 Samia Q. Khan
Samia Q. Khan Imran Khan
Imran Khan Vineet Gupta
Vineet Gupta- Drug Discovery Center, Department of Internal Medicine, Rush University Medical School, Chicago, IL, United States
Lupus nephritis (LN) is a common complication of systemic lupus erythematosus (SLE) with unclear etiology and limited treatment options. Immune cell infiltration into the kidneys, a hallmark of LN, triggers tissue damage and proteinuria. CD11b, the α-chain of integrin receptor CD11b/CD18 (also known as αMβ2, Mac-1, and CR3), is highly expressed on the surface of innate immune cells, including macrophages and neutrophils. Genetic variants in the human ITGAM gene, which encodes for CD11b, are strongly associated with susceptibility to SLE, LN, and other complications of SLE. CD11b modulates several key biological functions in innate immune cells, including cell adhesion, migration, and phagocytosis. CD11b also modulates other signaling pathways in these cells, such as the Toll-like receptor signaling pathways, that mediate generation of type I interferons, a key proinflammatory cytokine and circulating biomarker in SLE and LN patients. However, how variants in ITGAM gene contribute to disease pathogenesis has not been completely established. Here, we provide an overview of CD11b modulated mechanisms and the functional consequences of the genetic variants that can drive disease pathogenesis. We also present recent insights from studies after pharmacological activation of CD11b. These studies offer novel mechanisms for development of therapeutics for LN, SLE and other autoimmune diseases.
Introduction
Lupus nephritis (LN) is an inflammatory kidney disease that is a common complication of systemic lupus erythematosus (SLE), an autoimmune disease that affects women nine times more often than men and is characterized by aberrant autoimmune responses to self-antigens that can result in multiple end organ pathologies. Approximately 50–60% of SLE patients develop LN over the course of the disease, making LN the common cause of morbidity and mortality (1). There is considerable variation in the prevalence of SLE and the frequency of LN in different regions of the world and different races and ethnicities [reviewed in Ref. (2)]. In the United States and Canada, the prevalence of SLE ranges from 4.8 to 78.5 cases per 100,000 people (2, 3). LN cumulative incidence is higher in African descent (51%), Asian (55%), and Hispanic (43%) populations as compared with European ancestry (14%) (4–6). LN severity also differs among the ethnicities with more renal involvement in individuals of African lineage, Hispanics, and Asian populations (4–6). The discrepancies between ancestral backgrounds could be due to the complex interplay between genetic and environmental risk factors that drives kidney inflammation in LN. These factors could compromise mechanisms that normally assure immune tolerance to nuclear autoantigens leading to the activation and proliferation of immune cells, including monocytes, autoreactive B and T cells that are recruited to the kidney tissue where they produce destructive cytokines and chemokines, which in turn attract more infiltrating cells. This inflammatory cascade causes kidney damage and triggers proteinuria. Recent genome-wide association studies (GWAS) have revealed strong associations between single-nucleotide polymorphisms (SNPs) in the ITGAM gene (located at chromosome 16p11.2) and susceptibility to SLE and LN (7–9). Understanding how such mutations in the ITGAM gene change the molecular and cellular mechanisms that are modulated by integrin CD11b is essential for understanding the pathobiologic mechanisms driving this disease and for identifying novel therapeutics.
Mutations in the ITGAM Gene are Strong Genetic Risk Factors for LN
Recent GWAS have reported a significant association between three different variants in the ITGAM gene and LN susceptibility (7–9). Rs1143678, rs1143679, and rs1143683 are three non-synonymous ITGAM SNPs (10, 11) that translate into mis-sense mutations P1146S, R77H, and A858V, respectively, in the CD11b protein, and confer risk for SLE and SLE subphenotypes including LN, discoid rash and immunological manifestations [reviewed in Ref. (12)]. The rs1143678 (P1146S) and rs1143683 (A858V) SNPs have been found to be mostly in complete linkage disequilibrium (LD), forming a haplotype (10, 13, 14). Additionally, a strong LD has also been noticed between rs1143678 (P1146S) and rs1143679 (R77H) in SLE patients (9, 10, 15). The association between ITGAM SNPs and disease susceptibility holds among individuals across all different ancestries, including European ancestry, Hispanics, African Americans, and Asians (7–9, 13, 15). In particular, there is a robust association between ITGAM variants and renal disease in SLE patients (11, 13, 16–18). Higher prevalence of LN was reported in SLE patients carrying risk alleles rs1143679 (R77H) [odds ratio (OR) = 3.35, p = 0.029] or rs1143683 (A858V) (OR = 2.05, p = 0.022) in Hong Kong Chinese and Thai patients as compared to lupus patients without renal nephritis (13). Another study also demonstrated a strong genetic association of the rs1143679 (R77H) variant and LN (OR = 2.15, p = 4.69 × 10−21) in a large European ancestry SLE cohort where the risk allele frequency increased from 10.6% in healthy controls to 17% in SLE and 20.4% in LN (11).
Biological Functions of CD11b—Both Pro- and Anti-Inflammatory Roles
ITGAM encodes for CD11b, the 165-kDa alpha chain that non-covalently binds with its β-chain partner, CD18, to form the functional integrin heterodimer CD11b/CD18, also known as Mac-1, αMβ2, and CR3. This integrin is one of the four members of the hetrodimeric β2 integrin family, which share the common CD18 chain (19–22) (Figure 1). Integrin CD11b is primarily expressed on monocytes, macrophages, neutrophils, dendritic cells (DCs), NK cells, and a subset of B and T cells (23–28). It binds to a diverse array of ligands, including intercellular adhesion molecule family members, complement protein iC3b and fibrinogen (29–32), and mediates leukocyte adhesion, migration and tissue recruitment (Figure 1). CD11b participates in many proinflammatory biological processes primarily associated with controlling infection. For example, in response to inflammatory stimuli, it mediates leukocyte activation and accumulation at the sites of inflammation, by increasing leukocyte rolling, stable adhesion, crawling and transmigration across blood vessels (31, 33, 34). CD11b also plays a key role in the phagocytosis of opsonized particles, such as iC3b-coated bacteria, apoptotic cells and immune complexes (ICs), that is essential to limit unwanted inflammatory immune responses (34–36). Ligation of CD11b by iC3b also results in the production of anti-inflammatory cytokines such as IL-10 and TGF-β (37).
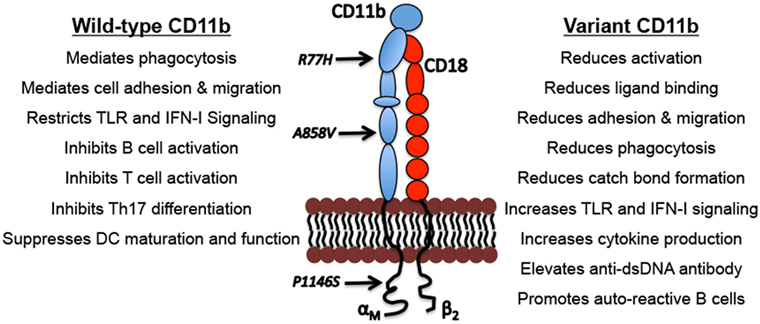
Figure 1. Biological functions of CD11b and the functional outcomes of CD11b variants. Indicated are examples of well described physiological functions of integrin CD11b and the functional consequences of the genetic variations that can drive lupus nephritis pathogenesis.
Integrin CD11b signaling also negatively regulates other proinflammatory signaling pathways, such as the toll-like receptor (TLR) and FcγR signaling pathways (14, 38–41). Under basal conditions, integrin CD11b is present in an inactive conformation on circulating leukocytes, but it is rapidly activated after TLR stimulation (42–44). Activation of CD11b, through intracellular signaling pathways including the PI(3)K and effector RapL pathways, after TLR stimulation, induces activation of Src and Syk, which phosphorylates MyD88 and TRIF leading to their ubiquitin-mediated degradation via Cbl-b (38). Consequently, there is reduction in activation of transcription factor NFκB, which correlates with reduced production of IL-6 and other proinflammatory proteins. Macrophages deficient in CD11b have reduced TLR-induced degradation of MyD88 and TRIF resulting in enhanced activation of NFκB and other TLR-dependent pathways and inflammatory cytokine production (14, 38). This suggests that CD11b negatively regulates innate immune cell activation downstream of TLR pathways, and misregulation of this key regulatory mechanism might underlie LN and SLE pathogenesis.
CD11b also negatively regulates other immune cell signaling pathways following ligand binding. Studies report that CD11b negatively regulates BCR signaling to maintain autoreactive B cell tolerance (38, 45), suppression of T-cell activation (46), negative regulation of Th17 cell development (47) and suppression of DC maturation and function (Figure 1). All together, these studies indicate that functional CD11b is crucial for maintaining homeostasis and tolerance and downregulating inflammatory mechanisms that contribute to LN development.
Functional Outcomes of CD11b Risk Variants
The three most common ITGAM SNPs are distributed in different CD11b domains across this large protein (Figure 1). All three variants result in functionally deficient CD11b protein on leukocytes, with limited effects on its surface expression levels (10, 14, 48–50) (Figure 1). Studies using cell lines expressing the R77H variant or monocytes/macrophages derived from R77H donors reveal that the mutation reduces the integrin’s ability to bind to ligands, including ICAM1 and iC3b, in static conditions as well as sheer flow conditions that closely mimic blood flow (48, 49). Similarly, firm adhesion of CD11b expressing neutrophils was significantly reduced in cells from individuals carrying any of the three common ITGAM SNPs (10). Integrin CD11b also contributes to the process of phagocytosis of opsonized particles that include apoptotic cells and ICs (36, 51, 52). CD11b-mediated phagocytosis was significantly reduced in neutrophils from donors carrying ITGAM variant alleles that was related to variations at rs1143679 (R77H), rs1143678 (P1146S), or rs1143683 (A858V) (10, 49, 50). Similarly, phagocytosis mediated by Fcγ receptors was also significantly reduced in cells from donors with ITGAM variant alleles (10). This defect is also apparent in cells that are heterozygous for the R77H allele, further implicating the importance of functional CD11b for normal phagocytosis (10, 49, 50). CD11b also plays an important role in downregulating TLR- and FcγR-dependent inflammatory pathways (38, 41, 53). Among these ITGAM risk variants, most of the functional studies to date have primarily been focused on the R77H variant, which was the first ITGAM variant identified. More studies are needed to fully define the structural and functional effects of all three variants on CD11b. The R77H substitution results in failure to downregulate IL-6, TNF-α, and IL-1β, expression in monocytes, and IL-6 expression in macrophages, indicating that the integrin is deficient in regulating proinflammatory cytokine production (40, 48, 49). The R77H substitution also abolishes the regulatory effect of CD11b on BCR signaling on B cells (45). Under normal conditions, CD11b colocalizes with BCR and CD22 to form a complex on the B cell surface hindering BCR-mediated signaling via the Lyn-CD22-SHP1 pathway to ensure auto-reactive B cell quiescence. Upon BCR ligation, CD11b-CD22 quickly dissociates from the activated BCR that results in a rapid increase of intracellular free calcium concentrations indicating activation. Interestingly, the R77H mutation in CD11b induces a drastic reduction in colocalization of BCR-CD11b with CD22, resulting in an abnormal influx of intracellular calcium and B cell hyperactivation. These studies suggest that the R77H substitution disrupts the direct binding of CD11b-CD22 thus abrogating the regulatory effect of CD11b on B cell activation (45). Rosetti et al. reported that the R77H mutation reduces affinity for ligand binding by CD11b and its ability to form catch-bonds, which strengthen receptor–ligand bonds under mechanical forces (54), How this decrease in catch bond formation plays a role in disease susceptibility is unclear, but it might be related to CD11b’s role in regulating the proinflammatory TLR and FcγR signaling in cells, which might be compromised due to reduction in mechanical forces in R77H mutant cells (54). We recently found that all three ITGAM variants show strong association with elevated type I interferons (IFN-I) serum activity in SLE patients (14). Transcriptional induction of IFN-I is mediated by transcription factors IRF3 and IRF7 that require phosphorylation for nuclear entry and activity (55). Under normal conditions, nuclear FOXO3 acts as a negative regulator of IFN-I pathway by imposing a brake on IRF7 (56). Upon TLR stimulation, FOXO3 is shuttled from the nucleus to the cytoplasm and degraded, via its AKT-mediated phosphorylation, consequently removing the brake on IRF7 and the IFN-I pathway. Interestingly, cells from donors carrying ITGAM variants showed increased transcript levels of IFNB and IRF7 and significantly reduced protein levels of FOXO3 in the nucleus (14), suggesting a key mechanism behind increased IFN I levels in SLE and LN patients—that the ITGAM variants likely reduce a CD11b-dependent tonic suppression of TLR signaling in cells, due to a failure of the mutant CD11b integrin in maintaining nuclear FOXO3 levels, resulting in reduced suppression of IRF7 (56) and increased IFN-I (14). Altogether, these studies indicate the importance of normal activity of CD11b for efficient clearance of dead cells and ICs, and for regulating proinflammatory pathways that, in cells expressing disease-associated ITGAM variants, elicits unchecked, chronic inflammation.
Pathogenic Mechanisms Driven by Deficient CD11b that Contributes to LN
How these ITGAM SNPs confer disease risk is not fully understood, but they have been shown to produce a dysfunctional CD11b protein that is deficient in many functions, as compared to the wild-type CD11b; activation, ligand binding and cell adhesion, phagocytosis, catch-bond formation and restricting TLR-signaling that mediate generation of IFN I (Figure 1). High levels of serum IFN-I is a heritable risk factor for SLE and is the main pathogenic mediator of disease (57). Elevated IFN-I makes lupus-prone mice more susceptible to disease, accelerates disease progression, and contributes to renal damage by directly damaging podocytes and inducing leukocyte infiltration of the kidney and other tissue (58, 59). IFN-I also activates adaptive immune responses, including B cells that secrete autoantibodies that form ICs that are deposited in the kidney, resulting in tissue scarring that leads to LN. Interestingly, studies show that CD11b downregulates TLR-induced pathways and B cell autoreactivity (38, 45), suggesting that SLE patients that carry CD11b variants have increased disease activity due to a reduction in CD11b’s normal anti-inflammatory signaling activities in immune cells. Perhaps because of these suppressive roles in immune system regulation, CD11b deficiency in murine systems results in higher susceptibility to autoimmune diseases including SLE [reviewed in Ref. (60)] and increased cellular infiltration of tissue and immune-mediated injury in lupus-prone mice (41, 61).
Although ample evidence indicates that ITGAM variants result in dysfunctional CD11b associated with SLE, it is still unknown how the mutations in CD11b contribute to intrarenal and systemic mechanisms that drive SLE and LN. Most of the current knowledge comes from studies performed in genetically modified and drug-induced mouse SLE models in the absence of CD11b (by blocking antibodies or CD11b knockout mice) and limited studies that report direct outcomes of ITGAM SNPs in SLE patients. Although CD11b deficiency has been reported to reduce renal accumulation of inflammatory neutrophils and complement-mediated proteinuria in an acute anti-glomerular basement membrane (GBM) nephritis model (62), it has also been reported to increase susceptibility to chronic inflammatory and autoimmune diseases (38, 41, 60, 61, 63–65) and to exacerbate disease that is associated with elevated neutrophil accumulation in tissue and elevated proteinuria (41, 61). Specifically, in lupus-prone MRL/lpr mice, CD11b deficiency resulted in severe glomerulonephritis (GN) as indicated by the accumulation of neutrophils in the kidney with elevated IC deposition, complement fixation and glomerular injury (61). Interestingly, transfer of human SLE sera into mice expressing the human FcγRIIA and FcγRIIIB receptors and lacking CD11b induced GN that was associated with elevated proteinuria, increased renal accumulation of neutrophils, and the presence of crescents (41). How CD11b deficiency results in proinflammatory or anti-inflammatory outcomes is not clear and needs further investigation. The differences in the disease models (acute vs. chronic) and/or the specific CD11b ligands involved in driving disease could possibly explain the discrepancies.
The decreased ability of mutant CD11b to bind to iC3b and subsequent reduction in phagocytosis and inflammatory responses may play a major role in driving disease. CD11b deficiency enhanced inflammation in a complement-induced tubulointerstitial nephritis model. In this study, Crry−/− C3−/− kidneys were transplanted into healthy WT mice that resulted in inflammation stemming from robust complement activation that leads to severe tubulointerstitial nephritis marked with leukocyte infiltration, tubular damage, and interstitial fibrosis. CD11b deficiency enhanced this phenotype, which was associated with an expansion of CD11b + F4/80 + macrophages. The in situ proliferation of CD11b+ F4/80+ macrophages suggests that the CD11b and iC3b interaction can limit the infiltration of monocytes into the kidney and subsequent differentiation/expansion of macrophages (66). CD11b deficiency in mice enhances humoral responses and circulating IC levels and reduces renal function in a chronic serum sickness model of IC-mediated GN (36). Altogether, these studies suggest that defective regulatory functions of CD11b (due to variants) in several processes known to be relevant in SLE and LN could have pathogenic outcomes in a permissive environment.
Deficient CD11b is unable to negatively modulate autoreactive B cells (45), which are expanded in SLE and secrete pathogenic autoantibodies, promote autoreactive T cells, and produce a wide variety of cytokines. Ding et al. recently reported that BCR crosslinking in CD11b deficient mice leads to strikingly enhanced B cell activation, differentiation, and survival and elevated autoantibody production and kidney Ig deposition. Studies in which B cells were transfected with WT or R77H CD11b suggest that the mutation completely abrogates the regulatory role of CD11b in maintaining auto-reactive B cell tolerance (45). More importantly, a GWAS reported that elevated anti-dsDNA autoantibody was strongly associated with ITGAM gene variants in lupus patients (67), which suggests that functional CD11b is required for suppressing B cells that drive SLE and LN pathogenesis.
CD11b plays an important role in negatively regulating TLR-induced signaling pathways and disruption of this anti-inflammatory property may affect processes that are implicated in driving disease. We recently reported that SLE subjects carrying ITGAM SNPs that produce defective CD11b have significantly elevated serum IFN-I activity, suggesting a strong direct link between reduced CD11b activity and the chronically increased inflammatory status in patients (14). This correlates with higher expression of IL-6, TNF-α, and IL-1β in R77H expressing cells, indicating that the integrin is deficient in regulating proinflammatory cytokine production (48, 49). Similarly, elevated levels of IL-6, TNF, and IFN-β are reported in CD11b deficient mice injected with TLR ligands (38). Interestingly, elevated levels of circulating IL-6 found in CD11b deficient mice promote the differentiation of naïve T cells into IL-17 producing helper T cells (47), which are involved in the development of many autoimmune diseases including LN (68). Altogether these studies suggests that variants in CD11b disrupts its modulatory mechanisms of suppressing TLR-induced inflammation and possibly restricting Th17 cells that are both important processes in lupus development.
Therapeutic Targeting of CD11b
Given that CD11b activity and function plays a vital role in reducing proinflammatory signaling pathways, and that LN-associated mutations in CD11b reduce its biological activity, it suggests therapeutic targeting of CD11b and its variants with CD11b agonists. Toward that, we utilized a novel small molecule CD11b agonist leukadherin-1 (LA1) that we recently discovered (69). LA1 activates CD11b and increases CD11b-dependent cell adhesion that results in decreased cell chemotaxis and transendothelial migration. We found that induction of disease in an anti-GBM nephritis model led to an influx of neutrophils into the kidney and elevated proteinuria, which were both significantly reduced with LA1 treatment (69). CD11b activation with LA1 significantly reduced TLR-dependent NFκB signaling and generation of proinflammatory cytokines IL-1β and IL-6 in primary neutrophils and macrophages (14). It also suppressed levels of IFN-I. More importantly, treatment with LA1 reduced IFN-I responses in WT but not CD11b-deficient mice, and protected lupus-prone mice from end-organ renal injury. Specifically, vehicle treated MRL/Lpr mice developed significant features consistent with lupus GN, including elevated proteinuria and IgG renal IC deposition, which were significantly reduced with treatment with LA1. CD11b activation via LA1 suppressed TLR dependent AKT/FOXO3/IRF7 pathway and IFN-I levels in vivo (14). Cells expressing either the wild-type CD11b or the R77H mutant CD11b displayed a similar level of LA1-mediated adhesion to immobilized ligand, suggesting that LA1 is able to bind and activate the WT and the CD11b variant integrin equally well (14). Furthermore, human macrophages from ITGAM variant carriers displayed elevated basal expression of IRF7 and IFN-β, as well as increased nuclear exclusion of FOXO3, which were further increased upon TLR-stimulation, and LA1 mediated activation of CD11b suppressed these proinflammatory changes (Figure 2). Altogether, these studies imply that CD11b activation via pharmacologic agents abrogates abnormal IFN-I signaling and is a novel therapeutic strategy for SLE and LN.

Figure 2. CD11b modulates toll-like receptor-induced type I interferon (IFN-I) pathway. A schematic showing the working model of how inactive CD11b due to ITGAM single nucleotide polymorphisms (SNPs) results in a deficit in the FOXO3-mediated IFN-I pathway, which can be reversed with pharmacological activation of CD11b via leukadherin-1 (LA1).
Conclusion
Genetic variants in ITGAM, the gene that encodes CD11b, confer high risk for SLE and LN in patients. The ITGAM variant produces dysfunctional CD11b that has defects in ligand binding, cell adhesion, phagocytosis, catch-bond formation, and downregulation of inflammatory cytokine production. These functional deficits result in pathological consequences that drive systemic and intrarenal mechanisms that contribute to LN. Pharmacologic activation of CD11b suppresses systemic increase in inflammatory cytokines and reduces tissue accumulation of activated leukocytes, thereby reducing disease. Small molecule agonists directed to the ligand binding αA/I-domain of CD11b are also able to efficiently rescue the functional defects in mutant CD11b commonly associated with LN. Future studies will determine how well such a strategy is able to ameliorate disease in LN patients.
Author Contributions
SK wrote the review article. IK designed and revised the figures in the article. SK and VG critically revised the article. SK, IK, and VG reviewed and approved the article for publication.
Conflict of Interest Statement
VG is a cofounder and a member of the scientific advisory board of Adhaere Pharmaceuticals Inc., a company developing novel therapeutics for inflammation, cancer, and autoimmune diseases. SK and IK declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by grants from the NIH (R01DK084195, R01DK106512, and R01DK107984) and resources from the Rush University Medical Center.
References
1. Ward MM, Pyun E, Studenski S. Mortality risks associated with specific clinical manifestations of systemic lupus erythematosus. Arch Intern Med (1996) 156(12):1337–44. doi:10.1001/archinte.156.12.1337
2. Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol (2017) 12(5):825–35. doi:10.2215/CJN.05780616
3. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus (2006) 15(5):308–18. doi:10.1191/0961203306lu2305xx
4. Bastian HM, Roseman JM, McGwin G Jr, Alarcon GS, Friedman AW, Fessler BJ, et al. Systemic lupus erythematosus in three ethnic groups. XII. Risk factors for lupus nephritis after diagnosis. Lupus (2002) 11(3):152–60. doi:10.1191/0961203302lu158oa
5. Seligman VA, Lum RF, Olson JL, Li H, Criswell LA. Demographic differences in the development of lupus nephritis: a retrospective analysis. Am J Med (2002) 112(9):726–9. doi:10.1016/S0002-9343(02)01118-X
6. Ortega LM, Schultz DR, Lenz O, Pardo V, Contreras GN. Review: Lupus nephritis: pathologic features, epidemiology and a guide to therapeutic decisions. Lupus (2010) 19(5):557–74. doi:10.1177/0961203309358187
7. International Consortium for Systemic Lupus Erythematosus Genetics, Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet (2008) 40(2):204–10. doi:10.1038/ng.81
8. Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med (2008) 358(9):900–9. doi:10.1056/NEJMoa0707865
9. Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet (2008) 40(2):152–4. doi:10.1038/ng.71
10. Zhou Y, Wu J, Kucik DF, White NB, Redden DT, Szalai AJ, et al. Multiple lupus-associated ITGAM variants alter Mac-1 functions on neutrophils. Arthritis Rheum (2013) 65(11):2907–16. doi:10.1002/art.38117
11. Kim-Howard X, Maiti AK, Anaya JM, Bruner GR, Brown E, Merrill JT, et al. ITGAM coding variant (rs1143679) influences the risk of renal disease, discoid rash and immunological manifestations in patients with systemic lupus erythematosus with European ancestry. Ann Rheum Dis (2010) 69(7):1329–32. doi:10.1136/ard.2009.120543
12. Fagerholm SC, MacPherson M, James MJ, Sevier-Guy C, Lau CS. The CD11b-integrin (ITGAM) and systemic lupus erythematosus. Lupus (2013) 22(7):657–63. doi:10.1177/0961203313491851
13. Yang W, Zhao M, Hirankarn N, Lau CS, Mok CC, Chan TM, et al. ITGAM is associated with disease susceptibility and renal nephritis of systemic lupus erythematosus in Hong Kong Chinese and Thai. Hum Mol Genet (2009) 18(11):2063–70. doi:10.1093/hmg/ddp118
14. Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, et al. CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest (2017) 127(4):1271–83. doi:10.1172/JCI88442
15. Han S, Kim-Howard X, Deshmukh H, Kamatani Y, Viswanathan P, Guthridge JM, et al. Evaluation of imputation-based association in and around the integrin-alpha-M (ITGAM) gene and replication of robust association between a non-synonymous functional variant within ITGAM and systemic lupus erythematosus (SLE). Hum Mol Genet (2009) 18(6):1171–80. doi:10.1093/hmg/ddp007
16. Sanchez E, Nadig A, Richardson BC, Freedman BI, Kaufman KM, Kelly JA, et al. Phenotypic associations of genetic susceptibility loci in systemic lupus erythematosus. Ann Rheum Dis (2011) 70(10):1752–7. doi:10.1136/ard.2011.154104
17. Bolin K, Sandling JK, Zickert A, Jonsen A, Sjowall C, Svenungsson E, et al. Association of STAT4 polymorphism with severe renal insufficiency in lupus nephritis. PLoS One (2013) 8(12):e84450. doi:10.1371/journal.pone.0084450
18. Chung SA, Brown EE, Williams AH, Ramos PS, Berthier CC, Bhangale T, et al. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J Am Soc Nephrol (2014) 25(12):2859–70. doi:10.1681/ASN.2013050446
19. Petty HR, Todd RF III. Receptor-receptor interactions of complement receptor type 3 in neutrophil membranes. J Leukoc Biol (1993) 54(5):492–4. doi:10.1002/jlb.54.5.492
20. Ross GD. Role of the lectin domain of Mac-1/CR3 (CD11b/CD18) in regulating intercellular adhesion. Immunol Res (2002) 25(3):219–27. doi:10.1385/IR:25:3:219
21. Hogg N, Berlin C. Structure and function of adhesion receptors in leukocyte trafficking. Immunol Today (1995) 16(7):327–30. doi:10.1016/0167-5699(95)80147-2
22. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell (1994) 76(2):301–14. doi:10.1016/0092-8674(94)90337-9
23. Springer T, Galfre G, Secher DS, Milstein C. Mac-1: a macrophage differentiation antigen identified by monoclonal antibody. Eur J Immunol (1979) 9(4):301–6. doi:10.1002/eji.1830090410
24. Anderson DC, Miller LJ, Schmalstieg FC, Rothlein R, Springer TA. Contributions of the Mac-1 glycoprotein family to adherence-dependent granulocyte functions: structure-function assessments employing subunit-specific monoclonal antibodies. J Immunol (1986) 137(1):15–27.
25. Hynes RO. Integrins: a family of cell surface receptors. Cell (1987) 48(4):549–54. doi:10.1016/0092-8674(87)90233-9
26. Kantor AB, Stall AM, Adams S, Herzenberg LA, Herzenberg LA. Differential development of progenitor activity for three B-cell lineages. Proc Natl Acad Sci U S A (1992) 89(8):3320–4. doi:10.1073/pnas.89.8.3320
27. Plow EF, Zhang L. A MAC-1 attack: integrin functions directly challenged in knockout mice. J Clin Invest (1997) 99(6):1145–6. doi:10.1172/JCI119267
28. McFarland HI, Nahill SR, Maciaszek JW, Welsh RM. CD11b (Mac-1): a marker for CD8+ cytotoxic T cell activation and memory in virus infection. J Immunol (1992) 149(4):1326–33.
29. Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, et al. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J Cell Biol (1990) 111(6 Pt 2):3129–39. doi:10.1083/jcb.111.6.3129
30. Gahmberg CG. Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr Opin Cell Biol (1997) 9(5):643–50. doi:10.1016/S0955-0674(97)80117-2
31. Beller DI, Springer TA, Schreiber RD. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. J Exp Med (1982) 156(4):1000–9. doi:10.1084/jem.156.4.1000
32. Altieri DC, Bader R, Mannucci PM, Edgington TS. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. J Cell Biol (1988) 107(5):1893–900. doi:10.1083/jcb.107.5.1893
33. Fagerholm SC, Varis M, Stefanidakis M, Hilden TJ, Gahmberg CG. alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation. Blood (2006) 108(10):3379–86. doi:10.1182/blood-2006-03-013557
34. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med (2006) 203(12):2569–75. doi:10.1084/jem.20060925
35. Dupuy AG, Caron E. Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J Cell Sci (2008) 121(11):1773–83. doi:10.1242/jcs.018036
36. Alexander JJ, Chaves LD, Chang A, Jacob A, Ritchie M, Quigg RJ. CD11b is protective in complement-mediated immune complex glomerulonephritis. Kidney Int (2015) 87(5):930–9. doi:10.1038/ki.2014.373
37. Amarilyo G, Verbovetski I, Atallah M, Grau A, Wiser G, Gil O, et al. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-kappaB-dependent blockade. Eur J Immunol (2010) 40(3):699–709. doi:10.1002/eji.200838951
38. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol (2010) 11(8):734–42. doi:10.1038/ni.1908
39. Means TK, Luster AD. Integrins limit the toll. Nat Immunol (2010) 11(8):691–3. doi:10.1038/ni0810-691
40. Roberts AL, Furnrohr BG, Vyse TJ, Rhodes B. The complement receptor 3 (CD11b/CD18) agonist Leukadherin-1 suppresses human innate inflammatory signalling. Clin Exp Immunol (2016) 185(3):361–71. doi:10.1111/cei.12803
41. Rosetti F, Tsuboi N, Chen K, Nishi H, Ernandez T, Sethi S, et al. Human lupus serum induces neutrophil-mediated organ damage in mice that is enabled by Mac-1 deficiency. J Immunol (2012) 189(7):3714–23. doi:10.4049/jimmunol.1201594
42. Altieri DC, Edgington TS. The saturable high affinity association of factor X to ADP-stimulated monocytes defines a novel function of the Mac-1 receptor. J Biol Chem (1988) 263(15):7007–15.
43. Ginsberg MH, Du X, Plow EF. Inside-out integrin signalling. Curr Opin Cell Biol (1992) 4(5):766–71. doi:10.1016/0955-0674(92)90099-X
44. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell (2002) 110(6):673–87. doi:10.1016/S0092-8674(02)00971-6
45. Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, et al. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat Commun (2013) 4:2813. doi:10.1038/ncomms3813
46. Varga G, Balkow S, Wild MK, Stadtbaeumer A, Krummen M, Rothoeft T, et al. Active MAC-1 (CD11b/CD18) on DCs inhibits full T-cell activation. Blood (2007) 109(2):661–9. doi:10.1182/blood-2005-12-023044
47. Ehirchiou D, Xiong Y, Xu G, Chen W, Shi Y, Zhang L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med (2007) 204(7):1519–24. doi:10.1084/jem.20062292
48. MacPherson M, Lek HS, Prescott A, Fagerholm SC. A systemic lupus erythematosus-associated R77H substitution in the CD11b chain of the Mac-1 integrin compromises leukocyte adhesion and phagocytosis. J Biol Chem (2011) 286(19):17303–10. doi:10.1074/jbc.M110.182998
49. Rhodes B, Furnrohr BG, Roberts AL, Tzircotis G, Schett G, Spector TD, et al. The rs1143679 (R77H) lupus associated variant of ITGAM (CD11b) impairs complement receptor 3 mediated functions in human monocytes. Ann Rheum Dis (2012) 71(12):2028–34. doi:10.1136/annrheumdis-2012-201390
50. Fossati-Jimack L, Ling GS, Cortini A, Szajna M, Malik TH, McDonald JU, et al. Phagocytosis is the main CR3-mediated function affected by the lupus-associated variant of CD11b in human myeloid cells. PLoS One (2013) 8(2):e57082. doi:10.1371/journal.pone.0057082
51. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med (1998) 188(12):2313–20. doi:10.1084/jem.188.12.2313
52. Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, et al. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood (2003) 101(2):611–20. doi:10.1182/blood-2002-06-1769
53. Xiong Y, Cao C, Makarova A, Hyman B, Zhang L. Mac-1 promotes FcgammaRIIA-dependent cell spreading and migration on immune complexes. Biochemistry (2006) 45(29):8721–31. doi:10.1021/bi060529u
54. Rosetti F, Chen Y, Sen M, Thayer E, Azcutia V, Herter JM, et al. A lupus-associated Mac-1 variant has defects in integrin allostery and interaction with ligands under force. Cell Rep (2015) 10(10):1655–64. doi:10.1016/j.celrep.2015.02.037
55. Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol (1998) 18(5):2986–96. doi:10.1128/MCB.18.5.2986
56. Litvak V, Ratushny AV, Lampano AE, Schmitz F, Huang AC, Raman A, et al. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature (2012) 490(7420):421–5. doi:10.1038/nature11428
57. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun (2007) 8(6):492–502. doi:10.1038/sj.gene.6364408
58. Liu Z, Bethunaickan R, Huang W, Lodhi U, Solano I, Madaio MP, et al. Interferon-alpha accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum (2011) 63(1):219–29. doi:10.1002/art.30087
59. Migliorini A, Angelotti ML, Mulay SR, Kulkarni OO, Demleitner J, Dietrich A, et al. The antiviral cytokines IFN-alpha and IFN-beta modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am J Pathol (2013) 183(2):431–40. doi:10.1016/j.ajpath.2013.04.017
60. Rosetti F, Mayadas TN. The many faces of Mac-1 in autoimmune disease. Immunol Rev (2016) 269(1):175–93. doi:10.1111/imr.12373
61. Kevil CG, Hicks MJ, He X, Zhang J, Ballantyne CM, Raman C, et al. Loss of LFA-1, but not Mac-1, protects MRL/MpJ-Fas(lpr) mice from autoimmune disease. Am J Pathol (2004) 165(2):609–16. doi:10.1016/S0002-9440(10)63325-1
62. Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, et al. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med (1997) 186(11):1853–63. doi:10.1084/jem.186.11.1853
63. Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest (2010) 120(6):1971–80. doi:10.1172/JCI40380
64. Liu JR, Han X, Soriano SG, Yuki K. The role of macrophage 1 antigen in polymicrobial sepsis. Shock (2014) 42(6):532–9. doi:10.1097/SHK.0000000000000250
65. Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat Med (2002) 8(4):373–8. doi:10.1038/nm0402-373
66. Chaves LD, Bao L, Wang Y, Chang A, Haas M, Quigg RJ. Loss of CD11b exacerbates murine complement-mediated tubulointerstitial nephritis. PLoS One (2014) 9(3):e92051. doi:10.1371/journal.pone.0092051
67. Chung SA, Taylor KE, Graham RR, Nititham J, Lee AT, Ortmann WA, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS Genet (2011) 7(3):e1001323. doi:10.1371/journal.pgen.1001323
68. Zhang Z, Kyttaris VC, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol (2009) 183(5):3160–9. doi:10.4049/jimmunol.0900385
Keywords: lupus nephritis, CD11b, ITGAM, type I interferon, leukadherin-1
Citation: Khan SQ, Khan I and Gupta V (2018) CD11b Activity Modulates Pathogenesis of Lupus Nephritis. Front. Med. 5:52. doi: 10.3389/fmed.2018.00052
Received: 16 December 2017; Accepted: 13 February 2018;
Published: 15 March 2018
Edited by:
Sandra Merscher, University of Miami, United StatesReviewed by:
Ana Claudia Zenclussen, Universitätsklinikum Magdeburg, GermanyDavid Andrew Fulcher, Australian National University, Australia
Copyright: © 2018 Khan, Khan and Gupta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samia Q. Khan, c2FtaWFfa2hhbkBydXNoLmVkdQ==;
Vineet Gupta, dmluZWV0X2d1cHRhQHJ1c2guZWR1