 Esther J. Nossent1,2*
Esther J. Nossent1,2* Alex R. Schuurman3,4
Alex R. Schuurman3,4 Tom D.Y. Reijnders3,4Anno Saris3,4
Tom D.Y. Reijnders3,4Anno Saris3,4 Ilse Jongerius5,6Siebe G. Blok1Heder de Vries2,7
Ilse Jongerius5,6Siebe G. Blok1Heder de Vries2,7 JanWillem Duitman3,4Anton Vonk Noordegraaf1,2Lilian J. Meijboom2,8
JanWillem Duitman3,4Anton Vonk Noordegraaf1,2Lilian J. Meijboom2,8 René Lutter1,4,9Leo Heunks2,7
René Lutter1,4,9Leo Heunks2,7 Harm Jan Bogaard1,2
Harm Jan Bogaard1,2 Tom van der Poll3,4,10 on behalf of the ArtDECO consortium and the Amsterdam UMC COVID study group
Tom van der Poll3,4,10 on behalf of the ArtDECO consortium and the Amsterdam UMC COVID study group- 1Department of Pulmonary Medicine, Amsterdam UMC, Free University Amsterdam, Amsterdam, Netherlands
- 2Amsterdam Cardiovascular Sciences Research Institute, Amsterdam UMC, Amsterdam, Netherlands
- 3Center for Experimental and Molecular Medicine, Amsterdam UMC, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 4Amsterdam Infection & Immunity Institute, Amsterdam UMC, Amsterdam, Netherlands
- 5Department of Immunopathology, Sanquin Research, Amsterdam and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 6Emma Children’s Hospital, Department of Pediatric Immunology, Rheumatology and Infectious Diseases, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 7Department of Intensive Care Medicine, Amsterdam UMC, Free University Amsterdam, Amsterdam, Netherlands
- 8Department of Radiology and Nuclear Medicine, Amsterdam UMC, Free University Amsterdam, Amsterdam, Netherlands
- 9Department of Experimental Immunology, Amsterdam UMC, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 10Department of Infectious Diseases, Amsterdam UMC, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
Rationale: Systemic activation of procoagulant and inflammatory mechanisms has been implicated in the pathogenesis of COVID-19. Knowledge of activation of these host response pathways in the lung compartment of COVID-19 patients is limited.
Objectives: To evaluate local and systemic activation of coagulation and interconnected inflammatory responses in critically ill COVID-19 patients with persistent acute respiratory distress syndrome.
Methods: Paired bronchoalveolar lavage fluid and plasma samples were obtained from 17 patients with COVID-19 related persistent acute respiratory distress syndrome (mechanical ventilation > 7 days) 1 and 2 weeks after start mechanical ventilation and compared with 8 healthy controls. Thirty-four host response biomarkers stratified into five functional domains (coagulation, complement system, cytokines, chemokines and growth factors) were measured.
Measurements and Main Results: In all patients, all functional domains were activated, especially in the bronchoalveolar compartment, with significantly increased levels of D-dimers, thrombin-antithrombin complexes, soluble tissue factor, C1-inhibitor antigen and activity levels, tissue type plasminogen activator, plasminogen activator inhibitor type I, soluble CD40 ligand and soluble P-selectin (coagulation), next to activation of C3bc and C4bc (complement) and multiple interrelated cytokines, chemokines and growth factors. In 10 patients in whom follow-up samples were obtained between 3 and 4 weeks after start mechanical ventilation many bronchoalveolar and plasma host response biomarkers had declined.
Conclusions: Critically ill, ventilated patients with COVID-19 show strong responses relating to coagulation, the complement system, cytokines, chemokines and growth factors in the bronchoalveolar compartment. These results suggest a local pulmonary rather than a systemic procoagulant and inflammatory “storm” in severe COVID-19.
Introduction
The severe acute respiratory syndrome corona virus (SARS-CoV)-2 pandemic has had a tremendous global impact. While most SARS-CoV-2 infections are mild, up to 20% of cases result in severe Coronavirus Disease (COVID)-19, particularly in the elderly and in patients with cardiopulmonary comorbidities (1). Severe COVID-19 is associated with respiratory failure, which has been considered a form of acute respiratory distress syndrome (ARDS), although its characteristics may differ from ARDS caused by other diseases (2, 3). Another distinctive feature of COVID-19 is the frequent occurrence of venous thrombo-embolic (VTE) events, which may be related to hypercoagulability (4, 5). These particularities of COVID-19 have raised considerable interest in the interactions between coagulation and the immune response triggered by this new disease.
The innate immune system is the first to respond to infection of the airways by SARS-CoV-2 (6). The respiratory epithelium and resident leukocytes release cytokines and chemokines that trigger recruitment of other immune cells to the primary site of infection. While this response is initiated to inhibit viral replication, unrestrained activation of inflammation can result in collateral tissue damage, which has been documented in ARDS associated with other types of lung infection (7). In addition, ARDS and pneumonia can result in aberrant activation of coagulation in the lung microenvironment. While this pulmonary coagulopathy is largely inflammation-driven, coagulation proteases in turn can amplify inflammation, resulting in an injurious vicious cycle (8–10).
Previous investigations have reported exuberant activation of the innate immune and coagulation systems in the systemic circulation of patients with COVID-19 (4, 11–14). Knowledge on local activation of these host response pathways in ventilated COVID-19 is limited. Autopsy studies have shown extensive alveolar damage accompanied by widespread inflammation and pulmonary in situ thrombosis in patients who succumbed to COVID-19 (2, 15). Recently, it was proposed that SARS-CoV-2 infection induces a process termed immunothrombosis, in which activated leukocytes interact with platelets and coagulation factors, leading to intravascular clot formation and microthrombotic complications in lungs and other organs (16). Here we set out to evaluate local and systemic activation of coagulation and interconnected inflammatory responses in critically ill patients with COVID-19 by measuring a large set of biomarkers in bronchoalveolar lavage fluid (BALF) and concurrently collected plasma. We hypothesized that severe COVID-19 would be associated with strong activation of coagulation especially locally in the lungs and that this would be associated with concurrent activation of host response pathways implicated in coagulation and lung injury. To test this we composed a set of 34 host response biomarkers reflection alterations in five pathophysiological domains, i.e., coagulation, the complement system, cytokines, chemokines and growth factors.
Methods
Study Design
This study was part of the Amsterdam Study for Deep Phenotyping of COVID-19 disease (ArtDECO) 1 study, a cohort study of all patients with PCR confirmed COVID-19 related persistent ARDS (mechanical ventilation > 7 days) admitted to the intensive care unit (ICU) of the Amsterdam University Medical Centers (Amsterdam UMC), location VUmc. ARDS was defined according to the Berlin criteria (17). Per clinical protocol all patients requiring more than 7 days of mechanical ventilation underwent video-assisted bronchoscopy BALF sampling. Left-over biological samples and clinical data were stored in the anonymized research Amsterdam UMC COVID-19 biobank (#2020-182) and database (Castor; castoredc.com). Informed consent for the use of samples and data was deferred until discharge from the ICU. In case of death, informed consent was requested from the patient’s relatives. The study procedure was approved by the Review Committee of the Amsterdam UMC Biobank (protocol number 2020-065). The study was performed in accordance with the declaration of Helsinki and adheres to Dutch regulations. The current investigation included patients from whom data and samples were available between March 27th and May 31st 2020. All available paired BALF-plasma samples harvested between 1 and 2 weeks after start invasive mechanical ventilation were used for this analysis; from a subset of patients obtained paired follow up samples were also analyzed. Techniques concerning blood and BALF sampling, and used assays, are described in the detail in the online data supplement. Biological samples were compared with samples of 8 healthy subjects (5/8 male, mean age 38.75 years, 1/8 ex-smoker) from whom BALF (8 x 20 ml 0,9% NaCl) and plasma was obtained as part of study protocols NL48912.018.14 (RILCA trial) and NL53354.018.15 (RILCO trial) approved by the institutional ethics committee after having given written informed consent.
Clinical Protocol
Per clinical protocol, all patients with PCR confirmed COVID-19 related persistent ARDS requiring more than 7 days of mechanical ventilation, admitted to the intensive care unit (ICU) of the Amsterdam University Medical Centers (Amsterdam UMC), location VUmc, underwent chest computed tomography (CT) without and with (when clinically indicated) intravenous contrast (CT pulmonary angiography (CTPA)) at fixed time-points. At the same time, respiratory mechanics were measured and video-assisted bronchoscopy BALF sampling was performed. These procedures were repeated on a weekly basis for as long as patients were intubated and did not show clinical improvement. Intravenous steroid treatment with 1mg/kg prednisone, with a maximum dose of 80 mg once a day, was started after fourteen days of mechanical ventilation, in absence of clinical improvement and after exclusion of pulmonary infectious complications. Steroids were tapered after 10 days. When clinically indicated, chest CT and bronchoscopy with BALF sampling were also performed in addition to these fixed time points.
Blood and BALF Sampling
Prior to diagnostic BALF sampling, venous blood was drawn in EDTA anticoagulated tubes. Blood was centrifuged 10 min at 1800g and supernatant plasma was collected and stored at -80°C. During bronchoscopy lungs were instilled with 2 x 20 ml 0,9% NaCl at a (sub)segmental level, each aspirated immediately with low suction for microbiological diagnostic purposes. Leftover BALF (3-20 ml) was centrifuged (300g, 10min, 4°C) and BALF supernatant was stored at -80°C until further analysis.
Assays
BALF was treated with 1% Triton-X100 for 2 hours before samples were used for the specific assays to eliminate all viable virus. D-dimer, soluble tissue factor, tissue type plasminogen activator (tPA), plasminogen activator inhibitor type I (PAI-1), soluble CD40 ligand (sCD40L) and soluble P-selectin (sP- selectin) were measured using Human Thrombosis LEGENDplex™ (#740892, BioLegend, Amsterdam, the Netherlands). Thrombin-antithrombin complexes (TATc) were measured by ELISA (TAT-EIA, Affinity Biologicals, Leiden, the Netherlands). Kallikrein-C1-inhibitor complexes (18) C3bc (19) C4bc (20) mannose binding lectin (MBL) (21) and C1-inhibitor (C1-INH) antigen (22) and activity (22) were measured by assays as described previously. All other mediators and growth factors were measured by Human XL Cytokine Magnetic Luminex Performance Assay (#LKTM014, R&D systems, Abingdon, United Kingdom) and were read on a Bioplex 200.
Statistical Analysis
All results are presented as numbers (percentages) for categorical variables, median and interquartile ranges (IQR, 25th and 75th percentiles) for non-parametric quantitative variables (boxplots) and mean ± standard deviation (SD) for parametric quantitative variables. Differences between groups were tested by Wilcoxon signed-rank test for paired data and Wilcoxon rank sum test for unpaired data. A p value ≤ 0.05 was considered statistically significant. All statistical analyses were performed in R (version 4.0.2; R Foundation for Statistical Computing, Vienna, Austria).
Results
Patients and Analysis of the Local and Systemic Host Response
Seventeen patients from whom paired BALF and plasma samples were harvested in parallel between 1 and 2 weeks after initiation of invasive mechanical ventilation were studied. Clinical characteristics are shown in Table 1. Patient demographics were comparable to previous reports on COVID-19 disease with a higher incidence in elderly, male subjects with an elevated BMI (1, 11, 12, 23). In eleven patients (64.7%) treatment with steroids was initiated. Ten patients started after baseline BALF and plasma sampling and 1 patient just prior to baseline sampling. Seven patients (41.2%) were treated with hydroxychloroquine, 2 patients (11.8%) with the tyrosine kinase inhibitor imatinib. All patients underwent CTPA within 7 days of ICU admission; ten patients (58.8%) were diagnosed with pulmonary embolism, for which therapeutic anticoagulation was initiated. Thirteen patients (76.5%) were treated for a possible secondary infectious complication during their stay in the ICU (Table 2). Four patients died during ICU stay (23.5%).

Table 1 Demographics and clinical characteristics of patients with COVID-19 related persistent acute respiratory distress syndrome at ICU admission.

Table 2 Secondary pulmonary infections during ICU stay.
Activation of procoagulant and immune responses in the bronchoalveolar compartment of critically ill COVID-19 patients were measured using a comprehensive set of biomarkers, stratified into five functional “domains”, i.e., coagulation, the complement system, cytokines, chemokines and growth factors. To obtain insight into the extent of compartmentalization of these responses, the same biomarkers were measured in plasma.
Coagulation Activation
Markers of coagulation activation (D-dimer, thrombin-antithrombin complexes (TATc) and soluble tissue factor) (9, 10) were strongly elevated in BALF of COVID-19 patients relative to levels measured in BALF from control subjects (Figure 1). Of interest, in plasma only D-dimer concentrations were elevated in patients, whereas the plasma levels of TATc and soluble tissue factor were not different from those in healthy controls. Kallikrein-C1-inhibitor complexes, reflecting activation of the contact system (24), was not detectable in BALF from either patients or controls, while plasma levels were not different between groups. C1-inhibitor (C1-INH) antigen and activity levels were elevated in both BALF and plasma from patients. The fibrinolysis markers tissue type plasminogen activator (tPA) and plasminogen activator inhibitor type I (PAI-1 (9) were markedly elevated in BALF and modestly in plasma of patients, when compared to heathy controls. tPA/PAI-1 ratio’s in BALF were not different between patients and controls, whilst in plasma these ratio’s were slightly higher in patients. Soluble CD40 ligand (sCD40L) and soluble P-selectin (sP-selectin), indicative of platelet activation (25), were increased in both BALF and plasma from patients when compared with controls.
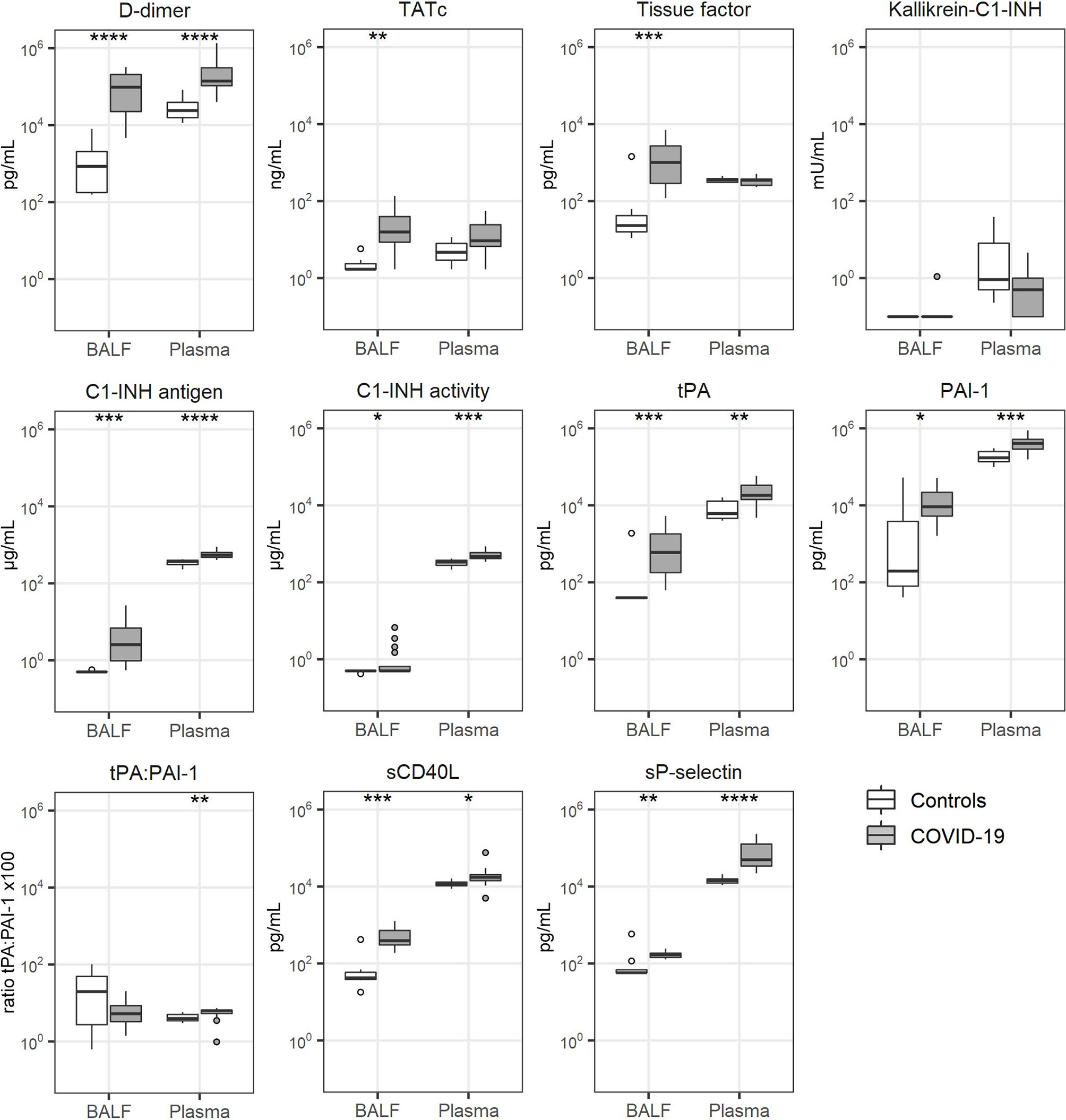
Figure 1 Coagulation activation. Bronchoalveolar lavage fluid and plasma were obtained from 17 critically ill COVID-19 patients who had been on mechanical ventilation for at least 7 days and 8 healthy control subjects. Data are expressed as box and whisker diagrams depicting the median and lower quartile, upper quartile, and their respective 1.5 interquartile range as whiskers (as specified by Tukey). Comparisons between groups were performed using the Wilcoxon rank sum test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p = ≤ 0.0001. BALF, bronchoalveolar lavage fluid; C1-INH, C1-inhibitor; Kallikrein-C1-INH, Kallikrein-C1-inhibitor complexes; PAI-1, plasminogen activator inhibitor type I; sCD40L, soluble CD40 Ligand; sP-selectin, soluble P-selectin; TATc, thrombin-antithrombin complexes; tPA, tissue type plasminogen activator.
Complement Activation
Tight interactions exist between coagulation and the complement system (26). BALF and plasma levels of C3bc (indicative of activation of the common pathway of complement) (27) and C4bc (indicative of activation of the classical and lectin pathways of complement) (27) were strongly increased in COVID-19 patients relative to controls (Figure 2). In contrast, mannose binding lectin (MBL) levels remained undetectable in BALF, while plasma MBL was higher in patients than in controls.
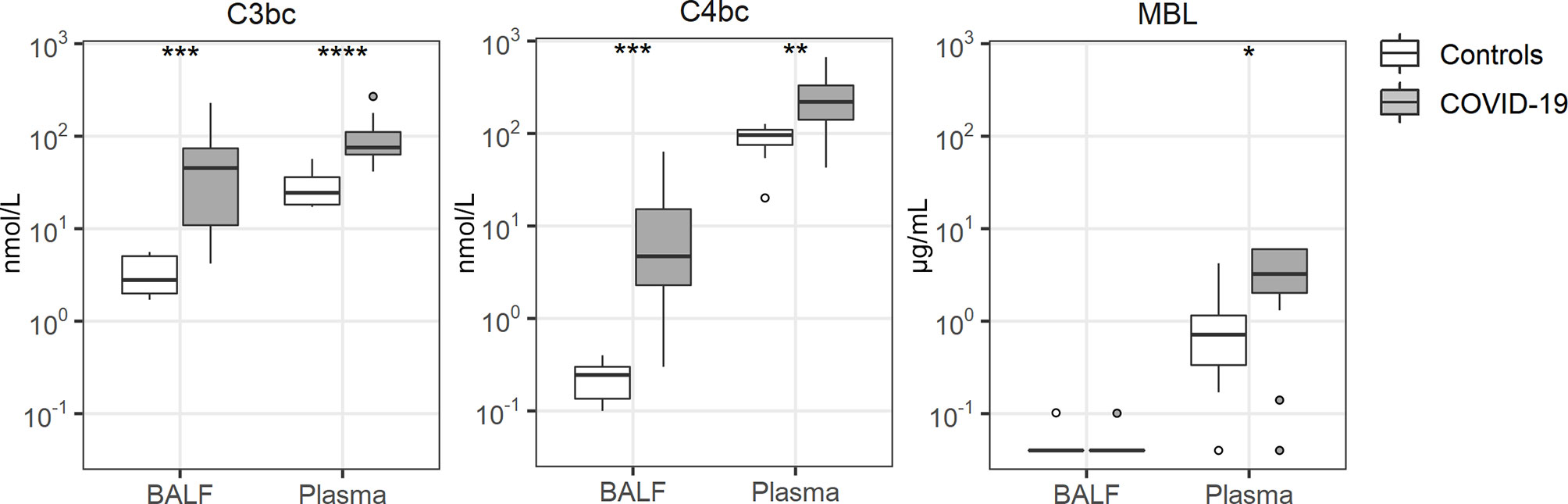
Figure 2 Complement activation. Bronchoalveolar lavage fluid and plasma were obtained from 17 critically ill COVID-19 patients who had been on mechanical ventilation for at least 7 days and 8 healthy control subjects. Data are expressed as box and whisker diagrams depicting the median and lower quartile, upper quartile, and their respective 1.5 interquartile range as whiskers (as specified by Tukey). Comparisons between groups were performed using the Wilcoxon rank sum test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. BALF, bronchoalveolar lavage fluid; C3bc, complement 3bc; C4bc, complement 4bc; MBL, mannose binding lectin.
Cytokine and Chemokine Release
Local cytokine release has been implicated in the pathogenesis of lung injury and pulmonary coagulopathy in patients with ARDS (7, 9) and several investigations reported a “cytokine storm” in patients with COVID-19, referring to elevated plasma concentrations of cytokines and chemokines (28, 29). Concentrations of cytokines were particularly elevated in BALF of patients with COVID-19 (Figure 3). This was true for proinflammatory cytokines (tumor necrosis factor (TNF)-α, interleukin (IL)-1α, IL-1β), anti-inflammatory cytokines (IL-1 receptor antagonist) as well as cytokines with a mixed functional profile (IL-6, IL-10, IL-33). The plasma levels of these cytokines were also higher in patients than controls. Likewise, all 10 chemokines measured, were elevated in BALF of patients when compared with that from controls; in plasma chemokine levels were more modestly (CXCL8, CX3CL, CCL3, CCL4, CCL18, CCL19, CCL20) or not elevated (CXCL1, CXCL2, CCL5) in patients (Figure 4).
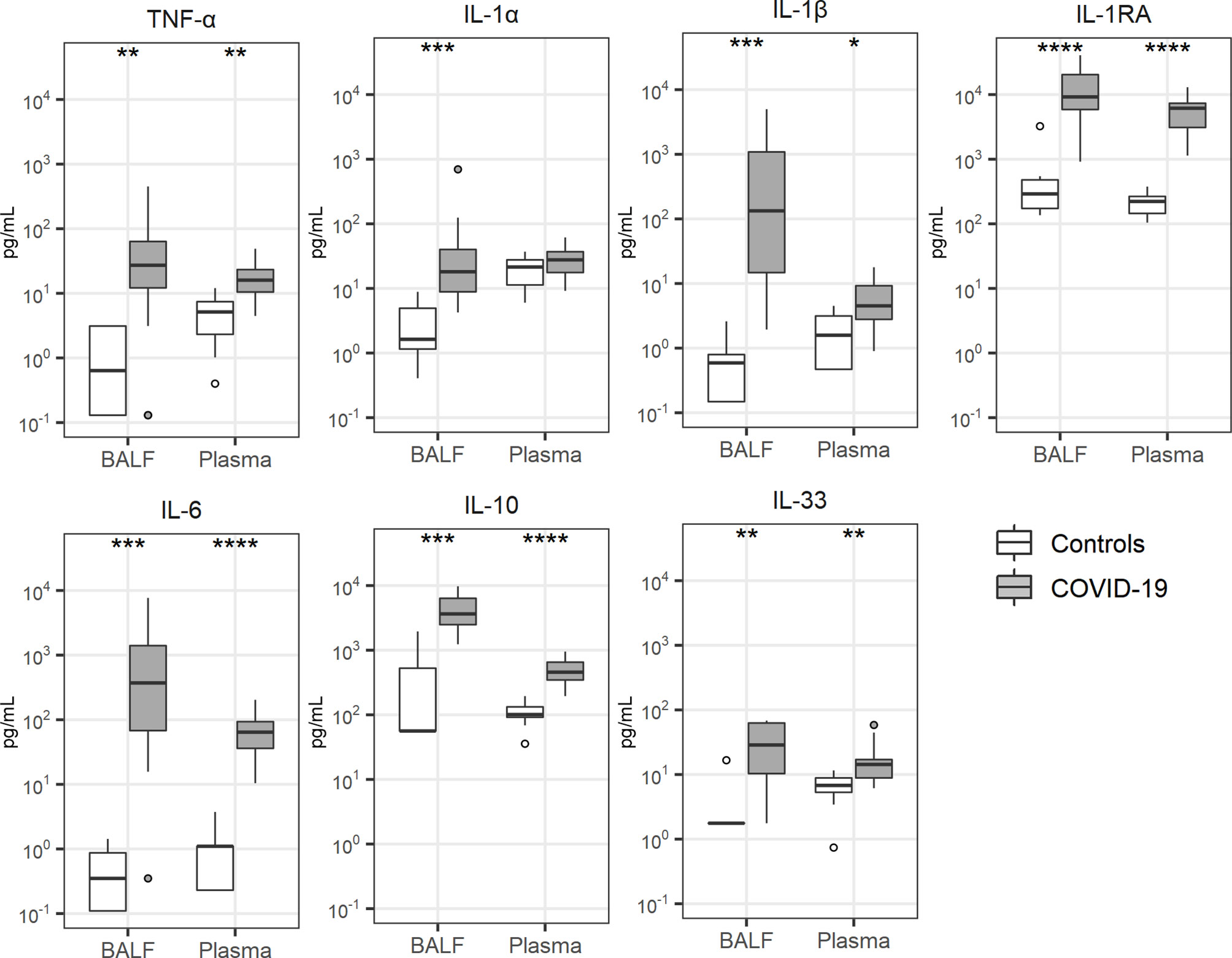
Figure 3 Cytokine release. Bronchoalveolar lavage fluid and plasma were obtained from 17 critically ill COVID-19 patients who had been on mechanical ventilation for at least 7 days and 8 healthy control subjects. Data are expressed as box and whisker diagrams depicting the median and lower quartile, upper quartile, and their respective 1.5 interquartile range as whiskers (as specified by Tukey). Comparisons between groups were performed using the Wilcoxon rank sum test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. BALF, bronchoalveolar lavage fluid; IL, interleukin; IL-1RA, interleukin-1 receptor antagonist; TNF-α, tumor necrosis factor-α.
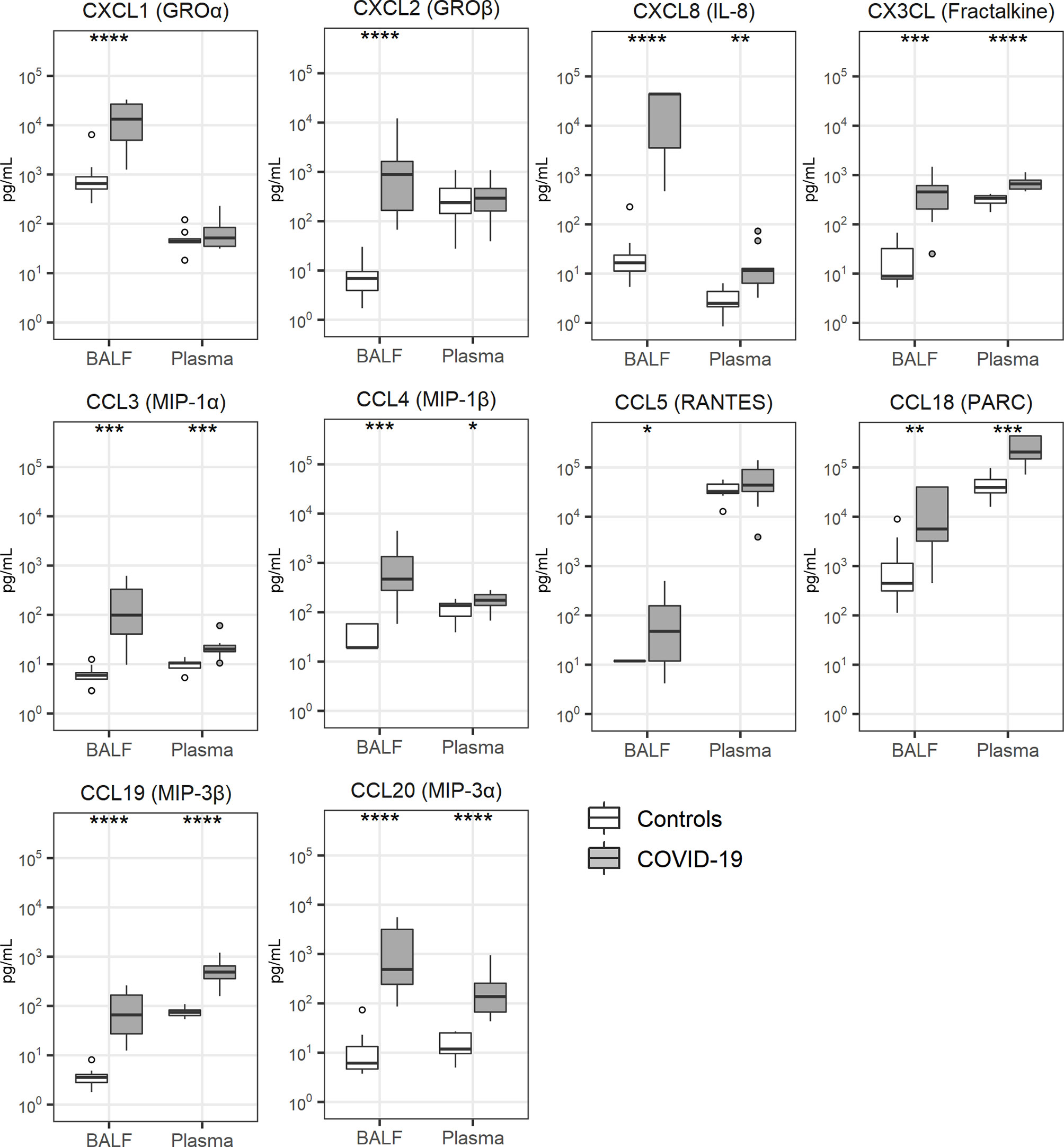
Figure 4 Chemokine release. Bronchoalveolar lavage fluid and plasma were obtained from 17 critically ill COVID-19 patients who had been on mechanical ventilation for at least 7 days and 8 healthy control subjects. Data are expressed as box and whisker diagrams depicting the median and lower quartile, upper quartile, and their respective 1.5 interquartile range as whiskers (as specified by Tukey). Comparisons between groups were performed using the Wilcoxon rank sum test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. BALF, bronchoalveolar lavage fluid; IL, interleukin; MIP, macrophage inflammatory protein; PARC, pulmonary and activation-regulated chemokine; RANTES, Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted.
Growth Factor Release
Several growth factors have been studied in the context of lung inflammation, including vascular endothelial growth factor (VEGF) (30), platelet derived growth factor (PDGF)-AA (31), PDGF-BB (31), fms like tyrosine kinase 3 ligand (FLT3L) (32) and granulocyte-macrophage colony stimulating factor (GM-CSF) (33). Of these, PDGF-AA and PDGF-BB were only elevated in BALF of patients with COVID-19, while FLTL3 and GM-CSF were elevated in BALF more so than in plasma (Figure 5). VEGF was only increased in plasma of patients.
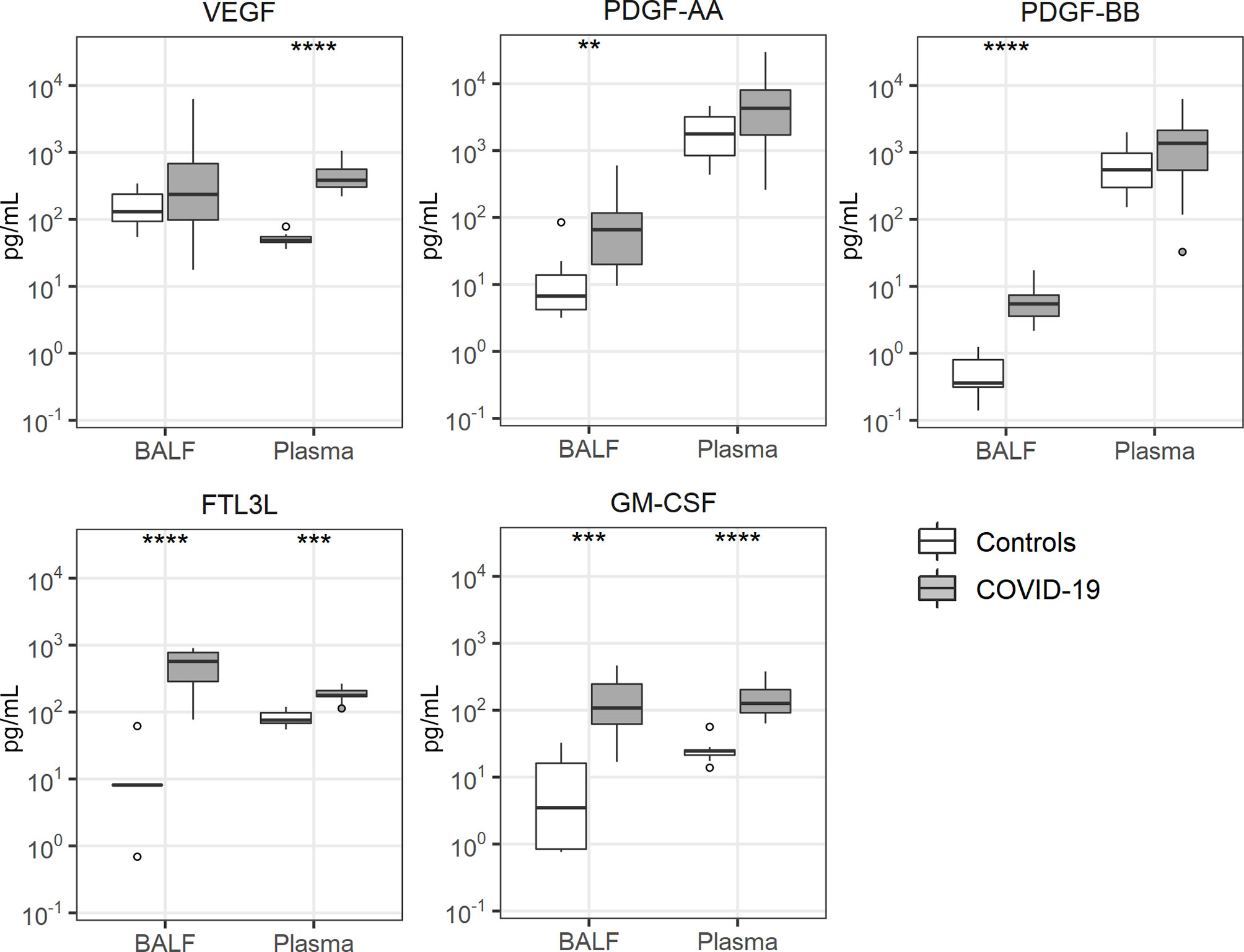
Figure 5 Growth factor release. Bronchoalveolar lavage fluid and plasma were obtained from 17 critically ill COVID-19 patients who had been on mechanical ventilation for at least 7 days and 8 healthy control subjects. Data are expressed as box and whisker diagrams depicting the median and lower quartile, upper quartile, and their respective 1.5 interquartile range as whiskers (as specified by Tukey). Comparisons between groups were performed using the Wilcoxon rank sum test. **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. BALF, bronchoalveolar lavage fluid; FTL3L, fms like tyrosine kinase 3 ligand; GM-CSF, granulocyte-macrophage colony-stimulating factor; PDGF, platelet derived growth factor; VEGF, vascular endothelial growth factor.
Temporal Changes in Local and Systemic Procoagulant and Immune Responses During ICU Stay
From ten patients follow up BALF and plasma samples were obtained 3 or 4 weeks after start invasive mechanical ventilation. Across all five functional domains many host response biomarker levels in BALF showed decreasing trends between week 1-2 and week 3-4, reaching statistical significance for soluble tissue factor, tPA, sCD40L, sP-selectin (coagulation, Supplementary Figure 1), TNF- α, IL-1α, IL-1β (cytokine release, Supplementary Figure 3), CXCL2, CXCL8, CCL3, CCL4 (chemokine release, Supplementary Figure 4), VEGF, PDGF-AA and FTL3L (growth factor release, Supplementary Figure 5). Complement activation products did not change over time in BALF (Supplementary Figure 2). In plasma, significant decreases were detected between week 1-2 and week 3-4 for TATc, PAI-1, IL-1 receptor antagonist, IL-6, IL-10, CXCL8, CCL19, CCL20, VEGF and GM-CSF (Supplementary Figures 6–10). There was no clear relationship between treatment with corticosteroids and changes in biomarker levels between 1-2 and 3-4 weeks (see color codes for absence or presence of corticosteroid treatment in individual patients in Supplementary Figures 1–10).
Differences Between Patients With and Without Pulmonary Embolism
Comparison of host response biomarkers between patients with pulmonary embolism (n = 10) and those without pulmonary embolism (n =7) revealed higher IL-6 plasma levels and in BALF lower C1-inhibitor activity, lower IL-10 and lower GM-CSF in patients with pulmonary embolism; Supplementary Table 1).
Discussion
Here we report an in depth biomarker analysis, both of the bronchoalveolar and systemic compartment, in consecutive ventilated critically ill COVID-19 patients admitted to the ICU who had needed mechanical ventilation for at least 7 days. Since the first description of COVID-19, many studies have documented activation of procoagulant and inflammatory pathways in the systemic circulation (4, 11, 14). We studied local activation of the coagulation system and interconnected inflammatory networks in the bronchoalveolar compartment during ICU stay. By measuring thirty-four host response biomarkers in paired BALF and plasma samples we demonstrate an especially strong response in the bronchoalveolar compartment of COVID-19 patients across five functional domains, i.e., coagulation, complement system, cytokines, chemokines and growth factors.
We show strong activation of coagulation in the respiratory system of COVID-19 patients, as reflected by elevated BALF levels of D-dimer and TATc, and in addition provide indirect evidence for a role for tissue factor herein, as suggested by highly elevated soluble tissue factor concentrations in BALF. Differences in these coagulation markers between patients and controls were much greater in BALF than in plasma, suggesting local activation of coagulation. SARS-CoV-2 may in part promote pulmonary coagulopathy by a direct effect on bronchial epithelial cells via activation of tissue factor signaling and impairment of epithelial anticoagulant mechanism (34). Notably, stronger activation of coagulation locally versus systemically in our ICU/ARDS cohort does not preclude a role for systemic coagulation activation in various thromboembolic and vascular events in COVID-19 in general (35). tPA/PAI-1 ratio’s were not different between groups in BALF and only modestly elevated in plasma of patients, arguing against a strongly disturbed fibrinolyic balance in COVID-19 patients and suggesting that elevated D-dimer levels reflect enhanced coagulation rather than hyperfibrinolysis. Besides via tissue factor-Factor VIIa, the coagulation system can be activated via the intrinsic pathway, which is tightly connected with the kallikrein-kinin system. Dysregulation of the kinin pathway has been suggested to contribute to pulmonary edema in COVID-19 and interventions inhibiting bradykinin activity or formation have been proposed as a potential therapy for COVID-19 (36). Activation of the kallikrein-kinin system in vivo is difficult to measure due to fast clearance of its proteases and protease-C1-inhibitor complexes (22). Thus, our results do not exclude activation of the kallikrein-kinin system, although one might argue that sufficient inhibitory capacity remained available in BALF and plasma, as indicated by elevated C1-inhibitor activity levels in COVID-19 patients.
Aberrant activation of the complement system has been implicated in COVID-19 coagulopathy and associated lung injury (37–41). We here provide evidence for not only activation of the complement system in the circulation, but also in the bronchoalveolar lumen of patients with COVID-19, as reflected by elevated C3bc and C4bc levels in BALF. In agreement, a recent study reported elevated C5a-desarg levels in BALF of four patients with ARDS due to COVID-19 (37). COVID-19 patients had elevated plasma MBL concentrations, suggesting involvement of the lectin pathway; in BALF MBL remained undetectable in both patients and controls. Of note, undetectable MBL levels in BALF of COVID-19 patients in the presence of high plasma MBL concentrations could reflect local activation of MBL, considering that this takes place at cell surfaces. Monocyte-derived macrophages from BALF of COVID-19 patients showed increased ficolin-1 mRNA expression, which may support local activation of the MBL pathway (42). Thus, the main route of complement activation in the lungs of COVID-19 patients remains to be determined. Besides complement products, proinflammatory cytokines such as TNF-α, IL-1α, IL-1β and IL-6 can activate coagulation, primarily via enhancing tissue factor expression (9, 10). We here report elevated concentrations of many proinflammatory cytokines, particularly in BALF, in COVID-19 patients
VEGF is a pluripotent glycoprotein that is constitutively expressed at high levels in the lung, where it may facilitate repair mechanisms following injury by epithelial regeneration (30). We found strongly elevated VEGF plasma levels in COVID-19 patients, while VEGF concentrations in BALF were highly variable and statistically not different from control values. VEGF may reduce vascular barrier function in the lungs and it has been postulated that angiotensin-converting enzyme (ACE)2 can antagonize this VEGF effect (43). Since SARS-CoV-2 cell invasion lowers expression of ACE2 (44), a possible detrimental role of VEGF vascular permeability could be enhanced during COVID-19; clearly, this hypothesis needs conformation in experimental settings. Elevated PDGF levels in BALF of COVID-19 patients may reflect activation of multiple cell types, including platelets, mast cells and the epithelium (31). Of interest, thrombin can induce PDGF release by human lung epithelial cells (45), pointing at a possible interaction between coagulation and PDGF production in the airways. Thus far, PDGF expression in ARDS or pneumonia has not been studied and its role in acute lung inflammatory conditions is speculative. Experimental studies have indicated that FLT3L may exert strong effects in the lung compartment. Pretreatment of mice with FLT3L increased lung injury during pneumococcal pneumonia, likely through inducing accumulation of proinflammatory dendritic cells (32), and pharmacological inhibition of FLT3 signaling attenuated LPS-induced lung injury and edema in mice (46). These studies suggest that the strongly elevated FLT3 levels in BALF of COVID-19 patients may contribute to lung injury.
Current knowledge of local activation of inflammatory mechanisms in the airways of patients with COVID-19 is limited. In severe COVID-19 lung macrophages displayed high expression of IL-1β, IL-6, TNF-α and various chemokines (CCL2, CCL3, CCL4, CCL7, CXCL9, CXCL10 and CXCL11). These patients had high IL-1β, IL-6 and IL-8 protein levels in BALF (42), a finding that was reproduced in a study entailing four patients with COVID-19 associated ARDS (37). We here expand these earlier reports to protein level and to procoagulant and inflammatory systems implicated in lung injury. We did not find evidence for a systemic “cytokine storm” in COVID-19 as plasma cytokine levels did not exceed 100 pg/mL. In line with previous reports (47, 48) our results show higher cytokine levels in the bronchoalveolar compartment suggesting an ongoing local hyperinflammatory state in severe COVID-19 patients rather than a systemic response. Our investigation adds information about a large set of biomarkers across five functional domains to these earlier studies (47, 48).
Our study has limitations. The first samples were obtained after at least 7 days on the ICU; admission samples would have provided insight into early activation of proinflammatory and procoagulant pathways during severe COVID-19. The sample size was relatively small although still considerably larger than previous studies evaluating host responses in the bronchoalveolar space of COVID-19 patients (37, 42, 47, 48). In accordance with previous studies (1, 49) we documented pulmonary emboli in 58.8% of the patients included in our investigation. It has been suggested that procoagulant and inflammatory responses (“thrombo-inflammation”) are involved herein (16, 50). Whilst we found limited differences in plasma and BALF biomarkers between patients with and without pulmonary embolism, our investigation was not designed or powered to detect such differences. Despite the selection of a specific population of COVID-19 patients there was still unavoidable heterogeneity within this observational cohort that may have affected biomarker levels, including steroid treatment in 64.7% of patients which per clinical protocol was started after two weeks of ICU stay and thus could have modified host response parameters in follow up samples obtained after 3-4 weeks. Finally, we used samples from healthy subjects who were not matched with regard to demographics, smoking or comorbidities as reference; our investigation did not entail critically ill patients without COVID-19, thereby precluding conclusions on the disease-specificity of the host response aberrations reported.
In conclusion, we report a strong local response in the lung compartment across multiple functional domains related to coagulation and inflammation in patients with persistent ARDS due to COVID-19. Early in the pandemic many studies suggested an important role for a systemic “cytokine storm” in the pathophysiology of severe COVID-19 (14, 28, 29). The current results suggest a local rather than a systemic procoagulant and inflammatory “storm” in these patients.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Review Committee of the Amsterdam UMC Biobank (protocol number 2020-065). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
EN, JD, LM, LH, HB, and TP contributed to conception and design of the present study. EN, ARS, TR, AS, IJ, SB, HV, JD, LM, RL, LH, HB, and TP contributed to the acquisition of data. EN, ARS, TR, AS, IJ, SB, HV, JD, AV, LM, RL, LH, HB, and TP contributed to analysis or interpretation of the data. EN, ARS, and TP drafted the manuscript and TR, AS, IJ, SB, HV, JD, AV, LM, RL, LH, and HB revised the article critically for intellectual content. All authors contributed to the article and approved the submitted version.
Funding
TR and AS are supported by the research program NACTAR (Novel Antibiotic Compounds and Therapies Antagonizing Resistance) and project multidrug resistant-phage (grant number 16447), which is financed by the Dutch Research Council (Nederlandse Organisatie voor Wetenschappelijk Onderzoek [NWO]). JD is supported by a personal grant of the Netherlands Organisation for Scientific Research (VENI grant 016.186.046). HV is supported by a research grant from Amsterdam Cardiovascular Sciences Research Institute.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Hella Aberson and Regina de Beer from the Center for Experimental and Molecular Medicine, Tamara Dekker and Barbara Dierdorp from the Department of Experimental Immunology for their technical support in the execution and analysis of the biomarker assays, Yanaika S. Sabogal Pineros from the Department of Experimental Immunology for performing the RILCO and RILCA studies, Gerard van Mierlo, Angela Kamp and Dorina Roem from Sanquin for their technical support in the execution and analysis of the complement assays, and also Jarne van Hattem and Frank van Someren from the department of Medical Microbiology for the clinical PCR data. On behalf of the ArtDECO consortium: EN, JD, AS, HV, LM, Lieuwe D. Bos, SB, AS, TR, Juan J. Garcia Vallejo, Hetty Bontkes, Alexander P.J. Vlaar, Joost Wiersinga, RL, TP, HB, and LH. On behalf of the Amsterdam UMC COVID study group: S. de Bruin, ARS, R. Koning, M. van Agtmael, A.G. Algera, F. van Baarle, D. Bax, M. Beudel, HB, M. Bomers, P.I. Bonta, L. Bos, M. Botta, J. de Brabander, G. de Bree, M. Bugiani, E. Bulle, O. Chouchane, A. Cloherty, D.A. Dongelmans, P. Elbers, L. Fleuren, S. Geerlings, B. Geerts, T. Geijtenbeek, A. Girbes, J.A. Goorhuis, M.P. Grobusch, F. Hafkamp, L. Hagens, J. Hamann, V. Harris, R. Hemke, S.M. Hermans, LH, M. Hollmann, J. Horn, J.W. Hovius, M.D. de Jong, M. van Mourik, J. Nellen, EN, F. Paulus, TR, TP, B. Preckel, J.M. Prins, J. Raasveld, M. Schinkel, M. Schultz, K. Sigaloff, M. Smit, C.S. Stijnis, W. Stilma, C. Teunissen, P. Thoral, A. Tsonas, M. van der Valk, D. Veelo, HV, M. van Vugt, D. Wouters, A.H. Zwinderman, M.C. Brouwer, W.J. Wiersinga, A.P.J. Vlaar and D. van de Beek.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.664209/full#supplementary-material
References
1. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA (2020) 324(8):782–93. doi: 10.1001/jama.2020.12839
2. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
3. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2020) 201(10):1299–300. doi: 10.1164/rccm.202003-0817LE
4. Connors JM, Levy JH. Covid-19 and its Implications for Thrombosis and Anticoagulation. Blood (2020) 135(23):2033–40. doi: 10.1182/blood.2020006000
5. Iba T, Levy JH, Levi M, Thachil J. Coagulopathy in COVID-19. J Thromb Haemost (2020) 18(9):2103–9. doi: 10.1111/jth.14975
6. McKechnie JL, Blish CA. The Innate Immune System: Fighting on the Front Lines or Fanning the Flames of COVID-19? Cell Host Microbe (2020) 27(6):863–9. doi: 10.1016/j.chom.2020.05.009
7. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute Respiratory Distress Syndrome. Nat Rev Dis Primers (2019) 5(1):18. doi: 10.1038/s41572-019-0069-0
8. Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks Between Inflammation and Coagulation. Respiration (2017) 93(3):212–25. doi: 10.1159/000453002
9. Glas GJ, Van Der Sluijs KF, Schultz MJ, Hofstra JJ, Van Der Poll T, Levi M. Bronchoalveolar Hemostasis in Lung Injury and Acute Respiratory Distress Syndrome. J Thromb Haemost (2013) 11(1):17–25. doi: 10.1111/jth.12047
10. van der Poll T. Tissue Factor as an Initiator of Coagulation and Inflammation in the Lung. Crit Care (2008) 12 Suppl 6:S3. doi: 10.1186/cc7026
11. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J Clin Invest (2020) 130(5):2620–9. doi: 10.1172/JCI137244
12. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
13. McElvaney OJ, McEvoy N, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med (2020) 202(6):812–21. doi: 10.1164/rccm.202005-1583OC
14. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine Storm and Leukocyte Changes in Mild Versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J Leukoc Biol (2020) 108(1):17–41. doi: 10.1002/JLB.3COVR0520-272R
15. Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H, et al. Histopathology and Ultrastructural Findings of Fatal COVID-19 Infections in Washington State: A Case Series. Lancet (2020) 396(10247):320–32. doi: 10.1016/S0140-6736(20)31305-2
16. Bonaventura A, Vecchie A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial Dysfunction and Immunothrombosis as Key Pathogenic Mechanisms in COVID-19. Nat Rev Immunol (2021) 21(5):319–29. doi: 10.1038/s41577-021-00536-9
17. Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA (2012) 307(23):2526–33. doi: 10.1001/jama.2012.5669
18. Nuijens JH, Huijbregts CC, Eerenberg-Belmer AJ, Abbink JJ, Strack van Schijndel RJ, Felt-Bersma RJ, et al. Quantification of Plasma Factor XIIa-Cl(-)-Inhibitor and Kallikrein-Cl(-)-Inhibitor Complexes in Sepsis. Blood (1988) 72(6):1841–8. doi: 10.1182/blood.V72.6.1841.1841
19. Hazelzet JA, de Groot R, van Mierlo G, Joosten KF, van der Voort E, Eerenberg A, et al. Complement Activation in Relation to Capillary Leakage in Children With Septic Shock and Purpura. Infect Immun (1998) 66(11):5350–6. doi: 10.1128/IAI.66.11.5350-5356.1998
20. Wolbink GJ, Bollen J, Baars JW, ten Berge RJ, Swaak AJ, Paardekooper J, et al. Application of a Monoclonal Antibody Against a Neoepitope on Activated C4 in an ELISA for the Quantification of Complement Activation Via the Classical Pathway. J Immunol Methods (1993) 163(1):67–76. doi: 10.1016/0022-1759(93)90240-8
21. Brouwer N, Frakking FN, van de Wetering MD, van Houdt M, Hart M, Budde IK, et al. Mannose-Binding Lectin (MBL) Substitution: Recovery of Opsonic Function In Vivo Lags Behind MBL Serum Levels. J Immunol (2009) 183(5):3496–504. doi: 10.4049/jimmunol.0900445
22. de Smet BJ, de Boer JP, Agterberg J, Rigter G, Bleeker WK, Hack CE. Clearance of Human Native, Proteinase-Complexed, and Proteolytically Inactivated C1-inhibitor in Rats. Blood (1993) 81(1):56–61. doi: 10.1182/blood.V81.1.56.56
23. Kox M, Waalders NJB, Kooistra EJ, Gerretsen J, Pickkers P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA (2020) 324(15):1565–7. doi: 10.1001/jama.2020.17052
24. Long AT, Kenne E, Jung R, Fuchs TA, Renne T. Contact System Revisited: An Interface Between Inflammation, Coagulation, and Innate Immunity. J Thromb Haemost (2016) 14(3):427–37. doi: 10.1111/jth.13235
25. Yun SH, Sim EH, Goh RY, Park JI, Han JY. Platelet Activation: The Mechanisms and Potential Biomarkers. BioMed Res Int (2016) 2016:9060143. doi: 10.1155/2016/9060143
26. Ekdahl KN, Teramura Y, Hamad OA, Asif S, Duehrkop C, Fromell K, et al. Dangerous Liaisons: Complement, Coagulation, and Kallikrein/Kinin Cross-Talk Act as a Linchpin in the Events Leading to Thromboinflammation. Immunol Rev (2016) 274(1):245–69. doi: 10.1111/imr.12471
27. Harboe M, Thorgersen EB, Mollnes TE. Advances in Assay of Complement Function and Activation. Adv Drug Delivery Rev (2011) 63(12):976–87. doi: 10.1016/j.addr.2011.05.010
28. Abdin SM, Elgendy SM, Alyammahi SK, Alhamad DW, Omar HA. Tackling the Cytokine Storm in COVID-19, Challenges and Hopes. Life Sci (2020) 257:118054. doi: 10.1016/j.lfs.2020.118054
29. Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front Immunol (2020) 11:1708. doi: 10.3389/fimmu.2020.01708
30. Barratt S, Medford AR, Millar AB. Vascular Endothelial Growth Factor in Acute Lung Injury and Acute Respiratory Distress Syndrome. Respiration (2014) 87(4):329–42. doi: 10.1159/000356034
31. Kardas G, Daszynska-Kardas A, Marynowski M, Brzakalska O, Kuna P, Panek M. Role of Platelet-Derived Growth Factor (PDGF) in Asthma as an Immunoregulatory Factor Mediating Airway Remodeling and Possible Pharmacological Target. Front Pharmacol (2020) 11:47. doi: 10.3389/fphar.2020.00047
32. Brumshagen C, Maus R, Bischof A, Ueberberg B, Bohling J, Osterholzer JJ, et al. FMS-Like Tyrosine Kinase 3 Ligand Treatment of Mice Aggravates Acute Lung Injury in Response to Streptococcus Pneumoniae: Role of Pneumolysin. Infect Immun (2012) 80(12):4281–90. doi: 10.1128/IAI.00854-12
33. Rosler B, Herold S. Lung Epithelial GM-CSF Improves Host Defense Function and Epithelial Repair in Influenza Virus Pneumonia-a New Therapeutic Strategy? Mol Cell Pediatr (2016) 3(1):29. doi: 10.1186/s40348-016-0055-5
34. FitzGerald ES, Jamieson AM. Unique Transcriptional Changes in Coagulation Cascade Genes in SARS-CoV-2-infected Lung Epithelial Cells: A Potential Factor in COVID-19 Coagulopathies. bioRxiv (2020) 2020.07.06.182972. doi: 10.1101/2020.07.06.182972
35. Godoy LC, Goligher EC, Lawler PR, Slutsky AS, Zarychanski R. Anticipating and Managing Coagulopathy and Thrombotic Manifestations of Severe COVID-19. CMAJ (2020) 192(40):E1156–61. doi: 10.1503/cmaj.201240
36. de Maat S, de Mast Q, Danser AHJ, van de Veerdonk FL, Maas C. Impaired Breakdown of Bradykinin and Its Metabolites as a Possible Cause for Pulmonary Edema in COVID-19 Infection. Semin Thromb Hemost (2020) 46(7):835–7. doi: 10.1055/s-0040-1712960
37. Carvelli J, Demaria O, Vely F, Batista L, Benmansour NC, Fares J, et al. Association of COVID-19 Inflammation With Activation of the C5a-C5aR1 Axis. Nature (2020) 588(7836):146–50. doi: 10.1038/s41586-020-2600-6
38. Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, et al. Complement Activation in Patients With COVID-19: A Novel Therapeutic Target. J Allergy Clin Immunol (2020) 146(1):215–7. doi: 10.1016/j.jaci.2020.05.006
39. Fletcher-Sandersjoo A, Bellander BM. Is COVID-19 Associated Thrombosis Caused by Overactivation of the Complement Cascade? A Literature Review. Thromb Res (2020) 194:36–41. doi: 10.1016/j.thromres.2020.06.027
40. Lo MW, Kemper C, Woodruff TM. COVID-19: Complement, Coagulation, and Collateral Damage. J Immunol (2020) 205(6):1488–95. doi: 10.4049/jimmunol.2000644
41. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
42. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
43. Yu X, Lin Q, Qin X, Ruan Z, Zhou J, Lin Z, et al. Ace2 Antagonizes Vegfa to Reduce Vascular Permeability During Acute Lung Injury. Cell Physiol Biochem (2016) 38(3):1055–62. doi: 10.1159/000443056
44. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e278. doi: 10.1016/j.cell.2020.02.052
45. Shimizu S, Gabazza EC, Hayashi T, Ido M, Adachi Y, Suzuki K. Thrombin Stimulates the Expression of PDGF in Lung Epithelial Cells. Am J Physiol Lung Cell Mol Physiol (2000) 279(3):L503–510. doi: 10.1152/ajplung.2000.279.3.L503
46. Li L, Dong L, Zhao D, Gao F, Yan J. Classical Dendritic Cells Regulate Acute Lung Inflammation and Injury in Mice With Lipopolysaccharideinduced Acute Respiratory Distress Syndrome. Int J Mol Med (2019) 44(2):617–29. doi: 10.3892/ijmm.2019.4208
47. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in COVID-19 Patients. Emerg Microbes Infect (2020) 9(1):761–70. doi: 10.1080/22221751.2020.1747363
48. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe (2020) 27(6):883–90.e882. doi: 10.1016/j.chom.2020.04.017
49. Liao SC, Shao SC, Chen YT, Chen YC, Hung MJ. Incidence and Mortality of Pulmonary Embolism in COVID-19: A Systematic Review and Meta-Analysis. Crit Care (2020) 24(1):464. doi: 10.1186/s13054-020-03175-z
Keywords: COVID-19, persistent ARDS, coagulation, innate immune response, bronchoalveolar space
Citation: Nossent EJ, Schuurman AR, Reijnders TDY, Saris A, Jongerius I, Blok SG, de Vries H, Duitman J, Vonk Noordegraaf A, Meijboom LJ, Lutter R, Heunks L, Bogaard HJ and van der Poll T (2021) Pulmonary Procoagulant and Innate Immune Responses in Critically Ill COVID-19 Patients. Front. Immunol. 12:664209. doi: 10.3389/fimmu.2021.664209
Received: 04 February 2021; Accepted: 30 April 2021;
Published: 14 May 2021.
Edited by:
Yoshiro Maru, Tokyo Women’s Medical University, JapanReviewed by:
Christopher Barrett, Massachusetts Institute of Technology, United StatesMarcel Levi, University College London, United Kingdom
Copyright © 2021 Nossent, Schuurman, Reijnders, Saris, Jongerius, Blok, de Vries, Duitman, Vonk Noordegraaf, Meijboom, Lutter, Heunks, Bogaard and van der Poll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esther J. Nossent, e.nossent@amsterdamumc.nl