 Jean-Pierre Pouget1,2,3,4*
Jean-Pierre Pouget1,2,3,4* Catherine Lozza1,2,3,4
Catherine Lozza1,2,3,4 Emmanuel Deshayes1,2,3,4
Emmanuel Deshayes1,2,3,4 Vincent Boudousq1,2,3,4
Vincent Boudousq1,2,3,4 Isabelle Navarro-Teulon1,2,3,4
Isabelle Navarro-Teulon1,2,3,4
- 1Institut de Recherche en Cancérologie de Montpellier, Montpellier, France
- 2INSERM, U1194, Montpellier, France
- 3Université de Montpellier, Montpellier, France
- 4Institut régional du Cancer de Montpellier, Montpellier, France
During the last decades, new radionuclide-based targeted therapies have emerged as efficient tools for cancer treatment. Targeted radionuclide therapies (TRTs) are based on a multidisciplinary approach that involves the cooperation of specialists in several research fields. Among them, radiobiologists investigate the biological effects of ionizing radiation, specifically the molecular and cellular mechanisms involved in the radiation response. Most of the knowledge about radiation effects concerns external beam radiation therapy (EBRT) and radiobiology has then strongly contributed to the development of this therapeutic approach. Similarly, radiobiology and dosimetry are also assumed to be ways for improving TRT, in particular in the therapy of solid tumors, which are radioresistant. However, extrapolation of EBRT radiobiology to TRT is not straightforward. Indeed, the specific physical characteristics of TRT (heterogeneous and mixed irradiation, protracted exposure, and low absorbed dose rate) differ from those of conventional EBRT (homogeneous irradiation, short exposure, and high absorbed dose rate), and consequently the response of irradiated tissues might be different. Therefore, specific TRT radiobiology needs to be explored. Determining dose–effect correlation is also a prerequisite for rigorous preclinical radiobiology studies because dosimetry provides the necessary referential to all TRT situations. It is required too for developing patient-tailored TRT in the clinic in order to estimate the best dose for tumor control, while protecting the healthy tissues, thereby improving therapeutic efficacy. Finally, it will allow to determine the relative contribution of targeted effects (assumed to be dose-related) and non-targeted effects (assumed to be non-dose-related) of ionizing radiation. However, conversely to EBRT where it is routinely used, dosimetry is still challenging in TRT. Therefore, it constitutes with radiobiology, one of the main challenges of TRT in the future.
Introduction
This article, which is part of the inaugural series for the launch of Frontiers in Nuclear Medicine, will discuss some of the main challenges of radiobiology in targeted radionuclide therapy (TRT). Investigating radiobiology and performing accurate dosimetry will contribute to the improvement of the therapeutic efficacy of TRT, especially in the case of solid tumors.
Radiobiology explores the biological effects of radiations. This research field was created following the description of the first cases of cutaneous erythema associated with the clinical use of radiation at the beginning of the twentieth century. The use of X-rays to treat patients with cancer was first experimented by V. Despeignes in Lyon in 1896, 6 months after their discovery by W. Roentgen (1). As early as 1902, the ability to quantify the delivered radiation dose (dosimetry) and to establish the dose–effect relationship led to a significant improvement of the patients’ outcome. In 1919, dose fractionation started to be investigated by C. Regaud, at the Curie Institute (Paris), who described how to treat tumors with high absorbed doses, while protecting healthy tissues (2).
Most of what we know about radiobiology concerns external beam radiation therapy (EBRT). Particularly, the therapeutic efficacy of low (X and γ rays, electrons) and high linear energy transfer (LET) radiation (neutrons, Auger electrons, protons, alpha-particles, and heavy ions) has been extensively investigated. This has been accompanied by the development of new techniques and technologies for dose delivery to the tumor (3). Besides total dose and LET, the biological effects of radiation depend on the absorbed dose rate, absorbed dose fractionation, tissue oxygenation, and volume of irradiated tissue. In addition, the cell response to radiation is highly dependent on the nature of the irradiated tissue (genetic background, cell proliferation rate) and its microenvironment.
Cellular Radiobiology
Initial Events
Ionizing radiations interact with biological substrates through direct and indirect mechanisms (4). Direct effects involve one-electron oxidation reactions, while indirect effects are mediated through water dissociation, leading to the production of reactive oxygen species (ROS), such as superoxide radicals and hydrogen peroxide (H2O2), the precursors of the highly damaging hydroxyl radicals (⋅OH). Noteworthy, these ROS are similar to those produced by endogenous sources, such as the mitochondrial oxidative metabolism (leading to formation during oxygen reduction), the plasma membrane-bound nicotinamide adenine dinucleotide phosphate [NAD(P)H] oxidases and lipoxygenases (5–7) and peroxisomes (formation of H2O2). Massive production of ROS and of reactive nitrogen species (RNS) is also mediated by activation of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), for instance, following induction of transcription factors involved in the inflammatory response, such as nuclear factor kappa B (NF-κB) or activator protein-1 (AP-1) (8). NF-κB is activated by ataxia telangiectasia mutated (ATM), the main protein involved in DNA damage recognition. COX-2 leads to production of prostaglandin-E2 and ROS that are released in the intra- and extra-cellular medium (9) and contribute to the inflammatory responses (8) (Figure 1). Activation of iNOS leads to the formation of nitric oxide (NO) that can react with superoxide anion to form RNS, such as peroxynitrite (10–12). and NO are produced by macrophages during inflammatory reactions, but they are also released by irradiated cells (12). can generate many of the degradation products observed with ⋅OH (9). Moreover, differently from ⋅OH that is very reactive and diffuses for only about 4 nm, can diffuse easily within cells and its highly oxidizing protonated form (ONOH) can cause DNA damage, cell death as well as protein and lipid peroxidation. H2O2 and NO can diffuse between cells (4).
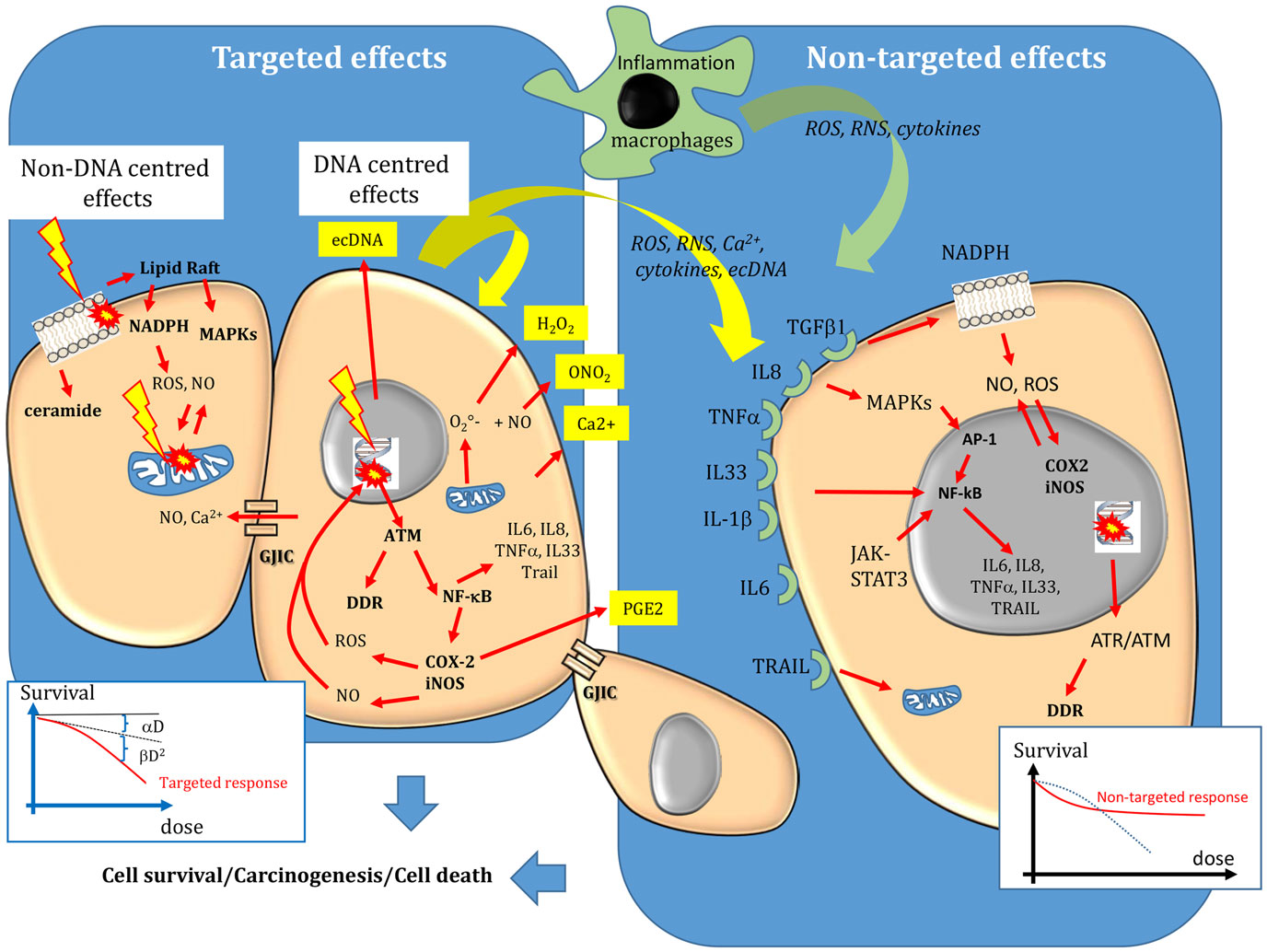
Figure 1. Targeted and non-targeted biological effects in conventional external beam radiotherapy. Targeted effects are caused by one or more particles traversing irradiated cells and can be divided in DNA and non-DNA-centered effects. Non-targeted effects describes the effects observed in cells that have not been directly traversed by particles but that are close to irradiated cells, as well as long-distance effects. DNA, mitochondria, and the cell membrane are the main sensitive targets of radiation. Following targeted and non-targeted effects, cells can survive (lesions are effectively repaired), they can die (lesions are not repaired) or they can be transformed. The dose–effect relationship of targeted effects is commonly fitted by linear or linear-quadratic models. A saturation of the response to non-targeted effects has been described. For more details, see the main text.
Therefore, ROS and RNS participate in physiological processes including cell signaling, immune response, inflammation, apoptosis, and cell growth, and also in the cell response to radiation (8). These endogenous and exogenous reactive species can cause cellular damage, when imbalance occurs between their production and their destruction by the cell enzymatic and non-enzymatic defense systems. For instance, can be reduced to H2O2 by the enzyme superoxide dismutase. H2O2 can in turn be reduced to water by the catalase or glutathione peroxidase enzymes, or can be used, in the presence of metal ions, such as Fe2+, to produce ⋅OH through the Fenton reaction. Superoxide dismutase, catalase, and glutathione peroxidase are part of the enzymatic defense system developed by cells to keep the level of these endogenous ROS as low as possible. Several intracellular components, particularly glutathione, urates, bilirubin, and vitamin E and C, can also act as radical scavengers. When the balance is tilted in favor of reactive species, all cell compartments (cell membrane, mitochondria, and particularly the nucleus) and constituents (DNA, lipids, and proteins) may be harmed and their functions altered.
DNA Damage
Radiation-induced oxidative DNA damage resulting from ⋅OH attack (indirect effect, water radiolysis) or from one-electron oxidation (direct effect) includes single-strand (SSB) and double-strand DNA breaks (DSB), DNA base damage (oxidized and abasic sites), and DNA–DNA or DNA–protein crosslinks.
The main reactions of radiation-induced radicals with DNA are hydrogen abstraction from deoxyribose molecules by ⋅OH (mainly from the C’ carbon of the sugar moiety) and ⋅OH addition to the π bonds of the bases [for reviews see Ref. (4, 13, 14)]. It is estimated that about 80% of ⋅OH radicals react with bases and the remaining 20% with sugar moieties. Hydrogen abstraction from the C2 of the sugar moiety can result in base loss or SSB formation through phosphate elimination. SSBs can also be produced after ⋅OH reaction with pyrimidine bases. DSBs can be caused by the attack of one or two ⋅OH radical species. However, it is very unlikely that a segment of a few DNA bases will be affected by two independent radiation events (between 10–6 and 10–4 Gy–1 for direct and indirect effects, respectively). Indeed, at biologically relevant doses, it is more probable that a single radiation track might induce ionization on opposite strands in close proximity. Therefore, DSBs can be produced after DNA denaturation consecutive to two SSBs occurring within a 20-bp stretch.
DNA damage incidence is proportional to the absorbed dose and is quantified per Gy and per cell, after exposure to low and high LET radiation (15). About 40 DSBs are formed per cell per Gy of low-LET radiation. DNA base lesions are the most abundant (about 1000/cell/Gy) and fourteen types of oxidative purine and pyrimidine DNA damage have been detected in cellular DNA, among which 8-oxo-7,8-dihydroguanine (8-oxoGua) and 5,6-dihydroxy-5,6-dihydrothymine (thymine glycol) are the most frequent (4). Although DSB frequency is quite low, cell survival and mutagenesis are highly dependent on DSB spatial distribution. About 150 DNA–protein and 30 DNA–DNA crosslinks are also estimated to be produced per Gy and per cell (15).
Accumulation of DNA damage can lead to even more complex lesions called locally multiply damaged sites (LMDS), also known as oxidative clustered DNA lesions (OCDLs) (16–18). LMDS occur when more than two lesions are produced within one or two helices, i.e., within 20 bp. These clustered DNA lesions include complex SSBs and also simple or complex DSBs. They may include up to 10 lesions in the case of low-LET radiation and even more complex damage with high LET radiation. Thus, although low and high LET radiation produce the same DNA lesions, their yield and spatial distribution are different.
DNA Damage Repair Systems
Cells have developed DNA damage repair (DDR) systems against DNA lesions. DNA damage activates sensor systems, such as ATM, Ataxia Telangiectasia, and Rad 3-related (ATR), which in turn induce signaling pathways involved in the cell response to radiation, including cell cycle arrest, DNA repair, or cell death (19–22). DNA base damage is mostly repaired by the base excision repair (BER) mechanism and to a lesser extent by the nucleotide excision repair (NER) mechanism (23, 24). DSB repair schematically involves two major recombination pathways. The first one is the homologous recombination (HR) system that is active specifically in the S and G2 phases of the cell cycle (25). It is an error-free mechanism because undamaged DNA (the sister chromatid) is used as template for DNA repair. The second mechanism is the non-homologous end-joining (NHEJ) system, which is active mainly in the G1 phase of the cell cycle (26). Although NHEJ is the main repair pathway used by irradiated cells, it is thought to be error-prone because the broken DNA ends are ligated without the need of an undamaged, homologous template.
When accurately repaired, DNA lesions have no effect on cell survival or in daughter cells. However, if the lesions are too complex or abundant, or if the cell DNA repair machinery is deficient, not all DNA lesions will be completely or correctly repaired. Unrepaired/misrepaired DNA damage can cause the formation of chromosomal aberrations when cells progress through the cell cycle, leading to mitotic catastrophe (cell death during mitosis) or to programed cell death (known as apoptosis). Not all unrepaired lesions are lethal for the cell. In this case, they may be passed to the daughter cells and could lead to mutations, genomic instability and eventually to cancer development (Figure 1).
A DNA-Centered Approach
According to the DNA-centered view of radiation-induced damage, unrepaired DNA lesions are the lethal event leading to cell death. The concerned DNA lesions are mainly DSBs, but more complex damage, involving OCDLs, may also constitute lethal lesions. As the aim of radiation therapy is to kill tumor cells or at least to prevent their division, the clonogenic assay, which was developed by Puck and Markus in 1956 to investigate the ability of a cell to form a new colony, has become the reference technique for assessing the cell response to radiation (27). This assay shows that the clonogenic survival of irradiated cells decreases exponentially as a function of the mean absorbed dose. Plotted on a graph, the survival logarithm can be experimentally fitted by a linear or a linear-quadratic regression model, depending on whether high or low-LET radiation is used. The linear part (αD) of the equation corresponds to single-hit killing events, while the quadratic part (βD2) requires two hits to kill cells (Figure 1). Thus, the overall cytotoxic effect is due to the sum of single and double hit events. This interpretation of cell cytotoxicity corresponds to the so-called target theory, which is an essential concept for understanding radiation biology. The fundamental principle of the target theory is that inactivation of the target(s) by a lethal event requires the cells to be crossed by radiation. The shape of the survival curve can be affected by the tissue radiation sensitivity that is defined by the α/β ratio and represents the cell capacity to repair damage. Radiation sensitivity, which was first hypothesized by Bergonié and Tribondeau in 1905 (28), is essentially explained by the patient’s genetic background concerning the DNA repair enzymes, antioxidant defenses and tissue proliferation. For instance, patients with pathologies like Ataxia Telangiectasia, Xeroderma Pigmentosum, Cockayne, or Nijmegen breakage syndromes have defects in proteins involved in DNA DSBs repair and show hypersensitivity to radiation (29–31). The shape of the survival curves is also affected by the LET, tissue hypoxia and dose fractionation, as shown in the 60s by Barenden and colleagues (32–36). The relevance of the α/β ratios determined in vitro in human cell lines for understanding the in vivo values has been extensively reviewed (37, 38).
New Paradigms in Radiation Biology: Non-DNA Centered and Non-Targeted Effects
For about a century, the paradigm of radiation biology has been that the biological effects of ionizing radiation occur only in the nucleus of cells crossed by particles and that cell death is strictly due to unrepaired or misrepaired DNA. Therefore, the biological effects of ionizing radiation should be strictly related to the energy absorbed by the tissues and the survival of irradiated cells, expressed as a function of the dose, should be strictly fitted by a linear or linear-quadratic curve explained by DNA hits. However, such DNA-centered approach is not fully satisfying. For instance, it cannot explain hypersensitivity to low doses and radiation sensitivity syndromes associated with mutation of cytoplasmic proteins. In addition, studies in cells, animal models, or patients treated by radiotherapy reported that biological effects could be observed also when only the cell cytoplasm was irradiated (known as non-DNA-centered effects) and in non-irradiated areas (known as non-targeted effects or bystander effects) (39–42). As early as 1922, the release of stress mediators in the serum of irradiated mice (43) or in blood samples from irradiated patients (44–46) was associated with long-distance bystander effects called abscopal effects. The concept of bystander effects emerged again in 1992 when Nagasawa and Little reported that in monolayer cell cultures exposed to alpha micro-beams, sister chromatid exchanges were observed in 30% of cells, although <1% of cells were crossed by particles (47). Since then, many studies have investigated the origin and nature of radiation-induced bystander effects that are defined as biological effects occurring in the neighborhood of irradiated cells (47).
Bystander effects include mutations, clastogenic effects, cell death, apoptosis, and cell transformation (6). They mainly occur after low dose (<1 Gy) or low dose-rate irradiation, although they have been observed in EBRT also after high absorbed dose (10 Gy). They involve signaling from irradiated cells toward non-irradiated cells. Specifically, stress mediators are transmitted to bystander cells by cell–cell interactions through gap junction intercellular communication (GJIC) when cells are in contact and the molecules are small (<1500 Da) (Figure 1), or by the release of soluble damage/stress signals that may have distant biological effects (abscopal effect) (6, 48–50). These mediators can be ROS/NO, cytokines (interleukin 8, interleukin 6, tumor necrosis factor, and interleukin-33), Ca2+, or extracellular DNA (ecDNA). They are produced by irradiated cells and are released in the extracellular environment (39), thus inducing oxidative stress in neighboring cells/tissues. However, they can be active also within the cell in an autocrine way. They can also activate immune cells (for instance, macrophages and T lymphocytes) that, in turn, release cytokines, leading to iNOS induction, and NO formation (51). Therefore, the inflammatory and radiation responses share common mechanisms to promote and perpetuate a harmful inflammatory/oxidative stress environment. This new paradigm also highlights the role of the tumor (or healthy tissue) microenvironment in the radiation response. The finding that ROS scavengers, such as DMSO, abolish the bystander response indicates that oxidative stress plays a major role in this phenomenon (52). While ⋅OH have a short life and interact within few nm, other species, such as H2O2 and NO, can migrate across the plasma membrane and cause oxidative damage in neighboring cells. Moreover, other systems leading to sustained intercellular production of reactive radicals can be activated in neighboring cells after cytokine release by irradiated cells or binding of immune cells to bystander cells. Interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-33 (IL-33), for instance, activate NF-κB that is involved in the expression of the COX-2 and iNOS genes, which participate in the inflammatory response and the local production of ROS and NO, respectively (Figure 1). Furthermore, TNF-α, interleukin 8, and transforming growth factor 1 b (TGFβ-1b) can activate the mitogen-activated protein kinase (MAPK) pathways [extracellular signal-related kinase (ERK), c-JUN N-terminal kinase (JNK), and p38] that participate in COX-2 and iNOS up-regulation (11, 53). Interleukin 6 is released by irradiated macrophages and activates Janus-kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3). STAT3 contributes to NFκ-B retention in the nucleus and thereby in the induction of COX-2 and iNOS expression (54, 55). TGFβ-1 secreted by irradiated cells can also activate NADPH oxidase, which is located at the cell membrane and is involved in bystander ROS and NO production. Therefore, bystander factors are involved in the long-term production of reactive radicals in a feed-forward and self-sustaining fashion and in the creation of an inflammatory environment, leading to the recruitment of immune cells (52). Noteworthy, the increase in intracellular oxidative stress leads to mitochondrial dysfunction that can further exacerbate oxidative processes by releasing ROS and RNS.
Finally, it has been shown that different signaling pathways are involved in DNA repair, depending on whether DNA damage is produced directly by irradiation, or indirectly by oxidative stress-induced bystander effects (56). Moreover, while targeted biological effects increase with the dose in EBRT, a saturation response is observed when non-targeted effects are involved, and above a certain dose, no additional effect is observed (Figure 1).
New Paradigms in Radiation Biology: Extra-Nuclear Targets
Cell Membrane
Although most attention has been focused on DNA as the main radiation target, the idea that the membrane could also be an important target was suggested in 1963 by Alper et al. (57). The cell membrane is now recognized as a key player in the radiation-induced biological effects. Its role in the cellular response to radiation may be explained by its function in many signaling pathways, including apoptosis (58–60). Radiation-induced hydroxyl radical molecules can attack not only nuclear DNA but also polyunsaturated fatty acid residues of membrane phospholipids. This results in the formation of malonedialdehyde or 4-hydroxynonenal that can induce DNA–protein crosslinks (58). Radiation can also cause activation of acid sphingomyelinase that hydrolyzes sphingomyelin in the cell membrane to produce ceramide and phosphorylcholine (59–62). Ceramide is a second messenger of apoptosis and is also involved, when associated with cholesterol, in the formation of ceramide-enriched platforms (also known as lipid rafts) containing signaling and transport proteins. These platforms play a central role in cellular functions such as cell signaling and trafficking. Specifically, plasma membrane activation stimulates diverse signaling pathways that are mediated by the MAPK superfamily, including ERK1/2, JNK, and p38. Moreover, lipid rafts contain NADPH oxidase that is involved in the sustained ROS/RNS production by bystander cells. NADPH can be activated by TGFβ secreted by irradiated cells after activation of cell membrane receptors by cytokines (TGFβ, TNFα, interleukins) and Ca2+ ion channels. These indirect effects can be partially inhibited by antioxidants, such as vitamins E and C, and by the enzymes superoxide dismutase, catalase, and glutathione peroxidase.
Mitochondria
Mitochondria are central cell organelles. They are involved in cell respiration by reducing O2 into during ATP production and constitute one of the main source of endogenous ROS and RNS (8, 63). They also play a role in radiation-induced cell signaling pathways, such as apoptosis (64). Indeed, one of the critical events is the change in mitochondrial membrane potential leading to leakage and release in the cytosol of pro-apoptotic proteins, including cytochrome C and apoptosis-inducing factor (10). This may be due to high ROS and NO production in response to direct irradiation or non-targeted effects. Moreover, ROS, such as superoxide anions, released by mitochondria can contribute to intracellular oxidative stress and non-targeted effects through their conversion into diffusible H2O2 molecules (52).
Mitochondrial DNA can also be altered after ROS attack or by direct radiation effects (65–67). Mitochondrial DNA is very sensitive to oxidative stress because it is not protected by histones, and mutations or deletions have been observed. When they concern the genes coding for mitochondrial ATPase, NADH dehydrogenase complex I and cytochrome c oxidase, they can lead to defects in the mitochondrial metabolism and the DNA repair efficiency as well as to the increase in ROS level. These effects have been observed both in directly irradiated cells and in non-targeted cells.
Targeted Radionuclide Therapy
Beside EBRT improvements, TRT has emerged as an attractive approach for treating tumors during the twentieth century. In TRT, a radionuclide is coupled to a vector [for instance, monoclonal antibodies (mAbs) or peptides] directed against cancer cells or their environment to specifically irradiate only the tumor targets. Therefore, TRT is particularly attractive when conventional EBRT (CEBRT) cannot be used due to unacceptable toxicities toward healthy tissues. This is the case of disseminated disease, metastases, or tumors located in close vicinity of sensitive organs. Compared to chemotherapy, it offers the possibility to specifically target tumor cells, thereby reducing the side effects, and also to treat distant tumor cells (which cannot be directly reached by the drug) through cross-fire irradiation (i.e., energy deposition in cells that are not specifically targeted) (Figure 2).
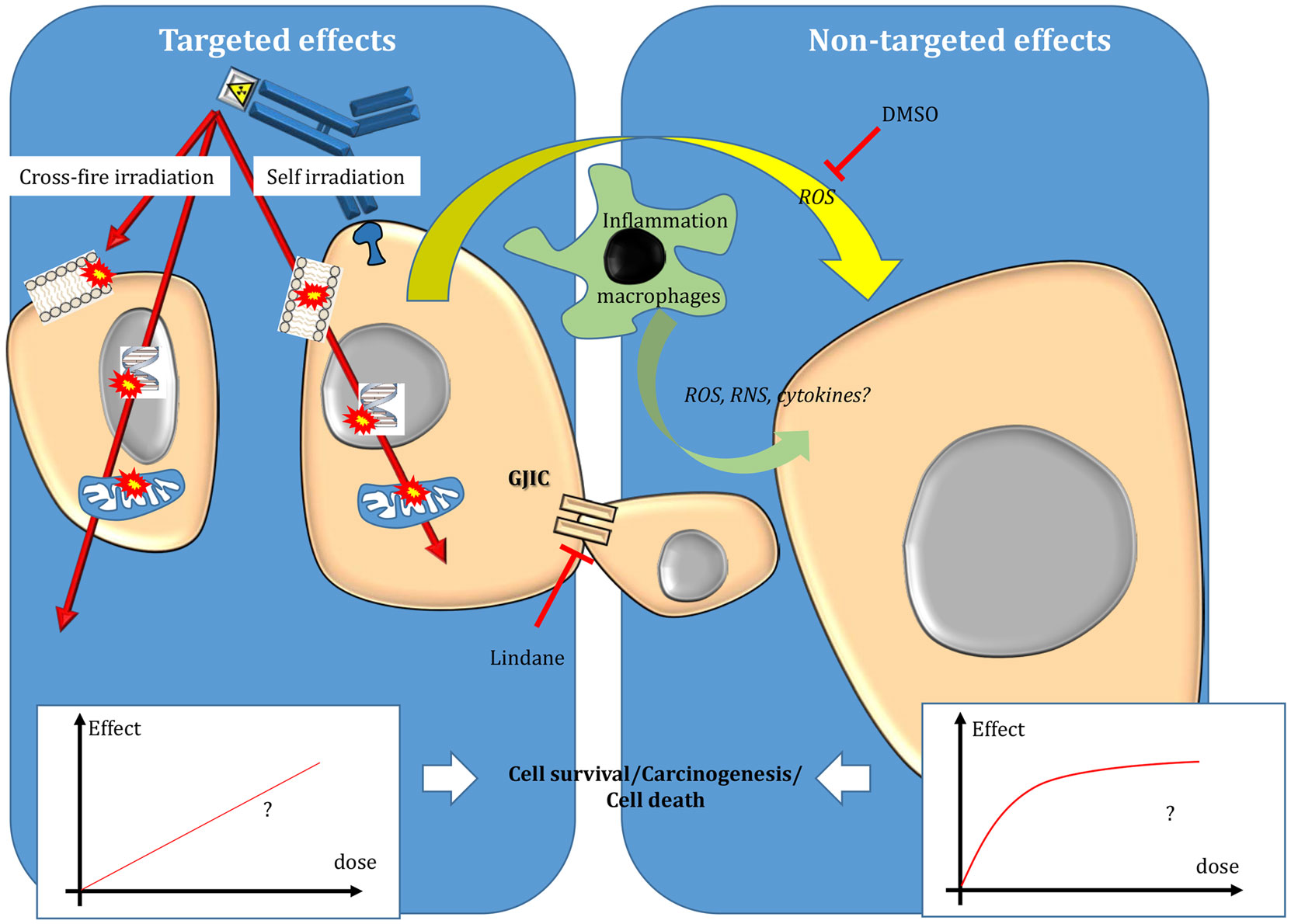
Figure 2. Targeted and non-targeted effects in targeted radionuclide therapy. Targeted effects are caused by one or more particles crossing irradiated cells and can be due to self-irradiation and cross-fire irradiation. Non-targeted effects include effects observed in cells close to irradiated cells and also long-distance effects. The nature of the dose–effect relationship resulting from targeted and non-targeted effects needs to be determined. For more details, see the main text.
Radionuclide therapy started in the 20–40s when injections of very simple chemical forms of a radionuclide (131I or 32P) were used to treat patients with differentiated thyroid or ovarian cancers, respectively. Other compounds have been largely employed in nuclear medicine, such as 89SrCl2, 153Sm-EDTMP, or 186/188Re–HEDP, for palliative treatment of bone metastases. Radio-embolization with 90Y microspheres has been used for the treatment of hepatocellular carcinoma. In 2013, 223Ra chloride (Xofigo®) was the first Food and Drug Administration (FDA)-approved unsealed α-emitting radiopharmaceutical for the treatment of patients with castration-resistant prostate cancer and metastatic bone disease. Radionuclides can also be bound to a vector that specifically targets tumor cells for TRT. Specific molecules, such as metaiodobenzylguanidine (mIBG) labeled with 131I for TRT of neuroblastoma and medullary thyroid cancer, have been progressively developed. Peptide receptor radionuclide therapy (PRRT) started to be assessed in the 70–80s and led to the development of 90Y-DOTATATE and 90Y-DOTATOC that are currently used for the treatment of neuroendocrine tumors. In radioimmunotherapy (RIT), antibodies against cancer cell antigens are used to target the radionuclide to cancer cells. In RIT, the radiation-induced biological effects may be combined with the antibody cytotoxic effect. The first RIT assays in patients using 131I-labeled polyclonal antibodies against cancer cell antigens were performed in 1953. The first clinical trials with mAbs (131I-labeled or 67Cu-labeled anti-HLA-DR mAbs) began in 1988 and 131I-labeled anti-CD20 antibodies were first used in patients with non-Hodgkin lymphoma (NHL) in 1993. Two radiopharmaceuticals for RIT have been approved by FDA (90Y-ibritumomab tiuxetan, Zevalin®, in 2002 and 131I–tositumomab, Bexxar®, in 2003) for the treatment of relapsed or refractory low-grade, follicular, or transformed B-cell lymphoma (68). NHL is undoubtedly the disease in which RIT has the highest success rates. Indeed, the overall response rate is between 60 and 83% in previously treated patients and of 95% in patients treated for the first time (69) compared to 56% in patients treated with rituximab immunotherapy (68). As the Zevalin® and Bexxar® activities administered to patients are currently based only on the patient’s weight (MBq/kg for Zevalin®) or on delivering 75 cGy to the whole body (Bexxar®), further improvements could be expected by planning patient-tailored treatments using a dosimetry approach.
On the other hand, TRT of solid tumors is more challenging, mainly because these tumors are more radiation-resistant than lymphomas. In such context, the approach consisting in one injection of the maximal tolerated activities of a radiopharmaceutical, based on the patient’s weight, is not sufficient and a more complex strategy is required (70–73) that takes into account both radiobiology and dosimetry data.
TRT Radiobiology Specificity
Although particles emitted by radionuclides produce similar physical events (ionization/excitation) as those described in EBRT, TRT radiobiology cannot be strictly extrapolated from the radiobiology developed for EBRT.
The main differences between EBRT and TRT concern the absorbed dose-rate and the spatial energy deposit (Figure 3). CEBRT produces homogeneous irradiation at high absorbed dose rate (about 1–2 Gy⋅min−1) of low-LET X-rays that target all the cells in the field with a total absorbed dose of 40–80 Gy, administered in fractions of 2 Gy, five fractions per week. Conversely, TRT is characterized by low absorbed dose rate (<1 Gy⋅h−1) with protracted, heterogeneous, and mixed irradiation. Indeed, vectors can be coupled to radionuclides that emit beta, alpha, or Auger electrons, associated or not with X or γ rays. In addition, when using alpha particle emitters, the decay spectrum of daughter radionuclides should also be taken into account. Therefore, LET ranges from 0.2 keV/μm for beta, X, and γ rays and 4–25 keV/μm for Auger electrons to 50–230 keV/μm for alpha particles (Figure 3). The particle path length will be also variable: few nm–μm for Auger electrons, fifty to about one hundred μm for alpha particles and from μm to mm for beta particles. Finally, the distribution of radiolabeled molecules is generally non-uniform and leads to strong heterogeneity in activity distribution, whatever scale considered (subcellular, organ, or organism). Therefore, some territories will be irradiated while others may partly escape. This phenomenon is even more marked for short range emitters, such as alpha particle and Auger electron emitters, because the path length of the emitted particles produces low cross-fire irradiation (i.e., cells are crossed by particles emitted by radiolabeled vectors bound to neighboring cells) (Figure 2). However, the highly localized energy deposition associated with Auger electrons and alpha particles is also an attractive tool to investigate the biological effects of ionizing radiations at the subcellular scale and has shown the weaknesses of a completely DNA-centered conception of TRT effects. Indeed, we and others have demonstrated that extra-nuclear targets are also involved in targeted and non-targeted responses (74–78). The existence of non-targeted effects may affect the absorbed dose–effect relationship.
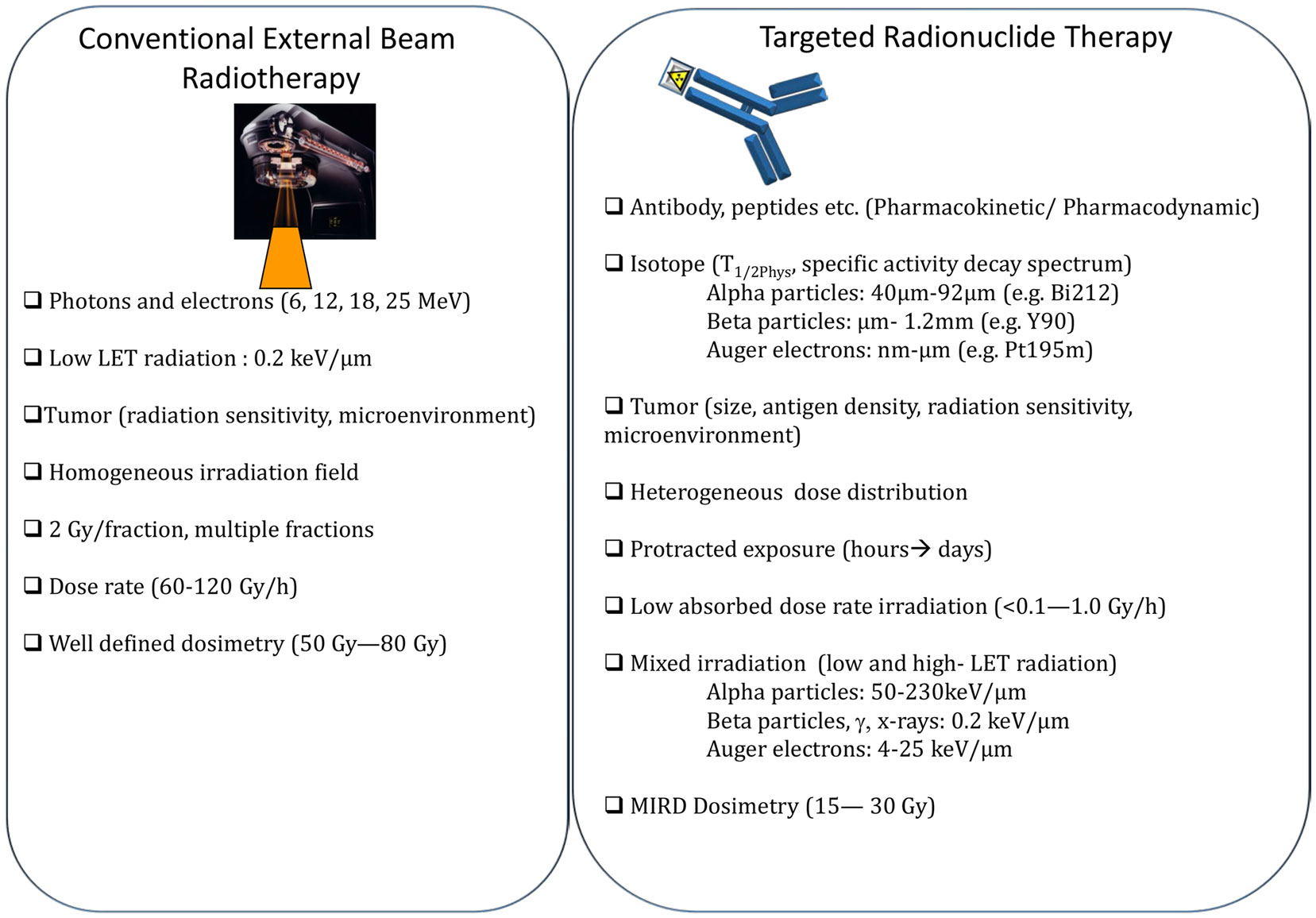
Figure 3. Comparison of conventional external beam radiotherapy and targeted radionuclide therapy.
Improving Dosimetry in TRT
One difficulty when investigating TRT radiobiology is that in many preclinical and clinical studies, dosimetry is not available or is not fully accurate. Biological effects are often related to the activities injected in patients/animal models (Bq/kg) or added to the medium of cultured cells in in vitro experiments (Bq/mL). However, like for EBRT, dose assessment is, at least theoretically, the only reference parameter common to all forms of treatment involving radiation and the dose–effect relationship provides useful information on how radiations act. In TRT, the biological effects observed in patients/animal models or in cultured cells will depend on the used activity, its distribution within the different compartments (organs, cell compartments), its pharmacokinetics/dynamics, the type of emission, and the target geometry (organ, cells, nucleus) and also, like for EBRT, on the nature of the targeted tissue and its microenvironment.
Therefore, dosimetry is a requirement for the radiobiologist; however, determining the absorbed dose is not straightforward in TRT and is often considered a tedious and unattractive task. The approach commonly used is based on the medical internal radiation dose (MIRD) formalism (79):
The MIRD formalism requires to determine the cumulative number of decays ÃrS, which is the time integrated activity (or total number of decays) occurring in a source region rS over the dose integration period TD, and the corresponding S values, which represent the absorbed dose in the target region rT per nuclear transformation in rS.
Determining the activity distribution and S values
The reliability of the radiation dose estimates in preclinical and clinical TRT studies is directly related to the accuracy of the activity assessment at each time point over the considered period. For instance, in in vitro experiments, the uptake of radioactivity per cell is assessed at various intervals after exposure to the radiolabeled vector. For this, the radioactivity distribution within the cell population needs to be considered (80). Moreover, the subcellular distribution may be different depending on the targeting vector and on the final source (cell membrane, cytoplasm, organelles, or nucleus) according to the MIRD formalism. For animal experiments, several methodologies are now available. The standard method relies on the determination of the average radioactivity in different organs after animal sacrifice at various time points following administration of the radiopharmaceutical. This approach can be completed by digital autoradiography that provides information about the spatio-temporal distribution of radioactivity in tissue cryosections (81). The development of small animal imaging techniques (micro-SPECT-CT or micro-PET-CT) provides an attractive alternative method by reducing the number of needed animals and allowing longitudinal studies (82, 83). However, because of the inherent limits of these imaging techniques, the determination of radioactivity is limited to organs showing specific targeting. In the clinic, dosimetry is routinely used in EBRT; conversely, TRT dosimetry is still in its early days and it is much more difficult. Initially, conventional methods based on planar imaging were used to assess the radioactivity distribution. However, it is now clear that, due to the inherent heterogeneity of the distribution of a radiopharmaceutical in tissues, three-dimensional anatomical (CT or MRI) and functional [PET or single-photon emission CT (SPECT)] imaging approaches are required and that the S value must be calculated using Monte Carlo simulations. However, these techniques are much more demanding in terms of human and methodological resources and uncertainties in the radiation dose estimation may still arise from incomplete kinetic assays and low accuracy in volume measurements and S value calculation (84). In both clinical and preclinical studies, S values (i.e., the absorbed dose per decay) can be determined using Monte Carlo codes that allow following the radiation transport and scoring the energy deposit (85–87). For this purpose, the cell, organ, or organism geometry must be known (87, 88). This can be done by microscopic observation of cells in vitro, and by using phantoms or the information provided by CT scans for animal models and patients.
Determining dose–effect relationship
Unfortunately, very few preclinical studies are available to demonstrate the validity in TRT of the linear quadratic or linear dose–effect relationship, as commonly observed in EBRT. In clinical TRT studies, interesting and encouraging results on the correlation between tumor-absorbed dose and treatment efficacy have been highlighted (89, 90), although no strong dose–effect relationship has been shown even in lymphoma (91), which is the disease most frequently treated by TRT. Strigari et al. reported that among 79 radiotherapy studies investigating dosimetry, an absorbed dose–effect correlation was found in 48 (92). Yet, the existence of a dose–effect relationship (efficacy/toxicity) in TRT is still a matter of debate. Besides performing accurate dosimetry, the most relevant biological endpoints must also be identified. Indeed, the follow-up of tumor shrinkage in patients is difficult because TRT is mainly used to treat small-volume solid tumors or disseminated disease. Therefore, other parameters, such as the clinical response or progression-free survival, should be investigated as endpoints to evaluate TRT efficacy and the dose–effect relationship (90, 93).
The link between absorbed dose and associated toxicities in healthy tissues (based on the creatinine level for kidney toxicity, blood cell count for bone marrow toxicity, or liver functional parameters) has also been studied in several targeting models, including PRRT (94). Therapeutic regimens could then be based on the maximal absorbed dose tolerated by healthy tissues rather than on the dose delivered to the tumor.
Investigating the “Inverse Dose Rate-Effect” Phenomenon
In TRT, the absorbed dose rate depends on the physical half-life of the radionuclide, its specific activity and the vector pharmacokinetics (transit, uptake, and clearance). Therefore, irradiation is usually protracted from hours to days and the generally low absorbed dose-rate values (<1 Gy⋅h−1) are expected to give cells time to repair damage. As the clonogenic survival of cells decreases when the dose or dose rate increases, TRT efficacy should therefore be very low. However, this is not true and TRT therapeutic efficacy per Gy, when accurate dose determination is available, is higher than that of CEBRT (95, 96). The observation that low absorbed dose rates are ultimately more cytotoxic per Gy than doses delivered at high dose rate is defined as the inverse dose rate-effect. It has been described also after low dose of EBRT and could contribute to the hypersensitivity to low doses (95, 97–99). Several hypotheses have been proposed to explain this effect, including TRT-mediated synchronization of cells in a radiosensitive cell cycle phase or defects in the detection of low levels of DNA damage. However, these mechanisms cannot be generalized (78) and TRT biology still needs to be investigated for each TRT situation. Models and notions have been developed to reduce the discrepancies between the theoretical dose–effect relationship and the real biological effects measured in human patients using clearly identified endpoints (creatinine values, for example). The notion of biological effective dose (BED) has been introduced to take into account the biological effects of low absorbed dose rates and the repair capacities allowed by protracted irradiation, as it is done for fractionation in EBRT. The concept of equivalent uniform biologically effective dose (EUBED) is used to take into account the dose distribution heterogeneity (100). However, these improvements might not be enough if all the aspects of radiobiology of the tissues treated by TRT have to be taken into account. For example, in most TRT dosimetric approaches, α/β are still extrapolated from EBRT data and these values should be confirmed (101, 102).
Investigating Non-Targeted Effects in TRT
As non-targeted effects have been described mostly after low doses of EBRT, their contribution in TRT (50, 103–106) should be more prominent because the dose is generally delivered at low dose rates (104). Therefore, the final TRT cytotoxicity could be the sum of both targeted effects (described by the absorbed dose–effect relationship) and non-targeted effects, which are likely to be described by the lack of absorbed dose–effect relationship and a saturated response. Therefore, the nature of the global absorbed dose–effect relationship, resulting from both phenomena, needs to be investigated.
Although the analysis of non-targeted effects in TRT is more challenging than in EBRT, several in vitro and in vivo studies have shown that beta-emitting particles, such as those released by 3H incorporated into DNA (thymidine (3H-dThd) or by 131I incorporated in metaiodobenzylguanidine (131I-MIBG), and Auger electrons emitted by 125I coupled to deoxyuridine (125IUdR) (26) or to antibodies (106) could lead to non-targeted effects. This was also observed when alpha-particle emitters (213Bi, 211At) were used to radiolabel mAbs (74, 75) or MIBG (105, 107), respectively. Moreover, drugs that interfere with gap junctions (for instance, lindane) or that scavenge ⋅OH radicals (DMSO) can abrogate the non-targeted response in a cell model of TRT (108).
The relative contribution of non-targeted effects, compared to the direct effects of radiation, may depend on the TRT nature and specifically on the absorbed dose rate and LET (105–107). One could expect TRT to behave like EBRT for long-range particles, thereby producing homogenous irradiation and for situations with high tumor uptake of radioactivity, thereby producing high dose–rate irradiation. However, this needs to be further confirmed.
Conclusion
Although the methodology for exploring radiation biology in conventional EBRT is well established, a specific method dedicated to TRT needs to be developed. This involves a solid dosimetry approach that takes into consideration the different TRT situations and that may become the reference. Moreover, the absorbed dose and absorbed dose rate are likely to be the critical parameters in the radiobiological response to TRT. The relative contribution of targeted and non-targeted effects in the organ and tissue responses to TRT needs also to be determined.
Preclinical experiments offer the possibility to study in simple models how TRT acts on cells and tissues with the aim of identifying the specific molecular and cellular mechanisms, as it has been done in EBRT. They might provide ways for improving TRT by taking into account both radiobiology and dosimetry to shift from a radioactive chemotherapy approach toward true targeted radiotherapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This paper was supported by Action Nu1.1 of Plan Cancer 2009–2013 (ASC 13038FSA).
References
1. Despeignes V. Observation concernant un cas de cancer de l’estomac traité par les rayons Roentge. In: Lyon Medical Lyon: Librairie Médicale de Louis Savy (1896). p. 428–30; 503–6.
2. Foray N. [Claudius Regaud (1870-1940): a pioneer of radiobiology and radiotherapy]. Cancer Radiother (2012) 16(4):315–21. doi: 10.1016/j.canrad.2012.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Wang CK. The progress of radiobiological models in modern radiotherapy with emphasis on the uncertainty issue. Mutat Res (2010) 704(1–3):175–81. doi:10.1016/j.mrrev.2010.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Cadet J, Ravanat JL, TavernaPorro M, Menoni H, Angelov D. Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer Lett (2012) 327(1–2):5–15. doi:10.1016/j.canlet.2012.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol (1994) 65(1):27–33. doi:10.1080/09553009414550041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Hamada N, Maeda M, Otsuka K, Tomita M. Signaling pathways underpinning the manifestations of ionizing radiation-induced bystander effects. Curr Mol Pharmacol (2011) 4(2):79–95. doi:10.2174/1874467211104020079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Droge W. Free radicals in the physiological control of cell function. Physiol Rev (2002) 82(1):47–95. doi:10.1152/physrev.00018.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med (2010) 49(11):1603–16. doi:10.1016/j.freeradbiomed.2010.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Hei TK, Zhou H, Chai Y, Ponnaiya B, Ivanov VN. Radiation induced non-targeted response: mechanism and potential clinical implications. Curr Mol Pharmacol (2011) 4(2):96–105. doi:10.2174/1874467211104020096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Hei TK, Zhou H, Ivanov VN, Hong M, Lieberman HB, Brenner DJ, et al. Mechanism of radiation-induced bystander effects: a unifying model. J Pharm Pharmacol (2008) 60(8):943–50. doi:10.1211/jpp.60.8.0001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Zhou H, Ivanov VN, Lien YC, Davidson M, Hei TK. Mitochondrial function and nuclear factor-kappaB-mediated signaling in radiation-induced bystander effects. Cancer Res (2008) 68(7):2233–40. doi:10.1158/0008-5472.CAN-07-5278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Matsumoto H, Tomita M, Otsuka K, Hatashita M, Hamada N. Nitric oxide is a key molecule serving as a bridge between radiation-induced bystander and adaptive responses. Curr Mol Pharmacol (2011) 4(2):126–34. doi:10.2174/1874467211104020126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Cadet J, Loft S, Olinski R, Evans MD, Bialkowski K, Richard Wagner J, et al. Biologically relevant oxidants and terminology, classification and nomenclature of oxidatively generated damage to nucleobases and 2-deoxyribose in nucleic acids. Free Radic Res (2012) 46(4):367–81. doi:10.3109/10715762.2012.659248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Pouget JP, Mather SJ. General aspects of the cellular response to low- and high-LET radiation. Eur J Nucl Med (2001) 28(4):541–61. doi:10.1007/s002590100484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol (1994) 65(1):7–17. doi:10.1080/09553009414550021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Georgakilas AG, O’Neill P, Stewart RD. Induction and repair of clustered DNA lesions: what do we know so far? Radiat Res (2013) 180(1):100–9. doi:10.1667/RR3041.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Sutherland BM, Georgakilas AG, Bennett PV, Laval J, Sutherland JC. Quantifying clustered DNA damage induction and repair by gel electrophoresis, electronic imaging and number average length analysis. Mutat Res (2003) 531(1–2):93–107. doi:10.1016/j.mrfmmm.2003.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Sage E, Harrison L. Clustered DNA lesion repair in eukaryotes: relevance to mutagenesis and cell survival. Mutat Res (2011) 711(1–2):123–33. doi:10.1016/j.mrfmmm.2010.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature (2000) 408(6811):433–9. doi:10.1038/35044005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Khanna KK, Lavin MF, Jackson SP, Mulhern TD. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ (2001) 8(11):1052–65. doi:10.1038/sj.cdd.4400874
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet (2001) 27(3):247–54. doi:10.1038/85798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol (2013) 14(4):197–210. doi:10.1038/nrm3546
23. Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis (2009) 30(1):2–10. doi:10.1093/carcin/bgn250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal (2011) 14(12):2491–507. doi:10.1089/ars.2010.3466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Jeggo PA, Geuting V, Lobrich M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother Oncol (2011) 101(1):7–12. doi:10.1016/j.radonc.2011.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:181–211. doi:10.1146/annurev.biochem.052308.093131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Puck TT, Marcus PI. Action of x-rays on mammalian cells. J Exp Med (1956) 103(5):653–66. doi:10.1084/jem.103.5.653
28. Vogin G, Foray N. The law of Bergonie and Tribondeau: a nice formula for a first approximation. Int J Radiat Biol (2013) 89(1):2–8. doi:10.3109/09553002.2012.717732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Lambert WC, Gagna CE, Lambert MW. Xeroderma pigmentosum: its overlap with trichothiodystrophy, Cockayne syndrome and other progeroid syndromes. Adv Exp Med Biol (2008) 637:128–37. doi:10.1007/978-0-387-09599-8_14
30. Jorgensen TJ, Shiloh Y. The ATM gene and the radiobiology of ataxia- telangiectasia. Int J Radiat Biol (1996) 69(5):527–37. doi:10.1080/095530096145535
31. Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature (2000) 405(6785):473–7. doi:10.1038/35013083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Barendsen GW, Beusker TL, Vergroesen AJ, Budke L. Effects of different radiations on human cells in tissue culture. II. Biological experiments. Radiat Res (1960) 13:841–9. doi:10.2307/3570858
33. Barendsen GW, Walter HM, Fowler JF, Bewley DK. Effects of different ionizing radiations on human cells in tissue culture. III. Experiments with cyclotron-accelerated alpha-particles and deuterons. Radiat Res (1963) 18:106–19. doi:10.2307/3571430
34. Barendsen GW. Dose-survival curves of human cells in tissue culture irradiated with alpha-, beta-, 20-kV. x- and 200-kV. x-radiation. Nature (1962) 193:1153–5. doi:10.1038/1931153a0
35. Barendsen GW. Modification of radiation damage by fractionation of the dose, anoxia, and chemical protectors in relation to let. Ann N Y Acad Sci (1964) 114:96–114. doi:10.1111/j.1749-6632.1964.tb53565.x
36. Barendsen GW, Walter HM. Effects of different ionizing radiations on human cells in tissue culture. iv. Modification of radiation damage. Radiat Res (1964) 21:314–29. doi:10.2307/3571569
37. Deschavanne PJ, Malaise EP. The relevance of alpha/beta ratios determined in vitro for human cell lines to the understanding of in vivo values. Int J Radiat Biol (1989) 56(5):539–42. doi:10.1080/09553008914551701
38. Thames HD, Bentzen SM, Turesson I, Overgaard M, van den Bogaert W. Fractionation parameters for human tissues and tumors. Int J Radiat Biol (1989) 56(5):701–10. doi:10.1080/09553008914551931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. De Ridder M, Jiang H, Van Esch G, Law K, Monsaert C, Van den Berge DL, et al. IFN-gamma+ CD8+ T lymphocytes: possible link between immune and radiation responses in tumor-relevant hypoxia. Int J Radiat Oncol Biol Phys (2008) 71(3):647–51. doi:10.1016/j.ijrobp.2008.03.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Morgan GW, Breit SN. Radiation and the lung: a reevaluation of the mechanisms mediating pulmonary injury. Int J Radiat Oncol Biol Phys (1995) 31(2):361–9. doi:10.1016/0360-3016(94)00477-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Calveley VL, Khan MA, Yeung IW, Vandyk J, Hill RP. Partial volume rat lung irradiation: temporal fluctuations of in-field and out-of-field DNA damage and inflammatory cytokines following irradiation. Int J Radiat Biol (2005) 81(12):887–99. doi:10.1080/09553000600568002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Shao C, Folkard M, Michael BD, Prise KM. Bystander signaling between glioma cells and fibroblasts targeted with counted particles. Int J Cancer (2005) 116(1):45–51. doi:10.1002/ijc.21003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Murphy JB, Liu JH, Sturm E. Studies on X-ray effects: Ix. The action of serum from X-rayed animals on lymphoid cells in vitro. J Exp Med (1922) 35(3):373–84. doi:10.1084/jem.35.3.373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol (1953) 26(305):234–41. doi:10.1259/0007-1285-26-305-234
45. Hollowell JG Jr, Littlefield LG. Chromosome damage induced by plasma of x-rayed patients: an indirect effect of x-ray. Proc Soc Exp Biol Med (1968) 129(1):240–4. doi:10.3181/00379727-129-33295
46. Emerit I, Levy A, Cernjavski L, Arutyunyan R, Oganesyan N, Pogosian A, et al. Transferable clastogenic activity in plasma from persons exposed as salvage personnel of the Chernobyl reactor. J Cancer Res Clin Oncol (1994) 120(9):558–61. doi:10.1007/BF01221035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Nagasawa H, Little JB. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res (1992) 52(22):6394–6.
48. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys (2004) 58(3):862–70. doi:10.1016/j.ijrobp.2003.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Demaria S, Formenti SC. Role of T lymphocytes in tumor response to radiotherapy. Front Oncol (2012) 2:95. doi:10.3389/fonc.2012.00095
50. Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer (2009) 9(5):351–60. doi:10.1038/nrc2603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood (2009) 114(3):589–95. doi:10.1182/blood-2009-02-206870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Havaki S, Kotsinas A, Chronopoulos E, Kletsas D, Georgakilas A, Gorgoulis VG. The role of oxidative DNA damage in radiation induced bystander effect. Cancer Lett (2015) 356(1):43–51. doi:10.1016/j.canlet.2014.01.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Andarawewa KL, Paupert J, Pal A, Barcellos-Hoff MH. New rationales for using TGFbeta inhibitors in radiotherapy. Int J Radiat Biol (2007) 83(11–12):803–11. doi:10.1080/09553000701711063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell (2009) 15(4):283–93. doi:10.1016/j.ccr.2009.02.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev (2010) 21(1):11–9. doi:10.1016/j.cytogfr.2009.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Burdak-Rothkamm S, Rothkamm K, Prise KM. ATM acts downstream of ATR in the DNA damage response signaling of bystander cells. Cancer Res (2008) 68(17):7059–65. doi:10.1158/0008-5472.CAN-08-0545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Alper T. Effects on irradiated micro-organisms of growth in the presence of acriflavine. Nature (1963) 200:534–6. doi:10.1038/200534a0
58. Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer (2003) 3(4):276–85. doi:10.1038/nrc1046
59. Corre I, Niaudet C, Paris F. Plasma membrane signaling induced by ionizing radiation. Mutat Res (2010) 704(1–3):61–7. doi:10.1016/j.mrrev.2010.01.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kolesnick RN, Haimovitz-Friedman A, Fuks Z. The sphingomyelin signal transduction pathway mediates apoptosis for tumor necrosis factor, Fas, and ionizing radiation. Biochem Cell Biol (1994) 72(11–12):471–4. doi:10.1139/o94-063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, et al. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med (1994) 180(2):525–35. doi:10.1084/jem.180.2.525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Haimovitz-Friedman A, Balaban N, McLoughlin M, Ehleiter D, Michaeli J, Vlodavsky I, et al. Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res (1994) 54(10):2591–7.
63. Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab (2009) 20(7):332–40. doi:10.1016/j.tem.2009.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Murphy JE, Nugent S, Seymour C, Mothersill C. Mitochondrial DNA point mutations and a novel deletion induced by direct low-LET radiation and by medium from irradiated cells. Mutat Res (2005) 585(1–2):127–36. doi:10.1016/j.mrgentox.2005.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev (2008) 129(7–8):383–90. doi:10.1016/j.mad.2008.03.002
66. Graziewicz MA, Day BJ, Copeland WC. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res (2002) 30(13):2817–24. doi:10.1093/nar/gkf392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Prithivirajsingh S, Story MD, Bergh SA, Geara FB, Ang KK, Ismail SM, et al. Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett (2004) 571(1–3):227–32. doi:10.1016/j.febslet.2004.06.078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Bodet-Milin C, Ferrer L, Pallardy A, Eugène T, Rauscher A, Faivre-Chauvet A, et al. Radioimmunotherapy of B-cell non-Hodgkin’s lymphoma. Front Oncol (2013) 3:177. doi:10.3389/fonc.2013.00177
69. Kaminski MS, Tuck M, Estes J, Kolstad A, Ross CW, Zasadny K, et al. 131I-tositumomab therapy as initial treatment for follicular lymphoma. N Engl J Med (2005) 352(5):441–9. doi:10.1056/NEJMoa041511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Pouget JP, Navarro-Teulon I, Bardiès M, Chouin N, Cartron G, Pèlegrin A, et al. Clinical radioimmunotherapy – the role of radiobiology. Nat Rev Clin Oncol (2011) 8(12):720–34. doi:10.1038/nrclinonc.2011.160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. DeNardo SJ, Denardo GL. Targeted radionuclide therapy for solid tumors: an overview. Int J Radiat Oncol Biol Phys (2006) 66(2 Suppl):S89–95. doi:10.1016/j.ijrobp.2006.03.066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Navarro-Teulon I, Lozza C, Pèlegrin A, Vivès E, Pouget JP. General overview of radioimmunotherapy of solid tumors. Immunotherapy (2013) 5(5):467–87. doi:10.2217/imt.13.34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Sharkey RM, Goldenberg DM. Cancer radioimmunotherapy. Immunotherapy (2011) 3(3):349–70. doi:10.2217/imt.10.114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Chouin N, Bernardeau K, Bardiès M, Faivre-Chauvet A, Bourgeois M, Apostolidis C, et al. Evidence of extranuclear cell sensitivity to alpha-particle radiation using a microdosimetric model. II. Application of the microdosimetric model to experimental results. Radiat Res (2009) 171(6):664–73. doi:10.1667/RR1536.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Chouin N, Bernardeau K, Davodeau F, Chérel M, Faivre-Chauvet A, Bourgeois M, et al. Evidence of extranuclear cell sensitivity to alpha-particle radiation using a microdosimetric model. I. Presentation and validation of a microdosimetric model. Radiat Res (2009) 171(6):657–63. doi:10.1667/RR1371.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Pouget JP, Santoro L, Raymond L, Chouin N, Bardiès M, Bascoul-Mollevi C, et al. Cell membrane is a more sensitive target than cytoplasm to dense ionization produced by auger electrons. Radiat Res (2008) 170(2):192–200. doi:10.1667/RR1359.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Freudenberg R, Runge R, Maucksch U, Berger V, Kotzerke J. On the dose calculation at the cellular level and its implications for the RBE of (99m)Tc and (1)(2)(3)I. Med Phys (2014) 41(6):062503. doi:10.1118/1.4876296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Piron B, Paillas S, Boudousq V, Pèlegrin A, Bascoul-Mollevi C, Chouin N, et al. DNA damage-centered signaling pathways are effectively activated during low dose-rate auger radioimmunotherapy. Nucl Med Biol (2014) 41(Suppl):e75–83. doi:10.1016/j.nucmedbio.2014.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Bolch WE, Eckerman KF, Sgouros G, Thomas SR. MIRD pamphlet No. 21: a generalized schema for radiopharmaceutical dosimetry – standardization of nomenclature. J Nucl Med (2009) 50(3):477–84. doi:10.2967/jnumed.108.056036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Neti PV, Howell RW. Log normal distribution of cellular uptake of radioactivity: implications for biologic responses to radiopharmaceuticals. J Nucl Med (2006) 47(6):1049–58.
81. Back T, Jacobsson L. The alpha-camera: a quantitative digital autoradiography technique using a charge-coupled device for ex vivo high-resolution bioimaging of alpha-particles. J Nucl Med (2010) 51(10):1616–23. doi:10.2967/jnumed.110.077578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Ritt P, Vija H, Hornegger J, Kuwert T. Absolute quantification in SPECT. Eur J Nucl Med Mol Imaging (2011) 38(Suppl 1):S69–77. doi:10.1007/s00259-011-1770-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Tornai MP, Jaszczak RJ, Turkington TG, Coleman RE, et al. Small-animal PET: advent of a new era of PET research. J Nucl Med (1999) 40(7):1176–9.
84. Lassmann M, Chiesa C, Flux G, Bardiès M, EANM Dosimetry Committee. EANM dosimetry committee guidance document: good practice of clinical dosimetry reporting. Eur J Nucl Med Mol Imaging (2011) 38(1):192–200. doi:10.1007/s00259-010-1549-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Stabin MG, Peterson TE, Holburn GE, Emmons MA, et al. Voxel-based mouse and rat models for internal dose calculations. J Nucl Med (2006) 47(4):655–9.
86. Bitar A, Lisbona A, Thedrez P, Sai Maurel C, Le Forestier D, Barbet J, et al. A voxel-based mouse for internal dose calculations using Monte Carlo simulations (MCNP). Phys Med Biol (2007) 52(4):1013–25. doi:10.1088/0031-9155/52/4/010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Goddu SM, Howell RW, Bouchet LG, Bolch WE, Rao DV. MIRD Cellular S Values: Self-Absorbed Dose Per Unit Cumulated Activity for Selected Radionuclides and Monoenergetic Electron and Alpha Particle Emitters Incorporated into Different Cell Compartments. Reston, VA: Society of Nuclear Medicine (1997).
88. Boutaleb S, Pouget JP, Hindorf C, Pelegrin A, Barbet J, Kotzki PO, et al. Impact of mouse model on pre-clinical dosimetry in targeted radionuclide therapy. Proc IEEE (2009) 87:2076–85. doi:10.1109/JPROC.2009.2026921
89. Flux GD, Haq M, Chittenden SJ, Buckley S, Hindorf C, Newbold K, et al. A dose-effect correlation for radioiodine ablation in differentiated thyroid cancer. Eur J Nucl Med Mol Imaging (2010) 37(2):270–5. doi:10.1007/s00259-009-1261-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Dewaraja YK, Schipper MJ, Shen J, Smith LB, Murgic J, Savas H, et al. Tumor-absorbed dose predicts progression-free survival following 131I-tositumomab radioimmunotherapy. J Nucl Med (2014) 55(7):1047–53. doi:10.2967/jnumed.113.136044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Dewaraja YK, Schipper MJ, Roberson PL, Wilderman SJ, Amro H, Regan DD, et al. 131I-tositumomab radioimmunotherapy: initial tumor dose-response results using 3-dimensional dosimetry including radiobiologic modeling. J Nucl Med (2010) 51(7):1155–62. doi:10.2967/jnumed.110.075176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Strigari L, Konijnenberg M, Chiesa C, Bardies M, Du Y, Gleisner KS, et al. The evidence base for the use of internal dosimetry in the clinical practice of molecular radiotherapy. Eur J Nucl Med Mol Imaging (2014) 41:1976–88. doi:10.1007/s00259-014-2824-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Sgouros G, Squeri S, Ballangrud AM, Kolbert KS, Teitcher JB, Panageas KS, et al. Patient-specific, 3-dimensional dosimetry in non-Hodgkin’s lymphoma patients treated with 131I-anti-B1 antibody: assessment of tumor dose-response. J Nucl Med (2003) 44(2):260–8.
94. Wessels BW, Konijnenberg MW, Dale RG, Breitz HB, Cremonesi M, Meredith RF, et al. MIRD pamphlet No. 20: the effect of model assumptions on kidney dosimetry and response – implications for radionuclide therapy. J Nucl Med (2008) 49(11):1884–99. doi:10.2967/jnumed.108.053173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Wessels BW, Vessella RL, Palme DF II, Berkopec JM, Smith GK, Bradley EW. Radiobiological comparison of external beam irradiation and radioimmunotherapy in renal cell carcinoma xenografts. Int J Radiat Oncol Biol Phys (1989) 17(6):1257–63. doi:10.1016/0360-3016(89)90534-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Ning S, Knox SJ. G2/M-phase arrest and death by apoptosis of HL60 cells irradiated with exponentially decreasing low-dose-rate gamma radiation. Radiat Res (1999) 151(6):659–69. doi:10.2307/3580204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Sgouros G, Knox SJ, Joiner MC, Morgan WF, Kassis AI. MIRD continuing education: bystander and low dose-rate effects: are these relevant to radionuclide therapy? J Nucl Med (2007) 48(10):1683–91. doi:10.2967/jnumed.105.028183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Knox SJ, Goris ML, Wessels BW. Overview of animal studies comparing radioimmunotherapy with dose equivalent external beam irradiation. Radiother Oncol (1992) 23(2):111–7. doi:10.1016/0167-8140(92)90342-R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Knox SJ, Sutherland W, Goris ML. Correlation of tumor sensitivity to low-dose-rate irradiation with G2/M-phase block and other radiobiological parameters. Radiat Res (1993) 135(1):24–31. doi:10.2307/3578392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Fowler JF. Radiobiological aspects of low dose rates in radioimmunotherapy. Int J Radiat Oncol Biol Phys (1990) 18(5):1261–9. doi:10.1016/0360-3016(90)90467-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Murtha AD. Radiobiology of low-dose-rate radiation relevant to radioimmunotherapy. Cancer Biother Radiopharm (2000) 15(1):7–14. doi:10.1089/cbr.2000.15.7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Murtha AD. Review of low-dose-rate radiobiology for clinicians. Semin Radiat Oncol (2000) 10(2):133–8. doi:10.1016/S1053-4296(00)80050-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Brady D, O’Sullivan JM, Prise KM. What is the role of the bystander response in radionuclide therapies? Front Oncol (2013) 3:215. doi:10.3389/fonc.2013.00215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Prise KM, Schettino G, Folkard M, Held KD. New insights on cell death from radiation exposure. Lancet Oncol (2005) 6(7):520–8. doi:10.1016/S1470-2045(05)70246-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Boyd M, Ross SC, Dorrens J, Fullerton NE, Tan KW, Zalutsky MR, et al. Radiation-induced biologic bystander effect elicited in vitro by targeted radiopharmaceuticals labeled with alpha-, beta-, and auger electron-emitting radionuclides. J Nucl Med (2006) 47(6):1007–15.
106. Paillas S, Boudousq V, Piron B, Kersual N, Bardiès M, Chouin N, et al. Apoptosis and p53 are not involved in the anti-tumor efficacy of (1)(2)(5)I-labeled monoclonal antibodies targeting the cell membrane. Nucl Med Biol (2013) 40(4):471–80. doi:10.1016/j.nucmedbio.2013.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Boyd M, Sorensen A, McCluskey AG, Mairs RJ. Radiation quality-dependent bystander effects elicited by targeted radionuclides. J Pharm Pharmacol (2008) 60(8):951–8. doi:10.1211/jpp.60.8.0002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: radiobiology, bystander effects, radioimmunotherapy, targeted radionuclide therapy
Citation: Pouget J-P, Lozza C, Deshayes E, Boudousq V and Navarro-Teulon I (2015) Introduction to radiobiology of targeted radionuclide therapy. Front. Med. 2:12. doi: 10.3389/fmed.2015.00012
Received: 06 August 2014; Accepted: 25 February 2015;
Published online: 17 March 2015.
Edited by:
Helmut Robert Maecke, University Hospital Freiburg, GermanyReviewed by:
Roger Schibli, Paul Scherrer Institute, SwitzerlandAbiraj Keelara, F. Hoffmann-La Roche Ltd., Switzerland
Helmut Robert Maecke, University Hospital Freiburg, Germany
Copyright: © 2015 Pouget, Lozza, Deshayes, Boudousq and Navarro-Teulon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Pierre Pouget, U1194, INSERM Institut de Recherche en Cancérologie, Montpellier Cedex 5 34298, France e-mail:amVhbi1waWVycmUucG91Z2V0QGluc2VybS5mcg==