 Xiaomeng Zhang1
Xiaomeng Zhang1 Jun Lv
Jun Lv- 1Agricultural and Rural Service Center of Moutai Town, Renhuai, China
- 2School of Food Engineering, Moutai Institute, Renhuai, Guizhou, China
- 3Kweichow Moutai Distillery (Group) Hongyingzi Science & Technology Development Co., Ltd, Renhuai, China
The red flour beetle Tribolium castaneum (Coleoptera: Tenebrionidae), a cosmopolitan stored-product pest frequently infesting sauce-flavor Daqu (a multi-microbial fermented starter), may experience mitochondrial genome variations under the selective pressure exerted by this enzyme-rich substrate. Here we test whether feeding on sauce-flavor Daqu is associated with mitogenomic differences in T. castaneum. We present the complete mitochondrial genome of T. castaneum from this environment: a 15,885 bp circular DNA (GenBank PV563855) retaining ancestral insect architecture with 71.81% A+T content and slight positive AT skew. The genome contains 37 functional elements: 22 tRNA genes (all exhibiting atypical cloverleaf structures except trnS1(AGN)), 13 protein-coding genes (PCGs), 2 rRNA genes, and a 1,238 bp A+T-rich control region (82.80% AT). Eleven PCGs initiate with ATN codons, while cox1 (CTG) and nad1 (TTG) show divergent initiation. Ten PCGs terminate with TAA/TAG codons. Gene order aligns with basal insect mitogenomes. Comparative analysis with Jiangsu (China) and California (USA) strains revealed conserved structural features, though sequence/assembly discrepancies require further investigation to assess potential pressure-induced mutations. While these differences may reflect adaptations to the enzyme-rich Daqu environment, technical and geographical factors could also contribute; further functional studies are needed to establish causal links.
1 Introduction
The red flour beetle, Tribolium castaneum (Coleoptera: Tenebrionidae), is a major pest of stored grains and grain products. In sauce-flavor baijiu production regions of China, T. castaneum is a predominant pest infesting sauce-flavor Daqu, the critical fermentation starter for this distinctive liquor. Sauce-flavor Daqu, a wheat-based fermentation starter for Chinese baijiu, differs from conventional stored grains by having a porous and thermophilic structure. This substrate supports a diverse microbial community (including Bacillus spp., Aspergillus fungi, and lactic acid bacteria) and accumulates metabolites like organic acids, and Maillard reaction products. These characteristics may have facilitated the adaptive evolution of T. castaneum. Mitochondrial variations could enhance survival in Daqu environments by improving heat tolerance during high-temperature fermentation (60-65 °C) and detoxification of organic acids. Such adaptations may increase infestation persistence, leading to microbial imbalance and starter quality deterioration. Identifying these genomic signatures could inform targeted pest management in baijiu production.
In eukaryotic cells, mitochondria serve as energy-producing organelles primarily through oxidative phosphorylation processes (1). The mitochondrial genome (mtDNA) of insects consists of a circular double-stranded DNA molecule measuring 15,000-18,000 base pairs (2). These maternally inherited genomes employ a variant genetic code for translation (3), maintaining high structural conservation across species. A typical insect mitochondrial genome contains 37 coding elements comprising 13 protein-coding genes, 2 ribosomal RNA (rRNA) genes, and 22 transfer RNA (tRNA) genes, accompanied by a non-coding control region responsible for transcription and replication initiation (2, 4). The non-protein coding components include tRNA genes (identified by their corresponding amino acid designations) and rRNA genes encoding both small (rrnS) and large (rrnL) mitochondrial ribosome subunits. Protein-coding sequences produce polypeptides essential for electron transport chain complexes: NADH dehydrogenase subunits (complex I, nad genes), cytochrome B (complex III, cob), cytochrome c oxidase subunits (complex IV, cox genes), and ATP synthase components (complex V, atp genes).
Prior to this study, two complete T. castaneum mitogenomes were available: a 15,881 bp genome from a California laboratory population (NC_003081; Friedrich et al., 2003) (5) and a 15,883 bp genome from a Jiangsu laboratory strain (KM009121; Liu et al., 2016) (6). Both share conserved features including 13 PCGs, 22 tRNAs, and 2 rRNAs. Here, we assemble a third mitogenome from a Guizhou population maintained on sauce-flavor Daqu, representing distinct geographical and dietary conditions. To investigate unresolved questions about substrate-specific variations (e.g., Daqu adaptation) and intercontinental micro-differentiation, we conducted comparative analyses among these three lineages: Guizhou (Daqu-fed), Jiangsu (grain-fed laboratory), and California (grain-fed laboratory).
2 Materials and methods
2.1 Sample collection
The T. castaneum specimens used in this study were collected in 2022 from infested sauce-flavor Daqu in Moutai Town, Guizhou Province, China (27°51′ N, 106°22′ E). These beetles have been maintained on sauce-flavor Daqu in the laboratory since collection. We pooled thirty adult beetles (15 males, 15 females) for analysis and stored them at -80 °C in the Entomology Laboratory of Moutai Institute.
2.2 Construction of the genomic library and sequencing
Total genomic DNA was extracted from insects using the TIANamp Genomic DNA Kit (Tiangen, Beijing, China) following the manufacturer’s protocol. DNA concentrations were standardized to 0.3 ng/μL for all samples. Whole genome shotgun (WGS) sequencing was conducted through next-generation sequencing (NGS) technology on the Illumina NovaSeq platform. A library with 400 bp inserts was constructed and sequenced using paired-end methodology at Personalbio (Nanjing, China). Raw sequencing data were transferred to a computer workstation for processing. Initial quality assessment was performed using FastQC v.0.11.9, followed by adapter removal with Trim Galore v.0.6.5. Final data validation was conducted through repeat FastQC analysis.
2.3 Genomic assembly
High-quality processed sequencing data were first assembled into Contigs using A5 miseq v20150522 (7), followed by Scaffold construction with SPAdes v3.9.0 (8). Gap filling between contigs was performed through collinearity analysis using Mummer v3.1 (9). Final mitochondrial sequence correction was completed using Pilon v1.18 (10). Assembly accuracy was validated through: (1) BLASTn-confirmed terminal overlaps for circularization, (2) uniform depth distribution averaging 1,580.97× (Supplementary Figure S1), and (3) >95% read concordance rate with Q≥30 scores derived from high-quality data (96.97% HQ reads).
2.4 Annotation and bioinformatic analysis
The assembled mitochondrial genome was annotated using the MITOS web server (http://mitos.bioinf.uni-leipzig.de) (11). Sequence alignment was performed with SnapGene software using sequences detailed in Table 1. The nad3 protein homology model was built with SWISS-MODEL (https://swissmodel.expasy.org/; accessed on 28 July 2025). Bar graphs were drawn using GraphPad Prism 8.0.
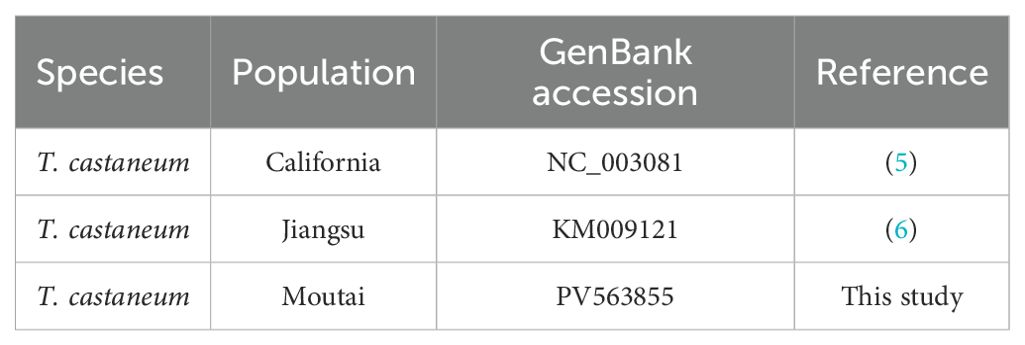
Table 1. Mitochondrial genomes used in this study.
3 Results
3.1 Genome organization and base composition
We sequenced the mitochondrial genome of the T. castaneum population from Moutai Town, Guizhou, China, reared on Sauce-flavor Daqu. The read sequencing produced 2.9 billion bases (Gb). MiSeq reads corresponding to the mitochondrial genome were separated from nuclear genomic reads (see Methods), with approximately 0.41% of the 19,670,196 total reads representing putative mitochondrial reads. The average sequencing coverage depth was 1580.97×(median = 1580) across the mitochondrial genome (Supplementary Figure S1). These reads produced a mitochondrial genome assembly (gMT) of the China isolate of T. castaneum with a total length of 15,885 bp (GenBank PV563855), sharing 98.86% identity with the California strain (5) and 98.87% with the Jiangsu strain (6). Features of the mitochondrial genome were annotated to identify 22 tRNA genes, 2 rRNA genes, 13 protein coding genes (7 nad subunits, 3 cox, 2 atp, and 1 cob), and a non-coding AT-rich control region (1238 bp) (Figure 1). The overall mitogenome exhibited a nucleotide composition of 39.80% A, 9.81% G, 18.38% C, and 32.01% T, with a markedly biased A+T content of 71.81% (Table 2). This A+T value slightly exceeds those reported for the California (5) population (71.68%) and Jiangsu (6) laboratory strain (71.00%), suggesting potential lineage-specific genomic adaptations.
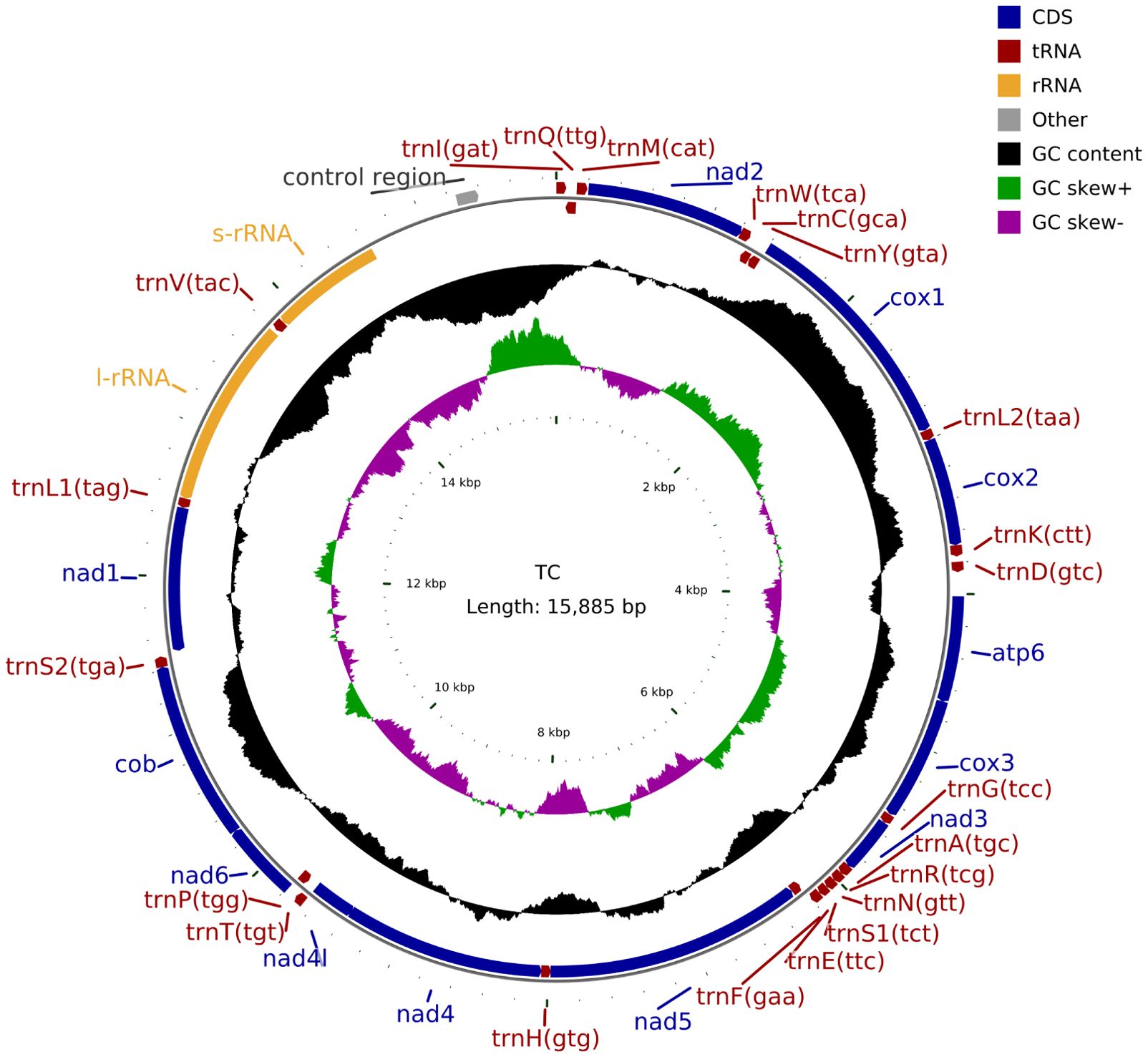
Figure 1. Mitochondrial genome map of Tribolium castaneum.
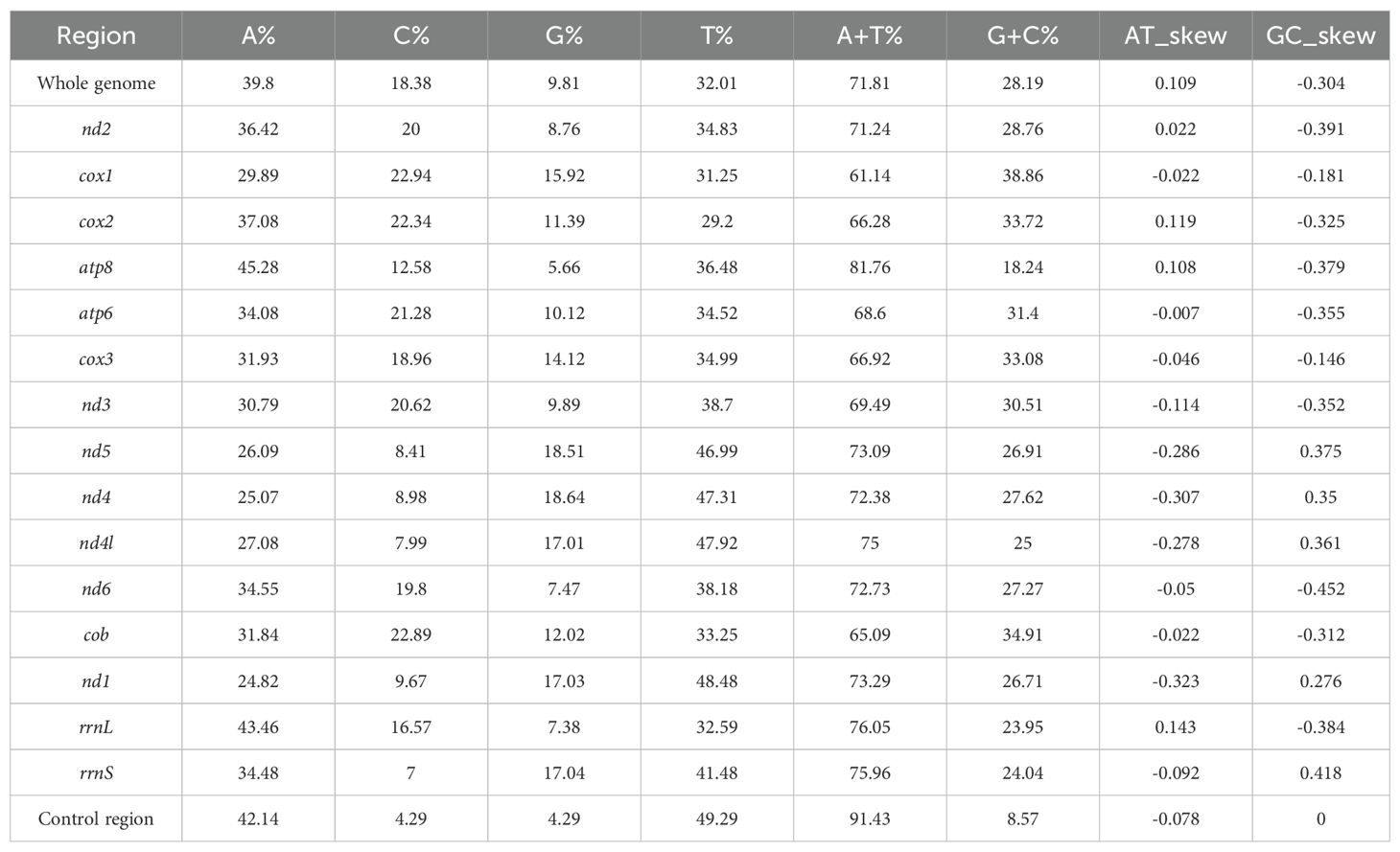
Table 2. Base composition of different regions of the mitochondrial genome of T. castaneum.
3.2 PCGs and codon usage
In PCGs, four (nad4, nad4l, nad5, nad1) of the 13 PCGs were coded on the N-strand, with the other nine genes (cox1, cox2, cox3, atp8, atp6, nad2, nad3, nad6, and cob) were coded on the C-strand. Among the 13 PCGs, the longest was the nad5 gene (1,713 bp) and the shortest was the atp8 gene (159 bp). The start codons of cox1 (CTG) and nad1 (TTG) in this study exhibit striking contrast with those of the Jiangsu and California populations (5, 6). Specifically, the Jiangsu population demonstrates AAT for cox1 and ATT for nad1, while the California population (5) utilizes AAA and ATA as start codons for cox1 and nad1, respectively (Table 3). Notably, the remaining 11 PCGs uniformly employ ATN start codons across all analyzed populations. Among the 13 PCGs, ten terminated with standard stop codons (TAA/TAG), while nad5 used ATT, and the remaining two ended with incomplete stops (single T) (Table 3). It is generally accepted that incomplete codon structures signal a halt of protein translation in insects and other invertebrates (12). With the exception of atp8, cox2, and cox3, all single-nucleotide variants (SNVs) resulted in nonsynonymous mutations (Figure 2). Notably, higher frequencies of nonsynonymous mutations were observed in nad5, nad4, nad4l, and nad1 (Figure 2).
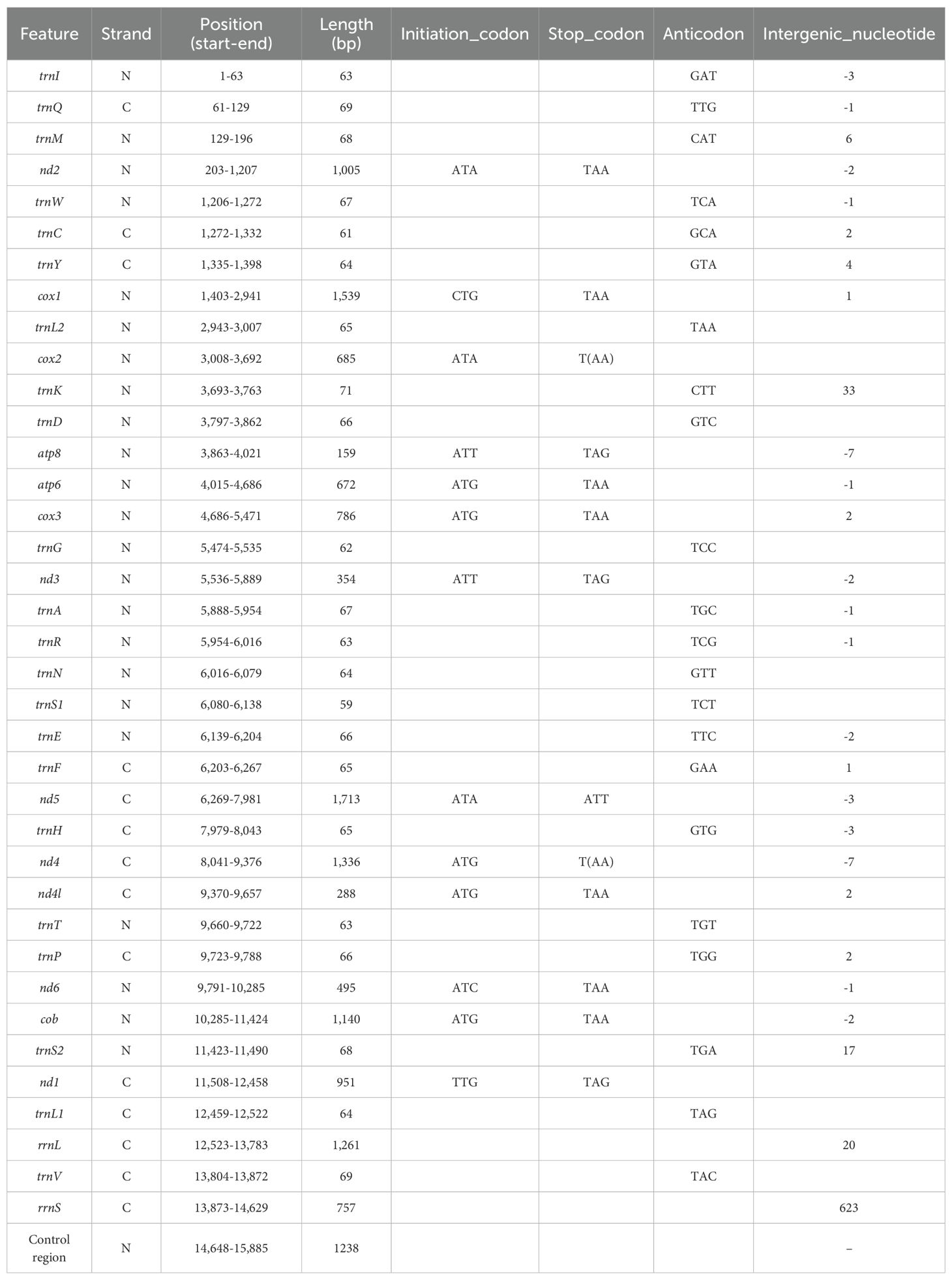
Table 3. Summary of the mitogenome of T. castaneum.
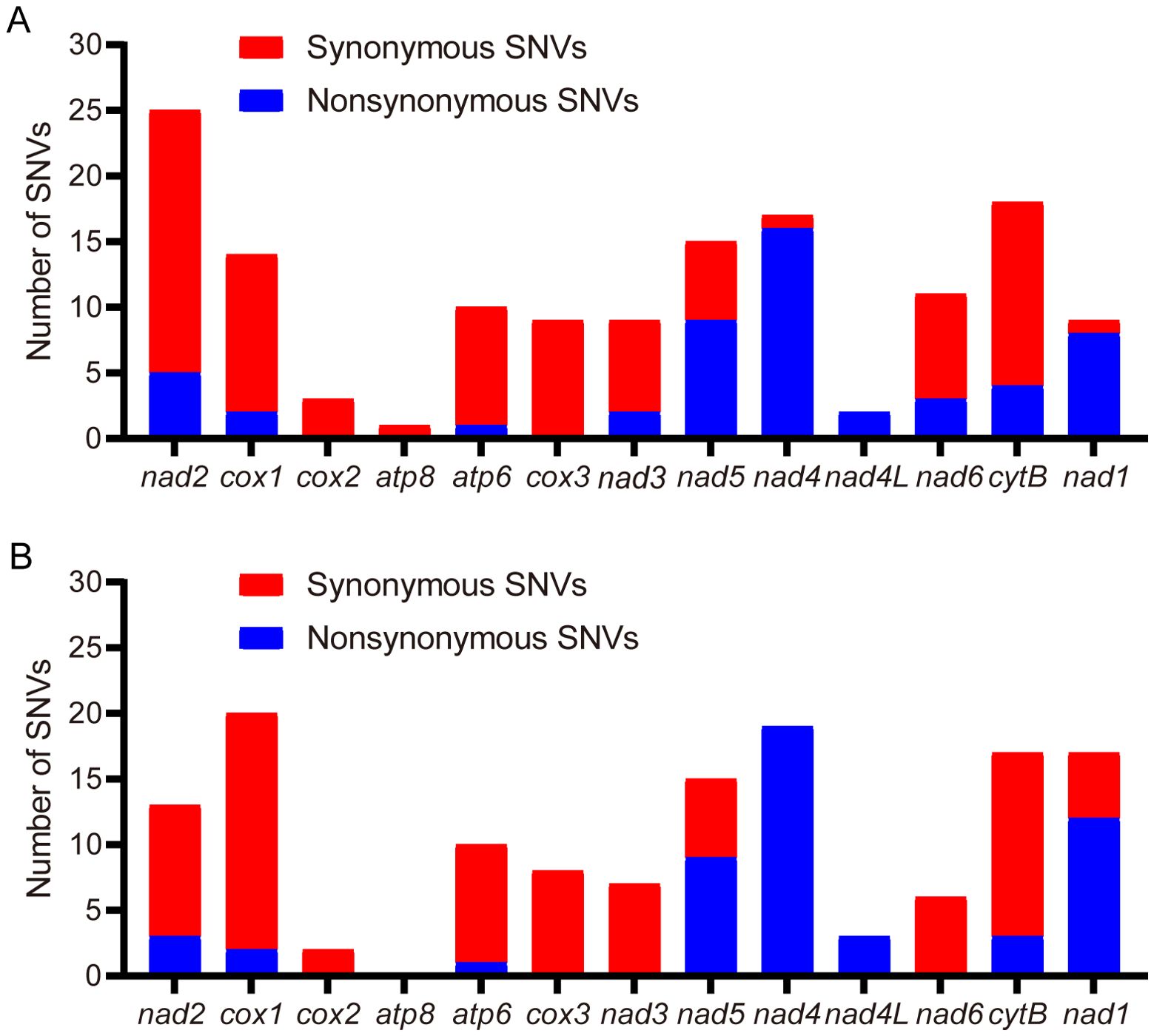
Figure 2. Distribution of single-nucleotide variants (SNVs) in protein-coding genes (PCGs) across T. castaneum populations. (A) SNV density versus Jiangsu strain (KM009121). (B) SNV density versus California strain (NC_003081).
In the present study, the nad3 gene comprises 354 nucleotides encoding 117 amino acids, contrasting with previously reported 360-nucleotide sequences encoding 119 amino acids in other populations (5, 6). Sequence alignment revealed a thymine insertion at position 5,594 within the nad3 locus (Figure 3). This single-nucleotide insertion induces a 7-bp frameshift (original initiation codon ATA at position 5,529 → revised ATT at position 5,536), resulting in complete divergence of the first 20 amino acid residues compared to published sequences. Crucially, this frameshift modifies the open reading frame (ORF), altering the protein’s tertiary structure at the N-terminus while preserving downstream structural domains (Figure 3C), suggesting potential compensatory mechanisms in mitochondrial translation.
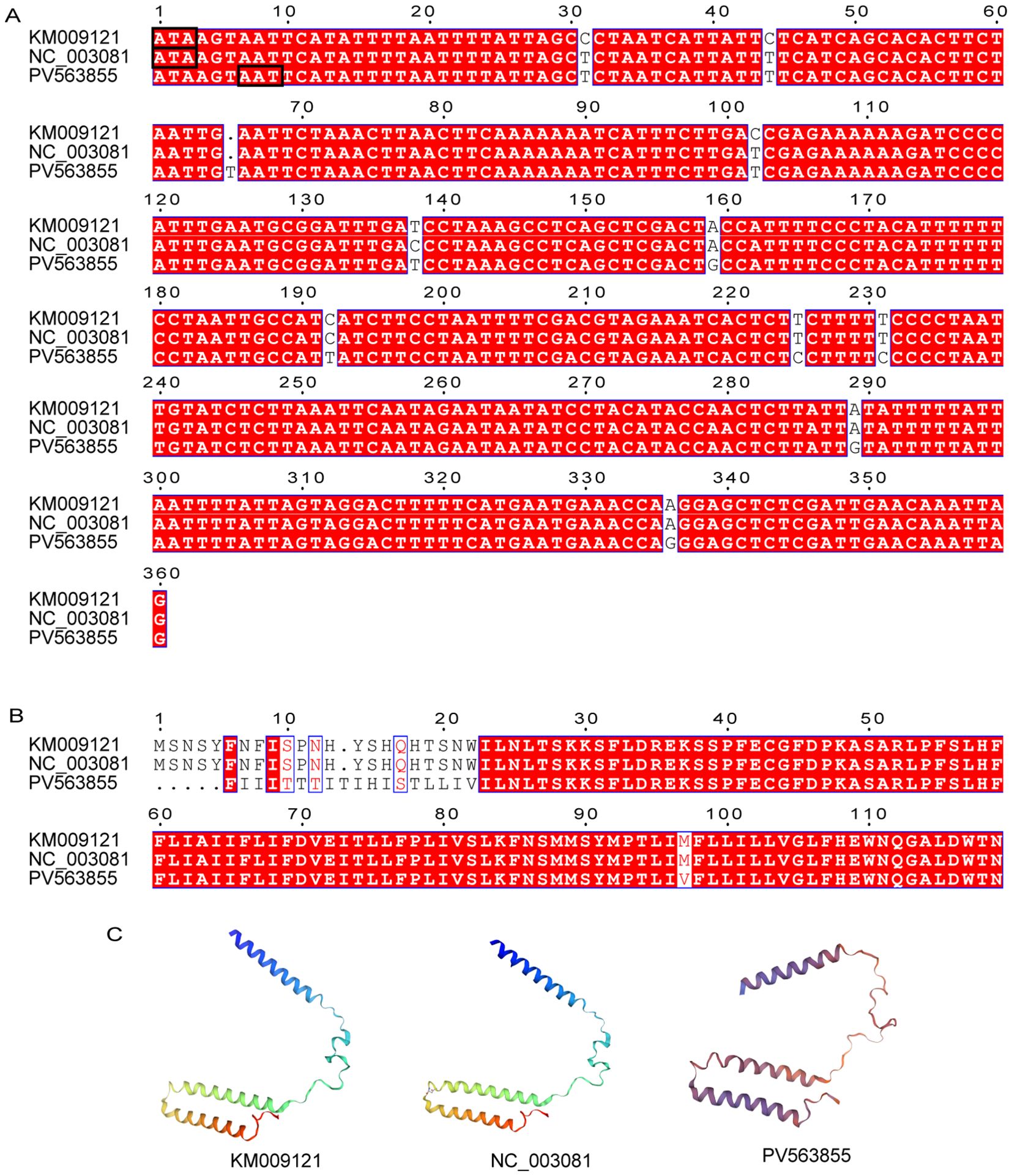
Figure 3. Alignment and sequence similarity analysis of nd3 gene. (A) Nucleic acid sequences. Black box indicates the promoter; (B) Amino acid sequences; (C) Comparison of nd3 protein 3D structure prediction models.
3.3 tRNA and rRNA genes
All 22 tRNA genes were identified, with a combined length of 1,435 bp (range: 59–71 bp; Table 3). Notably, only trnS1 (AGN) deviated structurally, lacking the dihydrouridine (DHU) arm characteristic of canonical cloverleaf folds (Supplementary Figure S2). The remaining 21 tRNAs exhibited standard cloverleaf secondary structures (Supplementary Figure S2). Gene boundaries were determined primarily by homology-based alignment using MITOS, with secondary-structure prediction (tRNAscan-SE) serving as validation. Eight tRNA genes were located on the minor strand, and fourteen on the major strand. The rrnL and rrnS genes measured 1,261 bp and 757 bp, respectively (Table 3).
3.4 Control region
The control region of T. castaneum, a highly variable segment critical for mitogenome replication and transcription initiation, spans 1,238 bp between trnI and rrnS (A+T content: 82.80%; AT-skew: -0.078; GC-skew: -0.001), with population-specific variations: 1,237 bp/82.30% A+T in the Jiangsu population and 1,239 bp/82.50% A+T in the California population (4). Sequence alignment revealed 26 single nucleotide variants (SNVs) between the reported mitochondrial genome control region and the Jiangsu population (6), 20 SNVs compared to the California population (5), including 7 shared SNVs (Supplementary Figure S3).
4 Discussion
The mitochondrial genome of T. castaneum from sauce-flavor Daqu, was sequenced as a circular molecule of 15,885 bp with a high A+T content (71.81%). It includes 13 protein-coding genes (PCGs), 22 tRNA genes, 2 rRNA genes, and a 1,238 bp control region (82.80% A+T). Key observations include unique initiation codons for cox1 (CTG) and nad1 (TTG), differing from previously reported populations, and a frameshift mutation in nad3 caused by a thymine insertion at position 5,594, which altered the open reading frame (ORF) while preserving downstream structural domains. The control region showed minor variations in length (1,238 bp) and A+T content (82.80%) compared to the Jiangsu (1,237 bp; 82.30% A+T) and California (1,239 bp; 82.50% A+T) populations (5, 6).
The PCGs of the mitochondrial genome reported in this study differ from those of two previously reported populations, including single nucleotide variants (SNVs) and insertions/deletions (indels). SNVs caused nonsynonymous mutations in all genes except atp6 and cox3. These mutations altered amino acid sequences, potentially modifying subunit secondary structures and interactions. We hypothesize that the observed SNVs may confer advantages in Daqu-adapted T. castaneum. This hypothesis requires validation through: (1) thermal tolerance tests comparing survival at 60-65 °C; (2) metabolic assays of detoxification enzymes; (3) fitness assessments tracking development and reproduction. A T-base insertion in the nad3 gene changed its initiation site, resulting in distinct differences in the first 20 amino acids compared to the nad3 genes of the Jiangsu (6) and California (5) populations, while subsequent sequences remained nearly identical. The nad3 frameshift may signify adaptive remodeling under Daqu’s unique stresses, mirroring T. castaneum mitochondrial adaptations to environmental pressures. For instance, in cold-adapted Laodelphax striatellus, cytB/nad5 mutations enhance ATP synthesis efficiency by 27% at 4°C despite reduced flight capacity (13); similarly, Curculio chinensis exhibits altitude-driven divergence in atp6/8 genes (dN/dS=0.38) to optimize hypoxia tolerance (14). Analogously, the nad3 frameshift mutation could reconfigure energy metabolism for Daqu’s high-temperature (60–65 °C) and acidic milieu, though functional validation remains essential.
The control region’s exceptional variability—demonstrated by length polymorphisms (1,238 bp vs. 1,237/1,239 bp in comparative strains) and 26 SNVs relative to the Jiangsu population (see Results 3.4)—aligns with its reduced selective constraints. This permits accumulation of neutral mutations through mechanisms like G-C tandem repeat expansion (15) and elevated mutation rates (16), paralleling patterns in Psittaciformes (17).
5 Conclusions
This study presents the first mitochondrial genome of T. castaneum adapted to sauce-flavor Daqu (15,885 bp; GenBank PV563855), revealing population-specific signatures including unique initiation codons (cox1 CTG, nad1 TTG), an nad3 frameshift mutation, and control region polymorphisms. While methodological variations may influence genomic interpretations, these features suggest potential responses to Daqu-specific conditions like organic acids and high-temperature fermentation. Future investigations could examine functional implications through enzyme activity measurements and expanded geographical sampling across Chinese baijiu production regions, providing deeper insights into pest adaptation mechanisms relevant to fermented food industries.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PV563855.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
XMZ: Writing – original draft. RH: Writing – original draft. YC: Writing – original draft. WL: Writing – original draft. XQZ: Writing – original draft. JY: Writing – original draft. JL: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was funded by the Guizhou Provincial Department of Education Youth Science and Technology Talents Growth Project (Qian jiao ji (2024) 264) and Guizhou Provincial Basic Research Program (Qian Ke He Basics-ZK (2025) General 066).
Conflict of interest
Author YC was employed by the company Kweichow Moutai Distillery Group Hongyingzi Science & Technology Development Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/finsc.2025.1621855/full#supplementary-material
Supplementary Figure 1 | The sequencing depth and coverage map for mitochondrial genomes.
Supplementary Figure 2 | Predicted secondary structures of tRNA products encoded by tRNA genes. (A) trnA (B) trnC (C) trnD (D) trnE (E) trnF (F) trnG (G) trnH (H) trnK (I) trnI (J) trnL1 (K) trnL2 (L) trnM (M) trnN (N) trnP (O) trnQ (P) trnR (Q) trnS1 (R) trnS2 (S) trnT (T) trnV (U) trnW (V) trnY.
Supplementary Figure 3 | Alignment and sequence similarity analysis of Control region.
References
1. T. JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. (1999) 9:103–23. doi: 10.1016/s0005-2728(98)00161-3
2. Cameron SL. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. (2014) 59:95–117. doi: 10.1146/annurev-ento-011613-162007
3. de Bruijn MH. Drosophila melanogaster mitochondrial DNA, a novel organization and genetic code. Nature. (1983) 304:234–41. doi: 10.1038/304234a0
4. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. (1999) 27:1767–80. doi: 10.1093/nar/27.8.1767
5. Friedrich M and Muqim N. Sequence and phylogenetic analysis of the complete mitochondrial genome of the flour beetle Tribolium castaneum. Mol Phylogenet Evol. (2003) 26:502–12. doi: 10.1016/S1055-7903(02)00335-4
6. Liu QN, Bian DD, Jiang SH, Li ZX, Ge BM, Xuan FJ, et al. The complete mitochondrial genome of the red flour beetle, Tribolium castaneum (Coleoptera: Tenebrionidae). Mitochondrial DNA A DNA Mapp Seq Anal. (2016) 27:1525–7. doi: 10.3109/19401736.2014.953122
7. Coil D, Jospin G, and Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. (2015) 31:587–9. doi: 10.1093/bioinformatics/btu661
8. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
9. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. (2004) 5:2004–5. doi: 10.1186/gb-2004-5-2-r12
10. Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One. (2014) 9:e112963. doi: 10.1371/journal.pone.0112963
11. Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. (2013) 69:313–9. doi: 10.1016/j.ympev.2012.08.023
12. Chen Y, Chen S, Kang J, Fang H, Dao H, Guo W, et al. Evolving molecular epidemiological profile of human immunodeficiency virus 1 in the southwest border of China. PloS One. (2014) 9. doi: 10.1371/journal.pone.0107578
13. Sun JT, Duan XZ, Hoffmann AA, Y.Liu MR, Chen L, Zhou JC, et al. Mitochondrial variation in small brown planthoppers linked to multiple traits and probably reflecting a complex evolutionary trajectory. Mol Ecol. (2019) 28:3306–23. doi: 10.1111/mec.15148
14. Zhang SK, Fang LX, Liu YN, Wang Y, Zhang W, Shu JP, et al. Genetic Differentiation and Structural Variation of ATP Synthase Gene of Curculio chinensis (Coleptera: Curculionidae) under Selection Pressure at Different Altitudes. Sci Silvae Sinicae. (2019) 55:65–73. doi: 10.11707/j.1001-7488.20190608
15. Lin ZJ, Wang X, Wang J, Tan Y, Tang X, Werren JH, et al. Comparative analysis reveals the expansion of mitochondrial DNA control region containing unusually high G-C tandem repeat arrays in Nasonia vitripennis. Int J Biol Macromol. (2021) 166:1246–57. doi: 10.1016/j.ijbiomac.2020.11.007
16. Lin CY, Tsai LC, Hsieh HM, Huang CH, Yu YJ, Tseng B, et al. Investigation of length heteroplasmy in mitochondrial DNA control region by massively parallel sequencing. Forensic Sci Int Genet. (2017) 30:127–33. doi: 10.1016/j.fsigen.2017.07.003
Keywords: Tribolium castaneum, mitochondrial genome, sauce-flavor Daqu, comparative genomics, genetic variation
Citation: Zhang X, Huang R, Chen Y, Li W, Zhang X, Yang J and Lv J (2025) Complete mitochondrial genome of Tribolium castaneum (Coleoptera: Tenebrionidae) reared on sauce-flavor Daqu. Front. Insect Sci. 5:1621855. doi: 10.3389/finsc.2025.1621855
Received: 02 May 2025; Accepted: 17 August 2025;
Published: 29 August 2025.
Edited by:
Wenjun Bu, Nankai University, ChinaReviewed by:
Jonathan J. Parrott, Arizona State University West Campus, United StatesYash Munnalal Gupta, Naresuan University, Thailand
Copyright © 2025 Zhang, Huang, Chen, Li, Zhang, Yang and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Lv, THl1ajEyM0BZZWFoLm5ldA==