 Dario Trujillo1
Dario Trujillo1 Thiago Mastrangelo2
Thiago Mastrangelo2 Consuelo Estevez de Jensen1Jose Carlos Verle Rodrigues3
Consuelo Estevez de Jensen1Jose Carlos Verle Rodrigues3 Roger Lawrie4
Roger Lawrie4 Steven E. Massey5*
Steven E. Massey5*- 1Department of Agro-Environmental Sciences, University of Puerto Rico - Mayaguez, Mayaguez, Puerto Rico
- 2Universidade de São Paulo, Centro de Energia Nuclear na Agricultura, Piracicaba, SP, Brazil
- 3USDA-APHIS-PPQ-S&T, Edinburg, TX, United States
- 4Center for Excellence in Quarantine and Invasive Species (CEQUIS), Estacion Experimental Agricola, San Juan, Puerto Rico
- 5Department of Biology, University of Puerto Rico - Rio Piedras, San Juan, Puerto Rico
Helicoverpa armigera, the cotton bollworm moth, is one of the world’s most important crop pests, and is spreading throughout the New World from its original range in the Old World. In Brazil, invasive H. armigera has been reported to hybridize with local populations of Helicoverpa zea. The correct identification of H. armigera-H. zea hybrids is important in understanding the origin, spread and future outlook for New World regions that are affected by outbreaks, given that hybridization can potentially facilitate H. zea pesticide resistance and host plant range via introgression of H. armigera genes. Here, we present a genome admixture analysis of high quality genome sequences generated from two H. armigera-H. zea F1 hybrids generated in two different labs. Our admixture pipeline predicts 48.8% and 48.9% H. armigera for the two F1 hybrids, confirming its accuracy. Genome sequences from five H. zea and one H. armigera that were generated as part of the study show no evidence of hybridization. Interestingly, we show that four H. zea genomes generated from a previous study are predicted to possess a proportion of H. armigera genetic material. Using unsupervised clustering to identify non-hybridized H. armigera and H. zea genomes, 8511 ancestry informative markers (AIMs) were identified. Their relative frequencies are consistent with a minor H. armigera component in the four genomes, however its origin remains to be established. We show that the size and quality of genomic reference datasets are critical for accurate hybridization prediction. Consequently, we discuss potential pitfalls in genome admixture analysis of H. armigera-H. zea hybrids, and suggest measures that will improve such analyses.
Introduction
Helicoverpa armigera, the Old World cotton bollworm, is an Old World species of moth, and one of the world’s most important plant pests, whose larvae consume plants belonging to at least 68 plant families (1). In the New World, H. armigera was initially observed in Brazil in 2013 (2), and has subsequently spread throughout much of Latin America (3, 4), appearing to have undergone multiple introduction events into South America from the Old World (5, 6). There has not yet been a formal identification of H. armigera in North America, although it has been intercepted at several ports (7). The potential economic damage that H. armigera could cause in North America is large: $78 billion worth of crops in the United States were estimated to be susceptible to the pest in 2015 (7).
A closely related species, Helicoverpa zea, is native to the New World, and does not have such a wide host range, feeding off over 110 host plant species (8). H. zea does not possess such a high degree of resistance to common pesticides as that observed in H. armigera (9) [although resistance to Bt-proteins has been widely documented in H. zea (10)], implying it does not pose such an economic threat as H. armigera.
H. armigera and H. zea diverged approximately 1.5 million years ago (11), and are able to produce viable hybrids (12). H. armigera-H. zea hybrids have been reported from Brazil (13–15), but have yet to be identified from elsewhere. Adult H. armigera are difficult to distinguish from H. zea on the basis of morphology, requiring dissection of genitalia (16). Identifying hybrids using such methods is impossible, while larvae of the two species are likewise indistinguishable using morphology (17). In addition, such methods are inappropriate for screening large numbers of animals. While pure H. armigera and H. zea can be differentiated using species-specific PCR of the ITS1 region, this method does not work for hybrids (18). Hence, genomic methods have great potential utility for accurate species and hybrid identification.
The occurrence of H. armigera-H. zea hybrids in Brazil (13–15) has implications for pest management programs. Adaptive introgression of genes from invasive pest species into related local species poses a significant threat to global agriculture (17). A primary reason for studying H. armigera-H. zea hybrids in the field is to monitor the adaptive introgression of pesticide resistance genes to H. zea from H. armigera, which has been subject to intense selective pressure from synthetic pesticides (19, 20). For example, there is evidence that the CYP337B3 gene, which confers resistance to pyrethroids, has already introgressed into H. zea populations in Brazil (15). The frequency of pesticide resistance genes in both H. zea and H. armigera populations has implications for the choice, duration and intensity of pesticide regimens dedicated to their control.
Genes in addition to those responsible for pesticide resistance may also have a propensity to introgress into local H. zea populations. For example, H. zea lacks genes for gustatory receptors and detoxification compared to H. armigera, which may help to explain its more limited range of host plant species (21). These genes may have the potential to introgress from H. armigera into H. zea, potentially increasing H. zea’s agricultural impact by increasing its range of host plants.
H. armigera has not yet been formally identified from North America, partly due to difficulties in distinguishing the species from H. zea. H. armigera was reported in Puerto Rico in 2014 and 2018, however since that time has not been reported again (22) The Caribbean represents a major transit route for pests and pathogens between North and South America (23), forming a ‘Caribbean corridor’, so Puerto Rico is a critical location for monitoring the potential spread of H. armigera from the South American continent into North America.
In this study, we implemented a bioinformatic pipeline to predict hybridization proportions by using whole genome sequences. We used the genomes of two lab generated H. armigera-H. zea F1 hybrids to confirm the accuracy of our admixture analysis procedure. We demonstrate that genomes from Puerto Rican and North American H. zea genomes generated as part of the study do not show evidence of hybridization with H. armigera. However, four attributed North American H. zea genomes from a previous study displayed potential evidence of hybridization, representing the potential early presence of H. armigera in North America. We show that high quality genome sequence data, reference genomic datasets and careful SNP filtration approaches are important for the accurate determination of hybridization proportions.
Methods
Collection and maintenance of parental species
Individual H. zea animals were collected by USDA APHIS collaborators (Todd Gilligan) and shipped to our lab in San Juan in ethanol from Colorado in 2015 (HzCol), Illinois in 2016 (HzIll), Maine in 2016 (HzMaine) and North Carolina in 2016 (HzNC). Species identifications were performed using species specific PCR of the ITS1 region, following the methods of (18).
All live Helicoverpa colonies were maintained under the following conditions: 25 ± 2°C, 57 ± 9% relative humidity, photoperiod of 15 hours of light and 9 hours of dark (15: 9 LD). Female pupae were placed in incubators at 22.7 ± 1.6°C, 82 ± 4% relative humidity, photoperiod of 15:9 LD, females were placed at a lower temperature to synchronize the emergence of adults with males (24). The larvae were fed with Gypsy Moth Diet (Frontier Agricultural Sciences, Product # F9630B, Newark DE): 140.2 g of dry mix, 20 g of fats and sugars, 1.6 g of vitamin mix, 0.8 g of aureomycin, 1000 ml of distilled water, with the addition of 12 ml of formaldehyde 1%, and 2.5 g of FABCO mold inhibitor (Frontier Agricultural Sciences, Product # F0018, Newark DE); the agar was dissolved, when the temperature was ~50°C the rest of the reagents were added. Each larva was maintained in transparent plastic cups of 30 ml containing diet. The pupae were maintained in the same cups.
Emerged adults and pupae near to emergence were placed in white plastic buckets of 18.9 l, the upper part of the buckets was covered with cheesecloth (DeRoyal, BIDF2012380-BX, Tennessee) for oviposition. Inside each bucket a Petri dish with autoclaved sand a potted tomato plant was placed to increase relative humidity. The adults received the following diet recipe modified from (25): 500 ml of distilled water, 50 ml of honey, 10 ml of solution 28% of Vanderzant vitamin mixture (Sigma, V1007, USA), 1 g of methyl-4-hydroxybenzoate (Sigma, H3647, USA), and 1 ml of ethanol 95%; methyl-4-hydroxybenzoate was dissolved in the 95% ethanol, then all the ingredients were mixed in the water and fed to adult moths using cotton wicks. The cheesecloth with the oviposited eggs was placed in Ziploc bags of 3.8 liters with fine strips of larval diet. Once larvae emerged, they were transferred to cups with diet. Prior to molecular work, all samples were stored in 90% ethanol in a –20 °C freezer until DNA extractions were performed.
In the Center for Excellence in Quarantine and Invasive Species (CEQUIS), separate colonies of H. armigera and H. zea were maintained. The colony of H. armigera was obtained from five larvae and 30 pupae from Brazil courtesy of Dr Thiago Mastrangelo, University of Sao Paolo. The insects were collected from Bahia (12°13’53’’S, 45°44’44’’W) in 2016 and were introduced to quarantine facilities of the CEQUIS on February 4, 2017, under Puerto Rico Department of Agriculture Permit number OV-1617-03 and USDA-APHIS Permit number P526P-15-04600 to Dr. José Carlos Verle Rodrigues. The initial colony of H. zea was obtained from larvae collected in Isabela, Puerto Rico, from pigeon peas on November 11, 2015. During the F9 generation, a reintroduction of insects was done, from larvae collected in Isabela in corn on November 22, 2016.
Breeding of the hybrids
The first hybrid included in the study (PRh) was generated in our lab from a male H. armigera from Brazil, and a female H. zea from Puerto Rico. Using the same rearing methods described above, 15 H. zea female pupa and 15 H. armigera male pupa were placed into a white plastic bucket with cheesecloth lid and allowed to emerge, mate, and oviposit. All surviving F1 hybrids resulting from this cross were labeled and stored in a –20°C freezer. The F1 hybrid that was sequenced (PRh) was a female.
Genome sequences were generated from parental animals. A sequence from a male H. armigera from Brazil (HaM) was generated. This animal was an adult male H. armigera from the H. armigera colony initiated in CEQUIS, and was one of the parents for the F1 hybrids generated in the lab. A sequence from a female H. zea from Puerto Rico (HzF) was also generated. HzF was reared following the conditions described above, and was a parent for the H. armigera-H. zea F1 hybrids (PRh in this study) generated in the lab.
The second hybrid included in the study (MAh) was generated from a female H. armigera from Portugal and a male H. zea from the mainland USA by the USDA APHIS Otis Lab in Buzzards Bay, Massachusetts in 2017 by Dr. Hannah Nadel. The Portugese H. armigera mother was reared from pupae supplied by Dr. Delia Munoz, Public University of Navarra, Spain in 2016. The H. zea father used in the MAh cross were supplied by Benzon Research Inc. (Carlisle, PA, USA). This hybrid was reared under the same rearing conditions described previously. Of potential interest, an asymmetrical hybridization between the Brazilian lineages of H. armigera and H. zea has been observed, where male H. zea and female H. armigera have a higher probability of generating F1 offspring (26). Both MAh and PRh F1 hybrids were females.
DNA extraction and sequencing
DNA samples were obtained from the animals using QIAGEN blood and tissue DNA extraction kits (QIAGEN INC., Cat No,/ID 69506) following the manufacturer’s protocol, with the exception of the Colorado, Illinois and North Carolina samples, which were extracted using the CTAB method (27). DNA quality was assessed using a NanoDrop 2000 (ThermoFisher Scientific, Waltham, MA) to assess DNA concentration (ng/uL) and absorbance (A260/280) and gel electrophoresis (1.5% agarose) to assess integrity and molecular weight. After checking DNA concentration and quality, the eight samples were shipped overnight on ice to the Rapid Genomics sequencing laboratory in Florida (www.rapid-genomics.com).
Paired end sequencing was conducted by Rapid Genomics on the Illumina HiSeq-X platform (sequencing statistics are displayed in Supplementary Table 1). The sequence data has been deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) under the Accession numbers SAMN35038651 (PRh), SAMN35038652 (MAh), SAMN35038653 (HzF), SAMN35038654 (HaM), SAMN35038647 (HzCol), SAMN35038648 (HzIll), SAMN35038649 (HzMaine), SAMN35038650 (HzNC). Additional genomic data was used in the analysis, consisting of 29 H. armigera, 9 H. zea and 9 H. armigera-H. zea hybrids, from (13) (Table 1). Raw sequence data for these animals were obtained from the Commonwealth Scientific and Industrial Research Organisation (CSIRO; https://data.csiro.au/collection/csiro:29053). Genome sequence analysis was performed on an Amazon Web Services c6g.4xlarge instance (comprising the AWS Graviton2 processor, 16 vCPUs, 32 Gb memory and Amazon Linux platform).
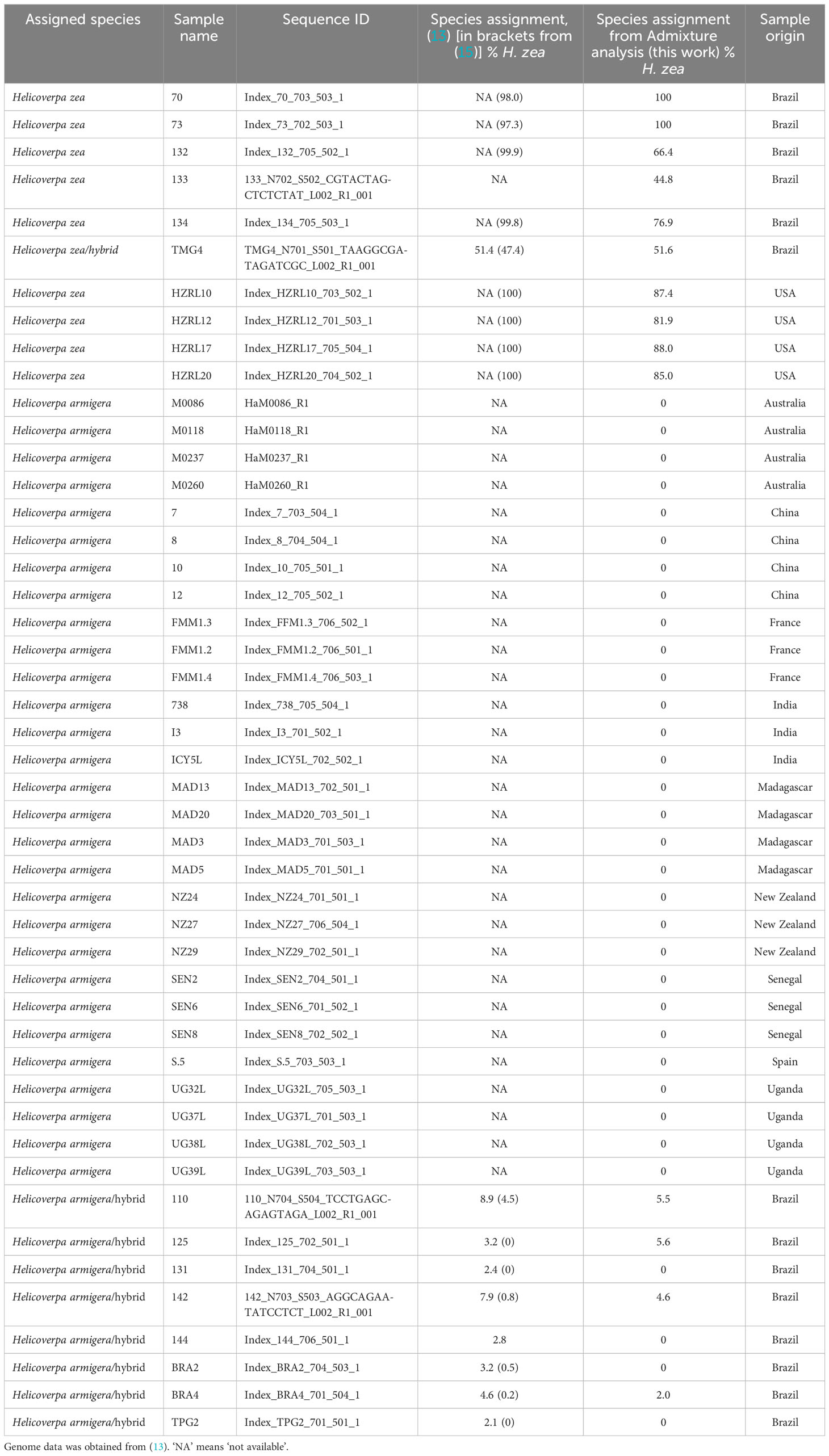
Table 1 Details of 47 additional Helicoverpa genomes used in the Admixture analysis.
Mapping and SNP calling procedure
Using fastp (28), sequences were removed if they did not fulfill the criteria of 95% nucleotides > Q20, 3’ trimming was conducted by quality, and polynucleotide runs (6 or more consecutive). Filtered and trimmed sequences were repaired using the repair.sh script of BBMap (v37.99) (sourceforge.net/projects/bbmap). They were then mapped to the H. armigera reference genome (21) (all-chr-r.fasta, obtained from CSIRO at https://data.csiro.au/collection/csiro:29053v1), using BBMap in paired-end mode. The resulting sam files were converted to bam files, and sorted using SAMtools (29).
BCFtools mpileup (29) was used for variant calling. Bam files for all Helicoverpa genomes in the study were processed together, to improve the accuracy of calls of SNPs shared across genomes. After SNP calling, the resulting vcf files were filtered using vcftools (30), removing those SNPs that possessed mean read depth (min-meanDP < 5), Q value (Q < 20) and minor allele frequency (MAF < 0.05).
Admixture analysis
Admixture v1.3.0 (29) was used for genome admixture analysis, and can be used to infer global ancestry proportions (31). Admixture assumes that genetic loci are independent. While linkage disequilibrium (LD) is not explicitly modeled by Admixture, it provides a useful approximation of global ancestry (29). In order to account for potential effects of LD, LD pruning was conducted using plink and an r2 threshold of 0.95, resulting in the removal of 277381 SNPs. Sex chromosomes (chromosome 1) were excluded from the analysis. Plink (32) was used to convert the combined vcf file into bed format, which was used as input for the Admixture analysis, which was run using K=2. Admixture output was visualized using the R ggplot2 package (https://github.com/tidyverse/ggplot2).
Identification of ancestry informative markers
34 H. armigera and 7 H. zea unhybridized genomes were identified using the unsupervised clustering approach of Admixture, described above. The genotype data from these genomes was then used to identify SNPs that possessed a minor allele count (MAC) of 7 for the H. zea genomes and 1 for H. armigera genomes, using vcftools. These were then pruned by removing all SNP positions where a SNP was completely absent (GT = 0/0) from one or more H. zea genome.
Results and discussion
Genome sequencing results are shown in Table 2, and show that the quality of the raw sequences was high for all eight genomes. For consistency, SNP calling was jointly conducted on the Helicoverpa raw sequence reads generated by (13), and on the sequences generated as part of this study. Filtering resulted in the removal of a large proportion of SNPs (83%); this might be reduced in future by increasing sequence depth in the overall dataset.
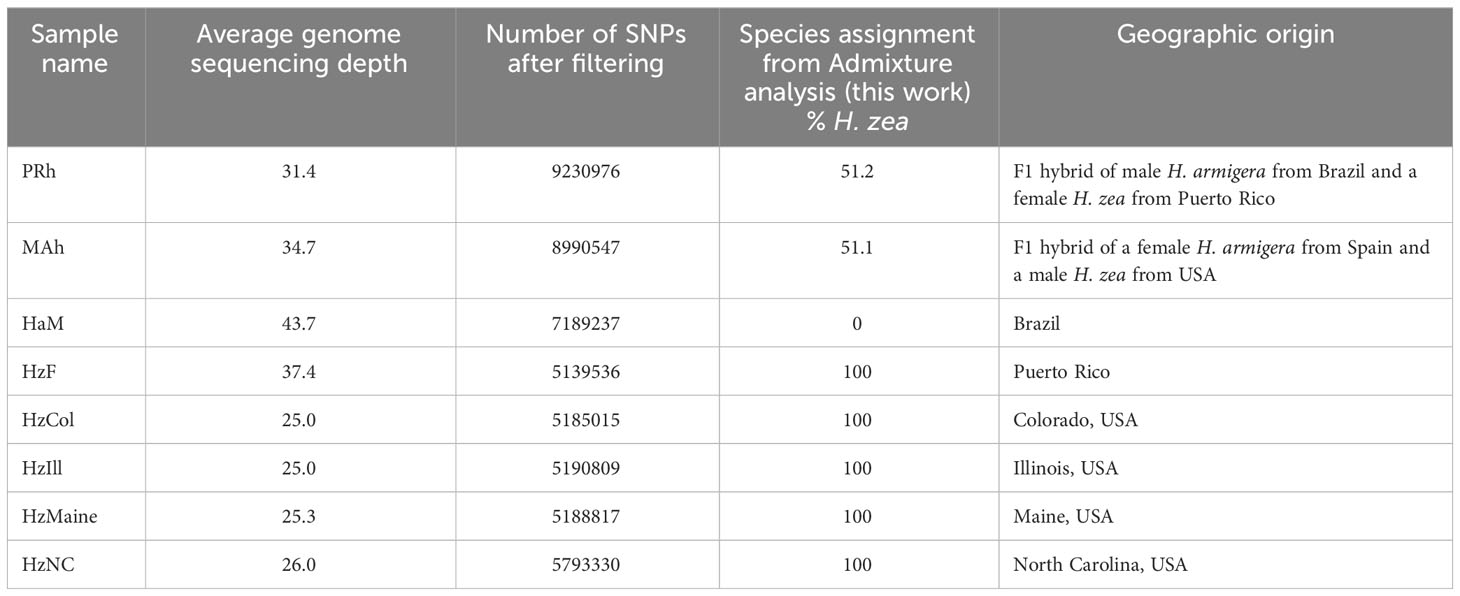
Table 2 Mapping and SNP statistics, and hybridization proportions, of eight new genome sequences generated in the study.
Predicted hybridization proportion of the two lab-reared F1 hybrids
In the hybrid animals, approximately equal proportions of the genome originate from both Helicoverpa species (51.1 % H. zea: 48.9% H. armigera in PRh and 51.2% H. zea: 48.8% H. armigera in MAh). These data are displayed on the Admixture plot (Figure 1). In both cases, the Admixture prediction is not exactly 50% H. zea: 50% H. armigera for either hybrid, even though in the case of PRh, genomes derived from the parental populations were 100% H. zea (HzF) and 100% H. armigera (HaM). This may be due to two reasons. Firstly, the (male) parental insect population may have possessed a degree of hybridization because they were collected originally from Brazil near where early hybrids have since been detected (15). However, it is notable that the MAh F1 hybrid also has a similar H. armigera: H. zea ratio (48.8%: 51.2%). This would mean that the H. zea from the USA, used to generate the hybrid would also have to have had a low level of H. armigera admixture; this seems more unlikely than for an H. zea insect from Brazil, where the presence of hybrids has been validated.
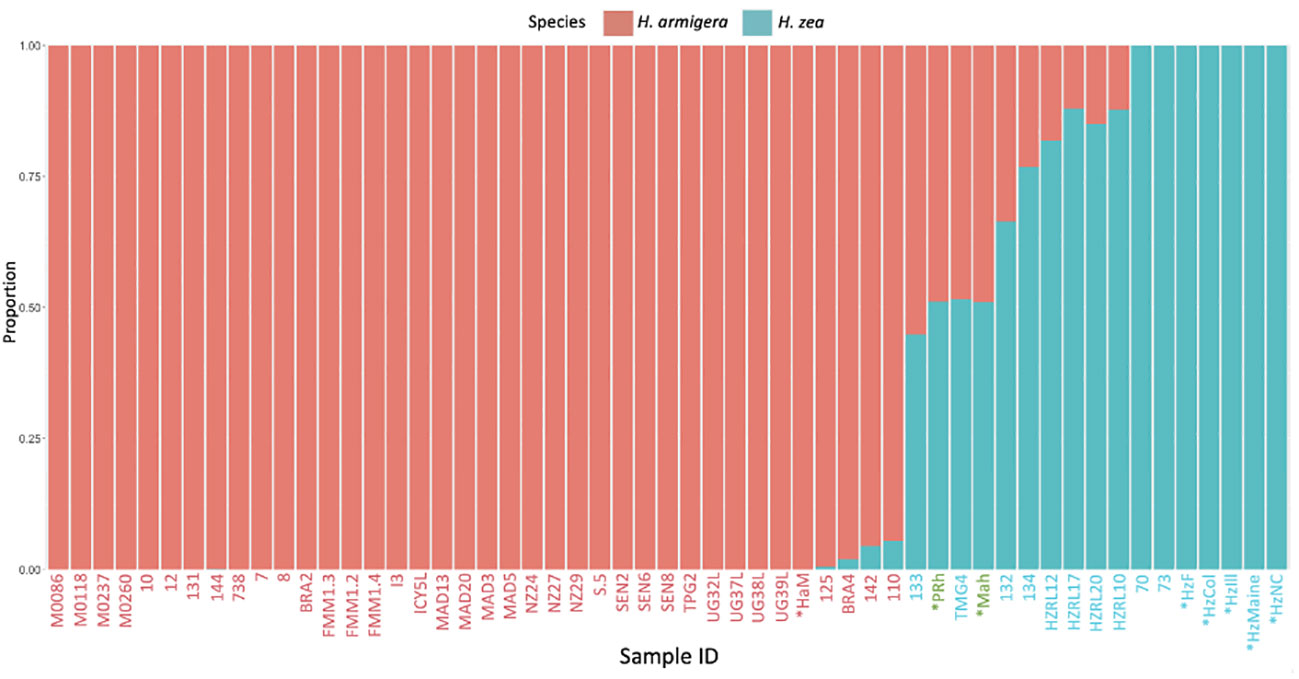
Figure 1 Admixture analysis of Helicoverpa armigera and Helicoverpa zea genomes. The bar plot shows the relative proportions of H. armigera and H. zea present in Helicoverpa genomes generated in this study (PRh, MAh, HaM, HzF, HzCol, HzIll, HzMaine, HzNC), and from (13). The Admixture analysis used K=2, and excluded sex chromosomes. The X-axis displays sample ID code, metadata for each sample is described in Tables 1 and 2. X-axis labels in red are H. armigera samples, labels in blue are H. zea samples, and labels in green are Helicoverpa zea-armigera F1 hybrids. X-axis labels with an asterisk are those that were sequenced during this study, the other samples are from previous studies. The Y-axis displays the proportion of SNPs specific to either H. zea (blue) or H. armigera (red).
Secondly, the Admixture analysis may lack exact precision. This may be the result of a limited number of pure H. zea in the dataset (seven), which means that the genetic diversity of the species is not adequately represented. This is supported by the observation that the H. armigera: H. zea ratio is the same for both F1 hybrids: this indicates a systemic bias in admixture prediction.
Encouragingly, even though H. armigera and H. zea are closely related species, the Admixture analysis is capable of accurately identifying the relative proportions present in an F1 hybrid genome. In future, accuracy may be improved by refinements in SNP calling, increasing the sequencing depth in the overall dataset and adding additional genomes, particularly from H. zea. Admixture analysis may be affected by a small sample size of one or more of the reference populations (33). In the analysis, even after the addition of five H. zea genomes generated in this study, only seven non-hybridized H. zea genomes were apparent.
Predicted hybridization proportions of other Helicoverpa spp. genomes
From the new genome data generated by the study, the analysis indicated that the animals identified as H. armigera (HaM), and H. zea (HzF, HzCol, HzIll, HzMaine, HzNC), were non-hybridized animals. All Old World H. armigera datasets from (13) were identified as non-hybridized, as expected.
The Admixture analysis reveals some discrepancies with those previously published for 47 previously sequenced Helicoverpa genomes (13), which were used as a reference dataset here and in other studies. Most of the animals previously identified as 100% H. zea (13, 15) are predicted in our analysis to have a H. armigera component (132, 133, 134, HZRL10, HZRL12, HZRL17, HZRL20), while several specimens previously identified as hybrids (13, 15) were identified here as 100% H. armigera (131, 144, BRA2, TPG2) (Table 1). TMG4, previously described as a H. zea hybrid (13, 15), is also predicted by our analysis as a hybrid and appears to be F1, given its predicted proportion of 48.9% H. armigera. Given that this animal was collected in August 2013, this implies that hybridization occurred one generation previous to the collection date.
A key difference between our study and (13, 15) is that our inclusion of two lab-reared F1 hybrids allows us to verify the accuracy of our analysis. Potential explanations for differences in predicted hybridization proportions reported in (13) may include lack of filtration after SNP calling, and the lower number of H. zea in the dataset (leading to a limited reference population for this species). In addition, in (13) SNPs were called on a dataset which included Helicoverpa punctigera, Helicoverpa gelotopoeon, Helicoverpa hardwicki and Helicoverpa assulta. In our method, our simultaneous SNP calling procedure only included H. armigera and H. zea datasets. In addition, in our analysis we chose not to include the Z sex chromosome (chromosome 1), focusing only on autosomes.
The reason for differences between our study and the predicted species proportions described in (15) is less clear, given that the authors used filtration criteria similar to our own, and only called SNPs against H. armigera and H. zea genomes, rather than including additional Helicoverpa spp. in their analyses. However, the ancestry proportions that they report in their Supplementary Table S4 are derived from ~1 million SNPs identified as segregating between the two species, whereas we base our ancestry proportions on Admixture analysis, consequently methodological differences may provide the source of the discrepancy.
Potential H. armigera hybridization detected in North American H. zea from 2005
The identification in the reference dataset of potential H. armigera-H. zea hybrids from North America (HZRL10, HZRL12, HZRL17, HZRL20), with predicted H. armigera proportions of 12.6%, 18.1%, 12% and 15%, respectively (Table 1) is interesting, given that H. armigera has not been formally identified in the mainland US, and that H. armigera was first detected in the Americas in 2013 in Brazil (2). This may therefore represent an early presence of H. armigera in the Americas.
The samples were originally described in a 2007 study that constructed a phylogeny of Helicoverpa spp. using mitochondrial DNA (11), and their genome sequences, used in the study described here, were described in (13). The samples are recorded as having been collected from ‘Riverland, NY’ (13), however this location is unclear. Dr Daniel Gilrein supplied the H. zea samples (11), and is based at the Long Island Horticultural Research and Extension Center (LIHREC), Riverhead, NY. The origin of the samples is confirmed as Riverhead, NY (personal communication, Dr Dan Gilrein).
The four samples were collected in 2005, in September/October (personal communication, Dr Dan Gilrein). Significantly, this date predates the first reports of H. armigera in the New World in 2013 in Brazil (2). In order to confirm this result, 8511 AIMs were identified, as described in Methods. Unsupervised clustering allowed the a priori identification of 34 H. armigera and 7 H. zea non-hybridized genomes (Table 1). These were used to identify SNPs that preferentially segregate in one species or the other (AIMS) (Supplementary Material). The 8511 AIMS thus identified indicate a H. armigera component ranging from 25.8 to 31.1% in the four genomes (Table 3). The predicted presence of a H. armigera component is consistent with the results from the Admixture analysis.
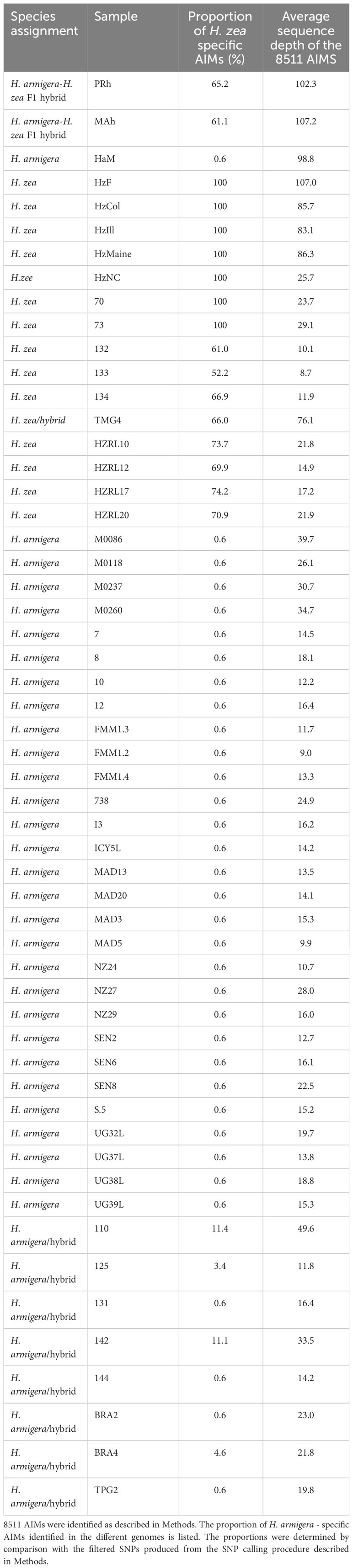
Table 3 Proportion of H. zea AIMs present in different genomic datasets.
Regarding the accuracy of this approach, using a reduced set of SNPs is not expected to give the same accuracy as the whole genome considerations utilized by Admixture, however the unsupervised clustering approach represents an independent manner of assessing a potential H. armigera contribution to H. zea genomic datasets. The H. armigera component is higher than predicted by the Admixture approach, which gives 12.6 to 18.1% H. armigera. Notably, the predicted H. armigera proportion for the two F1 hybrids is 34.8% (PRh) and 38.9% (MAh) (Table 3), which underestimates the true proportion of 50%. One potential source of error is uneven distribution of AIMs along the chromosomes. Another is that the H. zea dataset was limited in size, and so this reduces the accuracy in identifying species-specific AIMs. The low level (0.6%) of H. zea AIMs detected in most of the H. armigera genomes reflects the AIM selection approach: the H. zea AIMs were present in all 7 H. zea genomes, and were also found to be present in at most one H. armigera genome in the reference dataset.
Finally, it is possible that low sequencing depth may affect the predicted hybridization proportions. Given that the SNPs are called against a H. armigera reference genome, then if a SNP position has low or no read depth in a particular genome, the SNP calling software will call the H. armigera genotype at that position. This means a bias toward calling H. armigera AIMs when sequencing depth is low. For example, HZRL10 has 333 AIM positions where there is no sequence coverage, reflecting its low average sequencing depth of 21.8 for the AIM positions. In total there are 986 AIM positions where DP < 5, and so cannot be called with confidence; these constitute 11% of the total number of AIMs. The AIM positions where there is no sequence coverage are by default identified as H. armigera (reflecting the reference genome sequence at those positions). This therefore can account for a proportion of H. armigera AIMs in the HZRL10 genome sequence, but not all. This observation may also account for a proportion of the H. armigera ancestry in HZRL10 detected by the Admixture analysis.
Further work will be required to validate or discount these observations. In particular, the approaches described are not able to distinguish sample contamination from hybridization. Larger, high quality datasets will be necessary in order to distinguish these two alternative scenarios. Development of such fine-grained methods will have value in screening of historic samples and detection of contamination in hybridization studies. These are currently difficult to detect (a method developed by SEM for detecting contamination of NGS datasets, mitoscan https://github.com/semassey/Scanning-NGS-datasets-for-mitochondrial-and-coronavirus-contaminants/blob/main/mitoscan.sh, maps reads against all NCBI mitochondrial genomes, however it is not able to distinguish contamination by closely related species, due to cross-mapping between closely related mitochondria).
The use of genome admixture analysis for the identification and control of Helicoverpa infestations
We have shown the efficacy of genome admixture analysis for verifying the identity of Helicoverpa hybrids, which are morphologically cryptic, and so recalcitrant to traditional identification methods, as is the identification of the two Helicoverpa species themselves. We found that increasing the number of H. zea genomes in the analysis improved the accuracy of admixture prediction, for the H. zea and H. armigera genomes, and the two F1 hybrid genomes generated in the study. Likewise, filtering based on sequencing depth also had a similar effect, although we were restricted in increasing filtering stringency, given limitations in sequencing depth in the dataset. Future improvements in accuracy will arise from greater average sequencing depth in the reference genomes used in admixture analyses. Finally, for accurate hybrid identification, whole genome approaches are most likely to yield the precision necessary for understanding the dynamics of H. armigera invasivity in the field.
In addition to the indirect detection of H. armigera in a region via identification of H. armigera-H. zea hybrids, determining the presence of the hybrids will have utility for monitoring the occurrence and spread of pesticide resistance. This is desirable because H. armigera populations in the Old World have typically been subjected to significant pesticide exposure, thus leading to the evolution of resistance (15). Hybridization with local H. zea populations is expected to lead to the introgression of pesticide resistance genes from the H. armigera genomic component (15). The phenomenon of rapid introgression of pesticide resistance genes between sister species has been observed in Anopheles spp. exposed to selection pressure from pesticide exposure (34). The evolutionary dynamics would be expected to be rather similar in crop pests such as Helicoverpa spp.
Host plant preference is another agriculturally relevant phenotype that may be influenced by hybridization and gene introgression is that of host plant preference. H. armigera has a considerably more extensive plant host range than H. zea, apparently partly due to its larger number of gustatory receptor and detoxification genes compared to H. zea (21). Adaptive introgression of these genes from H. armigera into local populations of H. zea may cause changes in the host plant preferences of H. zea, a process consistent with the ‘hybrid bridge’ hypothesis of host shifting of herbivorous insect pests (35). Furthermore, increasing ease of H. armigera-H. zea hybrid detection will allow for the collection of empirical evidence for whether hybridization will influence changes in pesticide susceptibility or feeding behavior. Currently, because hybrids are extremely difficult to identify, empirical data for these phenotypic changes are near impossible to collect.
Puerto Rico is a stepping stone between North and South America, given its geographic location and possession of a major port in San Juan, through which agricultural produce enters and exits the United States. This transit route for agricultural pests and pathogens comprises part of a ‘Caribbean corridor’. So far, there are no reports in the literature on sustained H. armigera populations in North America or Puerto Rico. One potential route for the spread of H. armigera into North America from South America may be through Puerto Rico.
The detection of H. armigera-zea hybrids can reveal aspects of the population dynamics of both species and help inform control strategies. The accurate determination of hybrid proportions can also indicate whether species boundaries are maintained, given that hybridization is often maladaptive.
Accurate admixture prediction methods for Helicoverpa species are essential for the design of accurate high throughput hybrid identification tools, and so the datasets generated as part of this study will be useful in the development of tools for the rapid, economical and accurate identification of pure species or hybrids. Future detection of hybrids from Puerto Rico and potentially North America will help inform control regimens, facilitated by the development of rapid molecular tests to accurately determine hybrids. In particular, if there is detection of H. armigera in North America, screening of local H. zea populations for hybridization could be used to assess whether breeding has occurred.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
DT: Investigation, Writing – review & editing. TM: Investigation, Resources, Writing – review & editing. CE: Investigation, Funding acquisition, Project administration, Writing – review & editing. JV: Funding acquisition, Investigation, Project administration, Writing – review & editing, Conceptualization, Resources, Methodology. RL: Writing – review & editing, Visualization, Validation. SM: Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Software, Supervision, Validation, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by USDA/APHIS Agreement AP20PPQS&T00C161. It may not necessarily express APHIS’ views.
Acknowledgments
We would like to thank Dr Todd Gilligan, USDA-APHIS, for providing USA specimens, Dr Hannah Nadel, Otis Laboratory MA, for providing the MAh hybrid, Dr Fernando Rodrigues da Silva, University of Florida, for support during rearing insect colonies, and Patricia Caligari, UPR-CEQUIS, for assistance during DNA preparations for sequencing. We would also like to thank Dr Tom Walsh and Dr Tek Tay (CSIRO), and Dr Dan Gilrein (LIHREC, Cornell University, Riverhead, NY) for valuable discussion regarding the specimens collected from NY state. Lastly, we would like to thank an researcher, and two reviewers, for helpful comments prior to submission.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MT declared a shared affiliation with the author JV to the handling editor at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/finsc.2024.1339143/full#supplementary-material
References
1. Cunningham JP, Zalucki MP. Understanding heliothine (Lepidoptera: Heliothinae) pests: what is a host plant? J Economic Entomology. (2014) 107:881–965. doi: 10.1603/EC14036.
2. Czepak C, Albernaz KC, Vivan LM, Guimaraes HO, Carvalhais T. First reported occurrence of Helicoverpa armigera (Hubner) (Lepidoptera: Noctuidae) in Brazil. Pesquisa Agropecuaria Trop. (2013) 43:110–13. doi: 10.1590/S1983-40632013000100015.
3. Murúa MG, Scalora FS, Navarro FR, Cazado LE. First Record of Helicoverpa Armigera (Lepidoptera: Noctuidae) in Argentina. Florida, The Florida Entomologist (2014) 97, 854–6. Available at: https://bioone.org/journals/florida-entomologist/volume-97/issue-2/024.097.0279/First-Record-of-Helicoverpa-armigera-Lepidoptera-Noctuidae-in-Argentina/10.1653/024.097.0279.full.
4. Tembrock LR, Timm AE, Zink FA, Gilligan TM. Phylogeography of the recent expansion of Helicoverpa armigera (Lepidoptera: Noctuidae) in South America and the Caribbean basin. Ann Entomological Soc America. (2019) 112:388–4015. doi: 10.1093/aesa/saz019.
5. Arnemann JA, Roxburgh S, Walsh T, Guedes J, Gordon K, Smagghe G, et al. Multiple incursion pathways for Helicoverpa armigera in Brazil show its genetic diversity spreading in a connected world. Sci Rep. (2019) 9:193805. doi: 10.1038/s41598-019-55919-9.
6. Gonçalves RM, Mastrangelo T, Rodrigues JCV, Paulo DF, Omoto C, Corrêa AS, et al. Invasion origin, rapid population expansion, and the lack of genetic structure of cotton bollworm (Helicoverpa armigera) in the Americas. Ecol Evol. (2019) 9:7378–74015. doi: 10.1002/ece3.5123.
7. Kriticos DJ, Ota N, Hutchison WD, Beddow J, Walsh T, Tay WT, et al. The potential distribution of invading Helicoverpa armigera in North America: is it just a matter of time? PLoS One. (2015) 10:e01196185. doi: 10.1371/journal.pone.0133224.
8. Kogan M, Helm CG, Kogan J, Brewer E. Distribution and economic importance of Heliothis virescens and Heliothis zea in north, central, and South America and of their natural enemies and host plants. In: King EG, Jackson RD, editors. Proceedings of the Workshop on Biological Control of Heliothis: Increasing the Effectiveness of Natural Enemies, New Delhi, India 11 15 November 1985. Far Eastern Regional Research Office, Office of International Cooperation & Development, USDA, New Delhi, India (1989). p. 241–97.
9. da Silva FR, Trujillo D, Bernardi O, Rodrigues JCV, Bailey WD, Gilligan TM, et al. Comparative toxicity of Helicoverpa armigera and Helicoverpa zea (Lepidoptera: Noctuidae) to selected insecticides. Insects. (2020) 11:431. doi: 10.3390/insects11070431
10. Burd AD, Gould F, Bradley JR, Duyn JWV, Moar WJ. Estimated frequency of nonrecessive Bt resistance genes in bollworm, Helicoverpa zea (Boddie) (Lepidoptera: Noctuidae) in Eastern North Carolina. J Economic Entomology. (2003) 96:137–425. doi: 10.1093/jee/96.1.137.
11. Behere GT, Tay WT, Russell DA, Heckel DG, Appleton BR, Kranthi KR, et al. Mitochondrial DNA analysis of field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) and of its relationship to H. Zea. BMC Evolutionary Biol. (2007) 7:1175. doi: 10.1186/1471-2148-7-117.
12. Laster ML, Sheng CF. Search for hybrid sterility for Helicoverpa zea in crosses between the North-American Heliothis zea and Helicoverpa armigera (Lepidoptera, Noctuidae) from China. J Economic Entomology. (1995) 88:1288–91. doi: 10.1093/jee/88.5.1288.
13. Anderson CJ, Oakeshott JG, Tay WT, Gordon KHJ, Zwick A, Walsh TK. Hybridization and gene flow in the mega-pest lineage of moth, Helicoverpa. Proc Natl Acad Sci U S A. (2018) 115:5034–395. doi: 10.1073/pnas.1718831115.
14. Cordeiro EMG, Pantoja-Gomez LM, de Paiva JB, Nascimento ARB, Omoto C, Michel AP, et al. Hybridization and introgression between Helicoverpa armigera and H. Zea: an adaptational bridge. BMC Evolutionary Biol. (2020) 20:615. doi: 10.1186/s12862-020-01621-8.
15. Valencia-Montoya WA, Elfekih S, North HL, Meier JI, Warren IA, Tay WT, et al. Adaptive introgression across semipermeable species boundaries between local Helicoverpa zea and invasive helicoverpa armigera moths. Mol Biol Evol. (2020) 37:2568–83. doi: 10.1093/molbev/msaa108.
16. Pogue MG. A new synonym of Helicoverpa zea (Boddie) and differentiation of adult males of H. Zea and H. Armigera (Hübner) (Lepidoptera: Noctuidae: Heliothinae). Ann Entomological Soc America. (2004) 97:1222–26. doi: 10.1603/0013-8746(2004)097[1222:ANSOHZ]2.0.CO;2.
17. Tay WT, Gordon KHJ. Going global - genomic insights into insect invasions. Curr Opin Insect Sci. (2019) 31:123–30. doi: 10.1016/j.cois.2018.12.002.
18. Perera OP, Allen KC, Jain D, Purcell M, Little NS, Luttrell RG. Rapid identification of Helicoverpa armigera and Helicoverpa zea (Lepidoptera: Noctuidae) using ribosomal RNA internal transcribed spacer 1. J Insect Sci. (2015) 15:155. doi: 10.1093/jisesa/iev137
19. Walsh TK, Heckel DG, Wu Y, Downes S, Gordon KHJ, Oakeshott JG. Determinants of insecticide resistance evolution: comparative analysis among heliothines. Annu Rev Entomology. (2022) 67:387–406. doi: 10.1146/annurev-ento-080421-071655.
20. Jin M, North HL, Peng Y, Liu H, Liu B, Pan R, et al. Adaptive evolution to the natural and anthropogenic environment in a global invasive crop pest, the cotton bollworm. Innovation Journal: Public Sector Innovation J. (2023) 4:100454. doi: 10.1016/j.xinn.2023.100454.
21. Pearce SL, Clarke DF, East PD, Elfekih S, Gordon KHJ, Jermiin LS, et al. Genomic innovations, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive helicoverpa pest species. BMC Biol. (2017) 15:63. doi: 10.1186/s12915-017-0402-6
22. Flores-Rivera XL, Paula-Moraes SV, Johnson JJ, Jack CJ, Perera OP. Helicoverpa genus on the edge of the continental US: flight phenology, analysis of hybrid presence, and insecticide performance in high input field crops. Front Insect Sci. (2022) 2. doi: 10.3389/finsc.2022.1010310.
23. Waugh JD. Trade and invasive species in the caribbean: A universe of risk. In: The International Union for Conservation of Nature. Gland, Switzerland (2009).
24. Colvin J, Cooter RJ, Patel S. Laboratory Mating Behavior and Compatibility of Helicoverpa armigera (Lepidoptera: Noctuidae) Originating from Different Geographical Regions. Journal of Economic Entomology. (1994) 87:1502–1506. doi: 10.1093/jee/87.6.1502
25. Grzywacz D, Rossbach A, Rauf A, Russell DA, Srinivasan R, Shelton AM. Current control methods for diamondback moth and other brassica insect pestsand the prospects for improved management with lepidopteran-resistant Bt vegetable brassicas in Asia and Africa. Crop Protection. (2010) 29:68–79.
26. Corrêa AS, Cordeiro EMg, Omoto C. Agricultural insect hybridization and implications for pest management. Pest Manage Sci. (2019) 75:2857–645. doi: 10.1002/ps.5495.
27. Calderón-Cortés N, Quesada M, Cano-Camacho H, Zavala-Páramo G. A simple and rapid method for DNA isolation from xylophagous insects. Int J Mol Sci. (2010) 11:5056–645. doi: 10.3390/ijms11125056.
28. Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560.
29. Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. (2021) 10:giab008. doi: 10.1093/gigascience/giab008
30. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. (2011) 27:2156–58. doi: 10.1093/bioinformatics/btr330.
31. Tan T, Atkinson EG. Strategies for the genomic analysis of admixed populations. Annu Rev Biomed Data Sci. (2023) 6:105–27. doi: 10.1146/annurev-biodatasci-020722-014310.
32. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. (2007) 81:559–75. doi: 10.1086/519795.
33. Lawson DJ, Dorp Lv, Falush D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat Commun. (2018) 9:32585. doi: 10.1038/s41467-018-05257-7.
34. Norris LC, Main BJ, Lee Y, Collier TC, Fofana A, Cornel AJ, et al. Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc Natl Acad Sci U S A. (2015) 112:815–205. doi: 10.1073/pnas.1418892112.
Keywords: Helicoverpa zea, Helicoverpa armigera, hybrid, F1, admixture, genome, crop pest
Citation: Trujillo D, Mastrangelo T, Estevez de Jensen C, Verle Rodrigues JC, Lawrie R and Massey SE (2024) Accurate identification of Helicoverpa armigera–Helicoverpa zea hybrids using genome admixture analysis: implications for genomic surveillance. Front. Insect Sci. 4:1339143. doi: 10.3389/finsc.2024.1339143
Received: 15 November 2023; Accepted: 08 February 2024;
Published: 23 February 2024.
Edited by:
Michael Brewer, Texas A&M AgriLife Research, United StatesReviewed by:
Renata Bažok, University of Zagreb, CroatiaMackenzie Tietjen, Plains Area, Agricultural Research Service (USDA), United States
Copyright © 2024 Trujillo, Mastrangelo, Estevez de Jensen, Verle Rodrigues, Lawrie and Massey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven E. Massey, c3RldmVuLm1hc3NleUB1cHIuZWR1