas.93.9.4102
24. Foley MH, Forcier T, McAndrew E, Gonzalez M, Chen H, Juelg B, et al. High avidity CD8+ T cells efficiently eliminate motile HIV-infected targets and execute a locally focused program of anti-viral function. PLoS One (2014) 9:e87873. doi:10.1371/journal.pone.0087873
25. Keane NM, Roberts SG, Almeida CA, Krishnan T, Chopra A, Demaine E, et al. High-avidity, high-IFNgamma-producing CD8 T-cell responses following immune selection during HIV-1 infection. Immunol Cell Biol (2012) 90:224–34. doi:10.1038/icb.2011.34
26. Dudley ME, Nishimura MI, Holt AK, Rosenberg SA. Antitumor immunization with a minimal peptide epitope (G9-209-2M) leads to a functionally heterogeneous CTL response. J Immunother (1999) 22:288–98. doi:10.1097/00002371-199907000-00002
27. Dutoit V, Rubio-Godoy V, Dietrich PY, Quiqueres AL, Schnuriger V, Rimoldi D, et al. Heterogeneous T-cell response to MAGE-A10(254-262): high avidity-specific cytolytic T lymphocytes show superior antitumor activity. Cancer Res (2001) 61:5850–6.
28. Zeh HJ III, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol (1999) 162:989–94.
29. Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature (2009) 458:211–4. doi:10.1038/nature07657
30. Gallimore A, Dumrese T, Hengartner H, Zinkernagel RM, Rammensee HG. Protective immunity does not correlate with the hierarchy of virus-specific cytotoxic T cell responses to naturally processed peptides. J Exp Med (1998) 187:1647–57. doi:10.1084/jem.187.10.1647-b
31. Rosenberg SA, Dudley ME, Restifo NP. Cancer immunotherapy. N Engl J Med (2008) 359:1072. doi:10.1056/NEJMc081511
32. Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell (2013) 23:516–26. doi:10.1016/j.ccr.2013.03.018
33. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, et al. Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother (2009) 32:415–23. doi:10.1097/CJI.0b013e31819c8bda
34. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol (2008) 26:5233–9. doi:10.1200/JCO.2008.16.5449
35. Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res (2011) 17:5343–52. doi:10.1158/1078-0432.CCR-11-0503
36. Schwartzentruber DJ, Hom SS, Dadmarz R, White DE, Yannelli JR, Steinberg SM, et al. In vitro predictors of therapeutic response in melanoma patients receiving tumor-infiltrating lymphocytes and interleukin-2. J Clin Oncol (1994) 12:1475–83.
37. Zehn D, King C, Bevan MJ, Palmer E. TCR signaling requirements for activating T cells and for generating memory. Cell Mol Life Sci (2012) 69:1565–75. doi:10.1007/s00018-012-0965-x
38. Corse E, Gottschalk RA, Allison JP. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J Immunol (2011) 186:5039–45. doi:10.4049/jimmunol.1003650
39. Vigano S, Utzschneider DT, Perreau M, Pantaleo G, Zehn D, Harari A. Functional avidity: a measure to predict the efficacy of effector T cells? Clin Dev Immunol (2012) 2012:153863. doi:10.1155/2012/153863
40. Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology (2009) 126:165–76. doi:10.1111/j.1365-2567.2008.03015.x
41. Gannon PO, Wieckowski S, Baumgaertner P, Hebeisen M, Allard M, Speiser DE, et al. Quantitative TCR:pMHC dissociation rate assessment by NTAmers reveals antimelanoma T cell repertoires enriched for high functional competence. J Immunol (2015) 195(1):356–66. doi:10.4049/jimmunol.1403145
42. Hebeisen M, Schmidt J, Guillaume P, Baumgaertner P, Speiser DE, Luescher I, et al. Identification of rare high-avidity, tumor-reactive CD8+ T cells by monomeric TCR-ligand off-rates measurements on living cells. Cancer Res (2015) 75:1983–91. doi:10.1158/0008-5472.CAN-14-3516
43. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol (2012) 42:3174–9. doi:10.1002/eji.201242606
44. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi:10.1038/nri3667
45. Palmer E, Naeher D. Affinity threshold for thymic selection through a T-cell receptor-co-receptor zipper. Nat Rev Immunol (2009) 9:207–13. doi:10.1038/nri2469
46. Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, et al. Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol (1998) 16:523–44. doi:10.1146/annurev.immunol.16.1.523
47. Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol (2006) 24:175–208. doi:10.1146/annurev.immunol.24.021605.090733
48. Romero P, Valmori D, Pittet MJ, Zippelius A, Rimoldi D, Levy F, et al. Antigenicity and immunogenicity of Melan-A/MART-1 derived peptides as targets for tumor reactive CTL in human melanoma. Immunol Rev (2002) 188:81–96. doi:10.1034/j.1600-065X.2002.18808.x
49. Kyewski B, Klein L. A central role for central tolerance. Annu Rev Immunol (2006) 24:571–606. doi:10.1146/annurev.immunol.23.021704.115601
50. Kurts C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J Exp Med (1997) 186:239–45. doi:10.1084/jem.186.2.239
51. McMahan RH, Slansky JE. Mobilizing the low-avidity T cell repertoire to kill tumors. Semin Cancer Biol (2007) 17:317–29. doi:10.1016/j.semcancer.2007.06.006
52. Turner MJ, Jellison ER, Lingenheld EG, Puddington L, Lefrancois L. Avidity maturation of memory CD8 T cells is limited by self-antigen expression. J Exp Med (2008) 205:1859–68. doi:10.1084/jem.20072390
53. Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity (2006) 25:261–70. doi:10.1016/j.immuni.2006.06.009
54. Chlewicki LK, Holler PD, Monti BC, Clutter MR, Kranz DM. High-affinity, peptide-specific T cell receptors can be generated by mutations in CDR1, CDR2 or CDR3. J Mol Biol (2005) 346:223–39. doi:10.1016/j.jmb.2004.11.057
55. Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A (2000) 97:5387–92. doi:10.1073/pnas.080078297
56. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol (2005) 23:349–54. doi:10.1038/nbt1070
57. Dunn SM, Rizkallah PJ, Baston E, Mahon T, Cameron B, Moysey R, et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci (2006) 15:710–21. doi:10.1110/ps.051936406
58. Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat Immunol (2003) 4:55–62. doi:10.1038/ni863
59. Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol (2008) 180:6116–31. doi:10.4049/jimmunol.180.9.6116
60. Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol (2007) 179:5845–54. doi:10.4049/jimmunol.179.9.5845
61. Haidar JN, Pierce B, Yu Y, Tong W, Li M, Weng Z. Structure-based design of a T-cell receptor leads to nearly 100-fold improvement in binding affinity for pepMHC. Proteins (2009) 74:948–60. doi:10.1002/prot.22203
62. Malecek K, Grigoryan A, Zhong S, Gu WJ, Johnson LA, Rosenberg SA, et al. Specific increase in potency via structure-based design of a TCR. J Immunol (2014) 193:2587–99. doi:10.4049/jimmunol.1302344
63. Zoete V, Irving MB, Michielin O. MM-GBSA binding free energy decomposition and T cell receptor engineering. J Mol Recognit (2010) 23:142–52. doi:10.1002/jmr.1005
64. Zoete V, Irving M, Ferber M, Cuendet MA, Michielin O. Structure-based, rational design of T cell receptors. Front Immunol (2013) 4:268. doi:10.3389/fimmu.2013.00268
65. Irving M, Zoete V, Hebeisen M, Schmid D, Baumgartner P, Guillaume P, et al. Interplay between T cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8+ T cell responsiveness. J Biol Chem (2012) 287:23068–78. doi:10.1074/jbc.M112.357673
66. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol (2010) 184:4936–46. doi:10.4049/jimmunol.1000173
67. Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, et al. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest (2013) 123:1044–56. doi:10.1172/JCI65325
68. Alam SM, Davies GM, Lin CM, Zal T, Nasholds W, Jameson SC, et al. Qualitative and quantitative differences in T cell receptor binding of agonist and antagonist ligands. Immunity (1999) 10:227–37. doi:10.1016/S1074-7613(00)80023-0
69. Carreno LJ, Bueno SM, Bull P, Nathenson SG, Kalergis AM. The half-life of the T-cell receptor/peptide-major histocompatibility complex interaction can modulate T-cell activation in response to bacterial challenge. Immunology (2007) 121:227–37. doi:10.1111/j.1365-2567.2007.02561.x
70. Malherbe L, Hausl C, Teyton L, McHeyzer-Williams MG. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity (2004) 21:669–79. doi:10.1016/j.immuni.2004.09.008
71. Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, et al. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity (2001) 15:59–70. doi:10.1016/S1074-7613(01)00173-X
72. Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity (1996) 4:565–71. doi:10.1016/S1074-7613(00)80483-5
73. Aleksic M, Dushek O, Zhang H, Shenderov E, Chen JL, Cerundolo V, et al. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity (2010) 32:163–74. doi:10.1016/j.immuni.2009.11.013
74. Boulter JM, Schmitz N, Sewell AK, Godkin AJ, Bachmann MF, Gallimore AM. Potent T cell agonism mediated by a very rapid TCR/pMHC interaction. Eur J Immunol (2007) 37:798–806. doi:10.1002/eji.200636743
75. Carreno LJ, Riquelme EM, Gonzalez PA, Espagnolle N, Riedel CA, Valitutti S, et al. T-cell antagonism by short half-life pMHC ligands can be mediated by an efficient trapping of T-cell polarization toward the APC. Proc Natl Acad Sci U S A (2010) 107:210–5. doi:10.1073/pnas.0911258107
76. Chen JL, Morgan AJ, Stewart-Jones G, Shepherd D, Bossi G, Wooldridge L, et al. Ca2+ release from the endoplasmic reticulum of NY-ESO-1-specific T cells is modulated by the affinity of TCR and by the use of the CD8 coreceptor. J Immunol (2010) 184:1829–39. doi:10.4049/jimmunol.0902103
77. Corse E, Gottschalk RA, Krogsgaard M, Allison JP. Attenuated T cell responses to a high-potency ligand in vivo. PLoS Biol (2010) 8:e1000481. doi:10.1371/journal.pbio.1000481
78. Kalergis AM, Boucheron N, Doucey MA, Palmieri E, Goyarts EC, Vegh Z, et al. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat Immunol (2001) 2:229–34. doi:10.1038/85286
79. Liu B, Zhong S, Malecek K, Johnson LA, Rosenberg SA, Zhu C, et al. 2D TCR-pMHC-CD8 kinetics determines T-cell responses in a self-antigen-specific TCR system. Eur J Immunol (2014) 44:239–50. doi:10.1002/eji.201343774
80. Riquelme E, Carreno LJ, Gonzalez PA, Kalergis AM. The duration of TCR/pMHC interactions regulates CTL effector function and tumor-killing capacity. Eur J Immunol (2009) 39:2259–69. doi:10.1002/eji.200939341
81. Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin Exp Immunol (2015) 180:255–70. doi:10.1111/cei.12570
82. Tian S, Maile R, Collins EJ, Frelinger JA. CD8+ T cell activation is governed by TCR-peptide/MHC affinity, not dissociation rate. J Immunol (2007) 179:2952–60. doi:10.4049/jimmunol.179.5.2952
83. Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A (2013) 110:6973–8. doi:10.1073/pnas.1221609110
84. McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE. Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. J Clin Invest (2006) 116:2543–51.
85. Thomas S, Stauss HJ, Morris EC. Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology (2010) 129:170–7. doi:10.1111/j.1365-2567.2009.03227.x
86. Gonzalez PA, Carreno LJ, Coombs D, Mora JE, Palmieri E, Goldstein B, et al. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc Natl Acad Sci U S A (2005) 102:4824–9. doi:10.1073/pnas.0500922102
87. Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity (2003) 18:255–64. doi:10.1016/S1074-7613(03)00019-0
88. Chervin AS, Stone JD, Soto CM, Engels B, Schreiber H, Roy EJ, et al. Design of T-cell receptor libraries with diverse binding properties to examine adoptive T-cell responses. Gene Ther (2013) 20:634–44. doi:10.1038/gt.2012.80
89. Engels B, Chervin AS, Sant AJ, Kranz DM, Schreiber H. Long-term persistence of CD4(+) but rapid disappearance of CD8(+) T cells expressing an MHC class I-restricted TCR of nanomolar affinity. Mol Ther (2012) 20:652–60. doi:10.1038/mt.2011.286
90. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314:126–9. doi:10.1126/science.1129003
91. Borbulevych OY, Santhanagopolan SM, Hossain M, Baker BM. TCRs used in cancer gene therapy cross-react with MART-1/Melan-A tumor antigens via distinct mechanisms. J Immunol (2011) 187:2453–63. doi:10.4049/jimmunol.1101268
92. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114:535–46. doi:10.1182/blood-2009-03-211714
93. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi:10.1200/JCO.2010.32.2537
94. Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med (2003) 198:569–80. doi:10.1084/jem.20030590
95. Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, et al. Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of t cell-mediated vitiligo. J Exp Med (2000) 192:1637–44. doi:10.1084/jem.192.11.1637
96. Yeh S, Karne NK, Kerkar SP, Heller CK, Palmer DC, Johnson LA, et al. Ocular and systemic autoimmunity after successful tumor-infiltrating lymphocyte immunotherapy for recurrent, metastatic melanoma. Ophthalmology (2009) 116(981–989):e981. doi:10.1016/j.ophtha.2008.12.004
97. Schmitt TM, Aggen DH, Stromnes IM, Dossett ML, Richman SA, Kranz DM, et al. Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood (2013) 122:348–56. doi:10.1182/blood-2013-01-478164
98. Palmer DC, Chan CC, Gattinoni L, Wrzesinski C, Paulos CM, Hinrichs CS, et al. Effective tumor treatment targeting a melanoma/melanocyte-associated antigen triggers severe ocular autoimmunity. Proc Natl Acad Sci U S A (2008) 105:8061–6. doi:10.1073/pnas.0710929105
99. Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S, et al. TCR-engineered T cells meet new challenges to treat solid tumors: choice of antigen, T cell fitness, and sensitization of tumor milieu. Front Immunol (2013) 4:363. doi:10.3389/fimmu.2013.00363
100. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19:747–52. doi:10.1038/nm.3161
101. Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, et al. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci U S A (1995) 92:7976–80. doi:10.1073/pnas.92.17.7976
102. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi:10.1126/science.aaa4971
103. Stone JD, Harris DT, Kranz DM. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: impact on efficacy and toxicity. Curr Opin Immunol (2015) 33:16–22. doi:10.1016/j.coi.2015.01.003
104. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med (2013) 5:197ra103. doi:10.1126/scitranslmed.3006034
105. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi:10.1182/blood-2013-03-490565
106. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi:10.1097/CJI.0b013e3182829903
107. June CH, Maus MV, Plesa G, Johnson LA, Zhao Y, Levine BL, et al. Engineered T cells for cancer therapy. Cancer Immunol Immunother (2014) 63:969–75. doi:10.1007/s00262-014-1568-1
108. Hebeisen M, Oberle SG, Presotto D, Speiser DE, Zehn D, Rufer N. Molecular insights for optimizing T cell receptor specificity against cancer. Front Immunol (2013) 4:154. doi:10.3389/fimmu.2013.00154
109. Slansky JE, Jordan KR. The Goldilocks model for TCR-too much attraction might not be best for vaccine design. PLoS Biol (2010) 8:e1000482. doi:10.1371/journal.pbio.1000482
110. Fivash M, Towler EM, Fisher RJ. BIAcore for macromolecular interaction. Curr Opin Biotechnol (1998) 9:97–101. doi:10.1016/S0958-1669(98)80091-8
111. Bridgeman JS, Sewell AK, Miles JJ, Price DA, Cole DK. Structural and biophysical determinants of alphabeta T-cell antigen recognition. Immunology (2012) 135:9–18. doi:10.1111/j.1365-2567.2011.03515.x
112. Corr M, Slanetz AE, Boyd LF, Jelonek MT, Khilko S, al-Ramadi BK, et al. T cell receptor-MHC class I peptide interactions: affinity, kinetics, and specificity. Science (1994) 265:946–9. doi:10.1126/science.8052850
113. Edwards LJ, Zarnitsyna VI, Hood JD, Evavold BD, Zhu C. Insights into T cell recognition of antigen: significance of two-dimensional kinetic parameters. Front Immunol (2012) 3:86. doi:10.3389/fimmu.2012.00086
114. Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature (2010) 464:932–6. doi:10.1038/nature08944
115. Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature (2010) 463:963–7. doi:10.1038/nature08746
116. Pryshchep S, Zarnitsyna VI, Hong J, Evavold BD, Zhu C. Accumulation of serial forces on TCR and CD8 frequently applied by agonist antigenic peptides embedded in MHC molecules triggers calcium in T cells. J Immunol (2014) 193:68–76. doi:10.4049/jimmunol.1303436
117. Hong J, Persaud SP, Horvath S, Allen PM, Evavold BD, Zhu C. Force-regulated in situ TCR-peptide-bound MHC class II kinetics determine functions of CD4+ T cells. J Immunol (2015) 195:3557–64. doi:10.4049/jimmunol.1501407
118. Casas J, Brzostek J, Zarnitsyna VI, Hong JS, Wei Q, Hoerter JA, et al. Ligand-engaged TCR is triggered by Lck not associated with CD8 coreceptor. Nat Commun (2014) 5:5624. doi:10.1038/ncomms6624
119. Jiang N, Huang J, Edwards LJ, Liu B, Zhang Y, Beal CD, et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity (2011) 34:13–23. doi:10.1016/j.immuni.2010.12.017
120. Chen W, Zhu C. Mechanical regulation of T-cell functions. Immunol Rev (2013) 256:160–76. doi:10.1111/imr.12122
121. Dushek O, van der Merwe PA. An induced rebinding model of antigen discrimination. Trends Immunol (2014) 35:153–8. doi:10.1016/j.it.2014.02.002
122. Schmidt J, Dojcinovic D, Guillaume P, Luescher I. Analysis, isolation, and activation of antigen-specific CD4(+) and CD8(+) T cells by soluble MHC-peptide complexes. Front Immunol (2013) 4:218. doi:10.3389/fimmu.2013.00218
123. Zhu C, Jiang N, Huang J, Zarnitsyna VI, Evavold BD. Insights from in situ analysis of TCR-pMHC recognition: response of an interaction network. Immunol Rev (2013) 251:49–64. doi:10.1111/imr.12016
124. Angelov GS, Guillaume P, Cebecauer M, Bosshard G, Dojcinovic D, Baumgaertner P, et al. Soluble MHC-peptide complexes containing long rigid linkers abolish CTL-mediated cytotoxicity. J Immunol (2006) 176:3356–65. doi:10.4049/jimmunol.176.6.3356
125. Cebecauer M, Guillaume P, Mark S, Michielin O, Boucheron N, Bezard M, et al. CD8+ cytotoxic T lymphocyte activation by soluble major histocompatibility complex-peptide dimers. J Biol Chem (2005) 280:23820–8. doi:10.1074/jbc.M500654200
126. Dolton G, Lissina A, Skowera A, Ladell K, Tungatt K, Jones E, et al. Comparison of peptide-major histocompatibility complex tetramers and dextramers for the identification of antigen-specific T cells. Clin Exp Immunol (2014) 177:47–63. doi:10.1111/cei.12339
127. Kronig H, Hofer K, Conrad H, Guilaume P, Muller J, Schiemann M, et al. Allorestricted T lymphocytes with a high avidity T-cell receptor towards NY-ESO-1 have potent anti-tumor activity. Int J Cancer (2009) 125:649–55. doi:10.1002/ijc.24414
128. Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol (1999) 162:2227–34.
129. Bullock TN, Mullins DW, Colella TA, Engelhard VH. Manipulation of avidity to improve effectiveness of adoptively transferred CD8(+) T cells for melanoma immunotherapy in human MHC class I-transgenic mice. J Immunol (2001) 167:5824–31. doi:10.4049/jimmunol.167.10.5824
130. Hombrink P, Raz Y, Kester MG, de Boer R, Weissbrich B, von dem Borne PA, et al. Mixed functional characteristics correlating with TCR-ligand koff -rate of MHC-tetramer reactive T cells within the naive T-cell repertoire. Eur J Immunol (2013) 43:3038–50. doi:10.1002/eji.201343397
131. Wilde S, Sommermeyer D, Leisegang M, Frankenberger B, Mosetter B, Uckert W, et al. Human antitumor CD8+ T cells producing Th1 polycytokines show superior antigen sensitivity and tumor recognition. J Immunol (2012) 189:598–605. doi:10.4049/jimmunol.1102165
132. Dutoit V, Rubio-Godoy V, Doucey MA, Batard P, Lienard D, Rimoldi D, et al. Functional avidity of tumor antigen-specific CTL recognition directly correlates with the stability of MHC/peptide multimer binding to TCR. J Immunol (2002) 168:1167–71. doi:10.4049/jimmunol.168.3.1167
133. Laugel B, van den Berg HA, Gostick E, Cole DK, Wooldridge L, Boulter J, et al. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J Biol Chem (2007) 282:23799–810. doi:10.1074/jbc.M700976200
134. Wang XL, Altman JD. Caveats in the design of MHC class I tetramer/antigen-specific T lymphocytes dissociation assays. J Immunol Methods (2003) 280:25–35. doi:10.1016/S0022-1759(03)00079-6
135. Knabel M, Franz TJ, Schiemann M, Wulf A, Villmow B, Schmidt B, et al. Reversible MHC multimer staining for functional isolation of T-cell populations and effective adoptive transfer. Nat Med (2002) 8:631–7. doi:10.1038/nm0602-631
136. Neudorfer J, Schmidt B, Huster KM, Anderl F, Schiemann M, Holzapfel G, et al. Reversible HLA multimers (Streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor- and virus-derived antigens. J Immunol Methods (2007) 320:119–31. doi:10.1016/j.jim.2007.01.001
137. Guillaume P, Baumgaertner P, Angelov GS, Speiser D, Luescher IF. Fluorescence-activated cell sorting and cloning of bona fide CD8+ CTL with reversible MHC-peptide and antibody Fab’ conjugates. J Immunol (2006) 177:3903–12. doi:10.4049/jimmunol.177.6.3903
138. Schmidt J, Guillaume P, Irving M, Baumgaertner P, Speiser D, Luescher IF. Reversible major histocompatibility complex I-peptide multimers containing Ni(2+)-nitrilotriacetic acid peptides and histidine tags improve analysis and sorting of CD8(+) T cells. J Biol Chem (2011) 286:41723–35. doi:10.1074/jbc.M111.283127
139. Nauerth M, Weissbrich B, Knall R, Franz T, Dossinger G, Bet J, et al. TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci Transl Med (2013) 5:192ra187. doi:10.1126/scitranslmed.3005958
140. Nauerth M, Weissbrich B, Busch DH. The clinical potential for koff-rate measurement in adoptive immunotherapy. Expert Rev Clin Immunol (2013) 9:1151–3. doi:10.1586/1744666X.2013.855609
141. Artyomov MN, Lis M, Devadas S, Davis MM, Chakraborty AK. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc Natl Acad Sci U S A (2010) 107:16916–21. doi:10.1073/pnas.1010568107
142. Salter RD, Benjamin RJ, Wesley PK, Buxton SE, Garrett TP, Clayberger C, et al. A binding site for the T-cell co-receptor CD8 on the alpha 3 domain of HLA-A2. Nature (1990) 345:41–6. doi:10.1038/345041a0
143. Wooldridge L, van den Berg HA, Glick M, Gostick E, Laugel B, Hutchinson SL, et al. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor-antigen complexes at the cell surface. J Biol Chem (2005) 280:27491–501. doi:10.1074/jbc.M500555200
144. Cole DK, Laugel B, Clement M, Price DA, Wooldridge L, Sewell AK. The molecular determinants of CD8 co-receptor function. Immunology (2012) 137:139–48. doi:10.1111/j.1365-2567.2012.03625.x
145. Kersh GJ, Kersh EN, Fremont DH, Allen PM. High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity (1998) 9:817–26. doi:10.1016/S1074-7613(00)80647-0
146. Krogsgaard M, Davis MM. How T cells ‘see’ antigen. Nat Immunol (2005) 6:239–45. doi:10.1038/ni1173
147. Matsui K, Boniface JJ, Steffner P, Reay PA, Davis MM. Kinetics of T-cell receptor binding to peptide/I-Ek complexes: correlation of the dissociation rate with T-cell responsiveness. Proc Natl Acad Sci U S A (1994) 91:12862–6. doi:10.1073/pnas.91.26.12862
148. McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci U S A (1995) 92:5042–6. doi:10.1073/pnas.92.11.5042
149. Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature (1995) 375:148–51. doi:10.1038/375148a0
150. Govern CC, Paczosa MK, Chakraborty AK, Huseby ES. Fast on-rates allow short dwell time ligands to activate T cells. Proc Natl Acad Sci U S A (2010) 107:8724–9. doi:10.1073/pnas.1000966107
151. Lovgren T, Baumgaertner P, Wieckowski S, Devevre E, Guillaume P, Luescher I, et al. Enhanced cytotoxicity and decreased CD8 dependence of human cancer-specific cytotoxic T lymphocytes after vaccination with low peptide dose. Cancer Immunol Immunother (2012) 61:817–26. doi:10.1007/s00262-011-1140-1
152. Speiser DE, Wieckowski S, Gupta B, Iancu EM, Baumgaertner P, Baitsch L, et al. Single cell analysis reveals similar functional competence of dominant and nondominant CD8 T-cell clonotypes. Proc Natl Acad Sci U S A (2011) 108:15318–23. doi:10.1073/pnas.1105419108
153. Au-Yeung BB, Zikherman J, Mueller JL, Ashouri JF, Matloubian M, Cheng DA, et al. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc Natl Acad Sci U S A (2014) 111:E3679–88. doi:10.1073/pnas.1413726111
154. Bachmann MF, Sebzda E, Kundig TM, Shahinian A, Speiser DE, Mak TW, et al. T cell responses are governed by avidity and co-stimulatory thresholds. Eur J Immunol (1996) 26:2017–22. doi:10.1002/eji.1830260908
155. Faroudi M, Utzny C, Salio M, Cerundolo V, Guiraud M, Muller S, et al. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A (2003) 100:14145–50. doi:10.1073/pnas.2334336100
156. Fonteneau JF, Le Drean E, Le Guiner S, Gervois N, Diez E, Jotereau F. Heterogeneity of biologic responses of melanoma-specific CTL. J Immunol (1997) 159:2831–9.
157. Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, et al. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol (2013) 14:262–70. doi:10.1038/ni.2538
158. Valitutti S, Muller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med (1996) 183:1917–21. doi:10.1084/jem.183.4.1917
159. Wiedemann A, Depoil D, Faroudi M, Valitutti S. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc Natl Acad Sci U S A (2006) 103:10985–90. doi:10.1073/pnas.0600651103
160. La Gruta NL, Doherty PC, Turner SJ. A correlation between function and selected measures of T cell avidity in influenza virus-specific CD8+ T cell responses. Eur J Immunol (2006) 36:2951–9. doi:10.1002/eji.200636390
161. Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med (1999) 189:701–10. doi:10.1084/jem.189.4.701
162. Cukalac T, Chadderton J, Handel A, Doherty PC, Turner SJ, Thomas PG, et al. Reproducible selection of high avidity CD8+ T-cell clones following secondary acute virus infection. Proc Natl Acad Sci U S A (2014) 111:1485–90. doi:10.1073/pnas.1323736111
163. Dzutsev AH, Belyakov IM, Isakov DV, Margulies DH, Berzofsky JA. Avidity of CD8 T cells sharpens immunodominance. Int Immunol (2007) 19:497–507. doi:10.1093/intimm/dxm016
164. Liu J, Cao S, Peppers G, Kim SH, Graham BS. Clonotype-specific avidity influences the dynamics and hierarchy of virus-specific regulatory and effector CD4(+) T-cell responses. Eur J Immunol (2014) 44:1058–68. doi:10.1002/eji.201343766
165. Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity (1999) 10:485–92. doi:10.1016/S1074-7613(00)80048-5
166. Price DA, Brenchley JM, Ruff LE, Betts MR, Hill BJ, Roederer M, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med (2005) 202:1349–61. doi:10.1084/jem.20051357
167. Trautmann L, Rimbert M, Echasserieau K, Saulquin X, Neveu B, Dechanet J, et al. Selection of T cell clones expressing high-affinity public TCRs within human cytomegalovirus-specific CD8 T cell responses. J Immunol (2005) 175:6123–32. doi:10.4049/jimmunol.175.9.6123
168. Derby M, Alexander-Miller M, Tse R, Berzofsky J. High-avidity CTL exploit two complementary mechanisms to provide better protection against viral infection than low-avidity CTL. J Immunol (2001) 166:1690–7. doi:10.4049/jimmunol.166.3.1690
169. Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, et al. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med (2000) 192:1105–13. doi:10.1084/jem.192.8.1105
170. Turula H, Smith CJ, Grey F, Zurbach KA, Snyder CM. Competition between T cells maintains clonal dominance during memory inflation induced by MCMV. Eur J Immunol (2013) 43:1252–63. doi:10.1002/eji.201242940
171. Brentville VA, Metheringham RL, Gunn B, Durrant LG. High avidity cytotoxic T lymphocytes can be selected into the memory pool but they are exquisitely sensitive to functional impairment. PLoS One (2012) 7:e41112. doi:10.1371/journal.pone.0041112
172. Vigano S, Bellutti Enders F, Miconnet I, Cellerai C, Savoye AL, Rozot V, et al. Rapid perturbation in viremia levels drives increases in functional avidity of HIV-specific CD8 T cells. PLoS Pathog (2013) 9:e1003423. doi:10.1371/journal.ppat.1003423
173. Black CM, Armstrong TD, Jaffee EM. Apoptosis-regulated low-avidity cancer-specific CD8(+) T cells can be rescued to eliminate HER2/neu-expressing tumors by costimulatory agonists in tolerized mice. Cancer Immunol Res (2014) 2:307–19. doi:10.1158/2326-6066.CIR-13-0145
174. Bos R, Marquardt KL, Cheung J, Sherman LA. Functional differences between low- and high-affinity CD8(+) T cells in the tumor environment. Oncoimmunology (2012) 1:1239–47. doi:10.4161/onci.21285
175. Janicki CN, Jenkinson SR, Williams NA, Morgan DJ. Loss of CTL function among high-avidity tumor-specific CD8+ T cells following tumor infiltration. Cancer Res (2008) 68:2993–3000. doi:10.1158/0008-5472.CAN-07-5008
176. Weiss VL, Lee TH, Song H, Kouo TS, Black CM, Sgouros G, et al. Trafficking of high avidity HER-2/neu-specific T cells into HER-2/neu-expressing tumors after depletion of effector/memory-like regulatory T cells. PLoS One (2012) 7:e31962. doi:10.1371/journal.pone.0031962
Keywords: melanoma, immunotherapy, cytotoxic T cells, TCR affinity, TCR structural avidity, tumor antigens, T cell functionality, NTAmers
Citation: Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE and Rufer N (2015) Identifying Individual T Cell Receptors of Optimal Avidity for Tumor Antigens. Front. Immunol. 6:582. doi: 10.3389/fimmu.2015.00582
Received: 03 September 2015; Accepted: 30 October 2015;
Published: 18 November 2015
Edited by:
Eric Huseby, University of Massachusetts Medical School, USAReviewed by:
Salvatore Valitutti, INSERM, FranceViktor Umansky, German Cancer Research Center, Germany
Copyright: © 2015 Hebeisen, Allard, Gannon, Schmidt, Speiser and Rufer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathalie Rufer, nathalie.rufer@unil.ch
†Mathilde Allard and Philippe Gannon have contributed equally to this work.