 Juan Wang
Juan Wang Wenkang Wang2†
Wenkang Wang2† Hui Wang
Hui Wang- 1Department of Gastroenterology, Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 2Department of Critical Care Medicine of the Third Affiliated Hospital (The First People's Hospital of Zunyi City), Zunyi Medical University, Zunyi, China
Solute Carrier Family 26 (SLC26) is a conserved anion transporter family with 10 members in human (SLC26A1-A11, A10 being a pseudogene). All SLC26 genes except for SLC26A5 (prestin) are versatile anion exchangers with notable ability to transport a variety of anions. SLC26A6 has the most extensive exchange functions in the SLC26 family and is widely expressed in various organs and tissues of mammals. SLC26A6 has some special properties that make it play a particularly important role in ion homeostasis and acid-base balance. In the past few years, the function of SLC26A6 in the diseases has received increasing attention. SLC26A6 not only participates in the development of intestinal and pancreatic diseases but also serves a significant role in mediating nephrolithiasis, fetal skeletal dysplasia and arrhythmia. This review aims to explore the role of SLC26A6 in physiology and pathophysiology of relative mammalian organs to guide in-depth studies about related diseases of human.
Introduction
Transmembrane transport of anionic substrates is of crucially important for human-body water and electrolyte homeostasis, CO2 transport, pH modulation and buffering, absorption of nutrient and vitamin, absorption and secretion of fluid, osmoregulation of cells, neurotransmission, and metabolic processes. SLC4 family and SLC26 family have been reported to mediate the transport of cellular anions, and the SLC26 family serves as a more vital role in mediating the exchange of Cl−/ in the epithelial tissues. SLC26 family members are multifunctional transporters which transport monovalent and divalent anions in the body. Among the 10 members of this family, mutations and polymorphisms of several members have been correlated with human diseases (Table 1) (1–7). In addition, pathological phenotypes have been reported in knockout mice deficient in expression of other SLC26 isoforms for which human mutations have not yet been associated with diseases (Table 1) (8–11). As the member which has the most extensive transport functions in the family, SLC26A6 can mediate the transport of Cl−/ as well as other anions which include Cl−/formate, Cl−/oxalate, Cl−/nitrate, /oxalate and Cl−/OH− (12, 13). However, in non-epithelial tissues, SLC26A6 functions predominantly as Cl−/ and OH− exchangers (14). SLC26A6 is strongly expressed in the intercalated ducts of pancreas (15, 16), proximal tubules of kidney, and proximal small intestine (17, 18). The expression has also been found in the heart (19), reproductive system (20), placenta (21), parotid gland (22), esophagus (23), stomach (24), and even teeth (25, 26) of mammals. Systematic study on the distribution of SLC26A6 in human tissues is lacking, and it may be difficult to detect by Western blot and immunohistochemistry because the members of SLC26 family are heavily glycosylated and SLC26A6 band size varies in the analysis of Western blot (27), especially in other species than mouse, in which comparison with knockout tissue is beneficial. However, there have been reports confirming the expression of SLC26A6 mRNA in human pancreas (28, 29), duodenum (30), kidney (29, 31), and placenta (32). Recently, animal model studies have found that SLC26A6 is crucial significance for the physiology and normal functions of several organs (such as pancreas, intestine, heart, and kidney). However, the mechanism of SLC26A6 exerts in transport process still needs to be elucidated and little is known about the function and dysregulation of SLC26A6 contributes to the manifestations of commonly diagnosed human diseases. This review offers a glimpse into the function of SLC26A6 associated with anions transport and related diseases of mammalian relative organs.
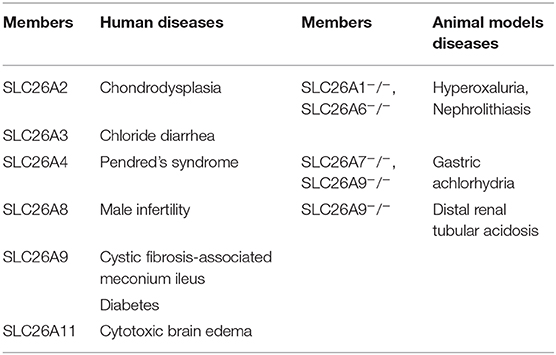
Table 1. SLC26A6 deletion related diseases.
Structural Features of SLC26A6
SLC26A6 is a transmembrane secondary transporter (symporters and exchangers) with a molecular mass of 83 kDa and consists of 759 amino acids, which is located on chromosome 3. The membrane-inserted domain of SLC26A6 consists of 14 variable-length α-helices, including two short helices (the 3rd and 10th helices) which do not span the entire width of the lipid bilayer (33). The 14 transmembrane segments are divided into two intertwined inverted repeats parts and 7 transmembrane segments each (33). The core domain contributes a pair of pseudosymmetry-related helices (α-helices 3 and 10) at the top of the cavity that point from opposite directions toward the hydrophobic center of the bilayer (33). There is a STAS (sulfate transporter and anti-sigma factor antagonist) domain at the C-terminus of SLC26A6 (14). The STAS domain is compact and contains a core which consists of just three α-helices and four β-strands (33) and is relevant for intracellular trafficking (34, 35) as well as protein–protein interactions (36, 37). Its deletion impairs substrate transport by the membrane domain (33). In addition, the C-terminus of SLC26A6 contains a consensus PDZ (PSD-95/Disc-large/ZO-1) interaction motif identical to that of the cystic fibrosis transmembrane conductance regulator (CFTR) (38) and the PDZ domains provide places for protein-protein interaction that plays an essential role in the assembly of multiprotein complexes and ultimately involves in regulation of membrane proteins, determining cell polarity, and plasma membrane targeting (39) (Figure 1). There are four isoforms of SLC26A6 have been demonstrated. SLC26A6A and SLC26A6B were cloned from the total RNA of the mouse intestine (13). SLC26A6A is a longer isoform, consisting of 758 amino acids, and SLC26A6B is the shorter isoform with 735 amino acids (28, 29). There are different opinions about which of these two isoforms correspond to the human isoform (28, 29). SLC26A6C lacks 38 amino acids by missing exon 6 and lacks 1 amino acid by using an alternative splice donor and acceptor site at the beginning of exon 17. SLC26A6D had unspliced intron after exon 16 resulting in frame-shift and early termination (38).
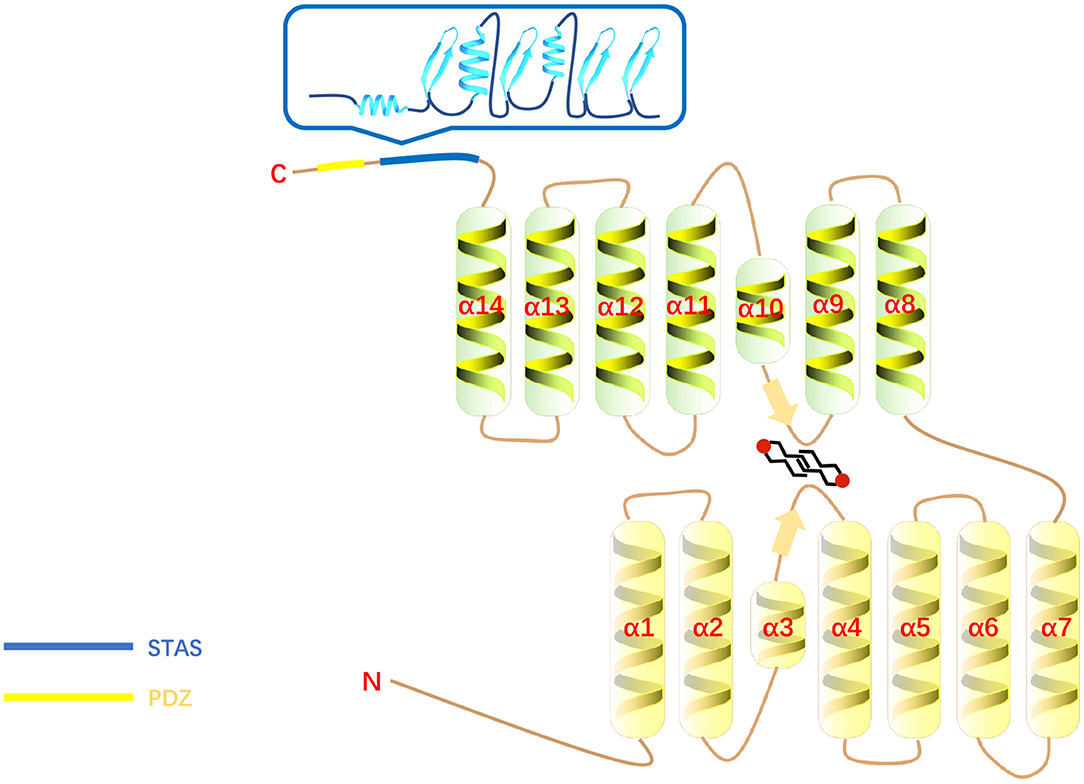
Figure 1. The structure of SLC26A6. The blue box is the STAS domain.
Electrogenic Property of SLC26A6
There is controversy about whether Cl−/ exchange mediated by SC26A6 is electrogenic. Initially, Ko et al. detected Cl− and transport in Xenopus oocytes expressed SLC26A6 and they demonstrated that the transporter is electrogenic, with unique property (40). Then they expressed SLC26A6 in Xenopus oocytes and HEK293 cells again to confirm that SLC26A6 behaves as an electrogenic transporter based on electrophysiological and pH fluorescence experiments (41). Xie et al. thought that both SLC26A6 and SLC26A3 are electrogenic, but with opposite polarities. SLC26A6 appears to have a Cl−: stoichiometry of 1:2, while the ratio for SLC26A3 is 2:1 (13). In the case of SLC26A3 depletion, the inward current may originate from the SLC26A6. The expression of two members in the same cells (to be precise in the case of epithelia and in the same membrane domain), one with the Cl−/ stoichiometry of SLC26A6 and one with that of SLC26A3, results in electroneutral Cl−/ exchanges apparently (40). This indicates that the stoichiometry of the two transporting Cl−/ is opposite, which may lead to the formation of reverse current. The opposite is that Chernova et al. expressed the homolog of SLC26A6 in Xenopus oocytes and concluded that both human and mouse SLC26A6 mediate the electroneutral Cl−/ exchange (42). In addition, SLC26A6-null mouse intestinal transepithelial studies also show that Cl−/ exchange by SLC26A6 is electroneutral. Similarly, the current electricity generation and transport stoichiometry of human and mouse SLC26A3 are also controversial and most studies agree that not only human but also mouse SLC26A3 Cl−/ exchanges are electroneutral (stoichiometry 1Cl−/1) exchange (43–45). Interestingly, Chernova et al. considered that mouse SLC26A6 mediated Cl−/oxalate exchange was apparently electrogenic, whereas that mediated by human SLC26A6 appeared electroneutral at the same time (42). However, Cl−/oxalate exchange in the apical membrane vesicles of rat mediated by SLC26A6 is also thought to be electrogenic in another study (46). On the contrary, unlike Cl−/oxalate exchange, Cl−/formate exchange is electroneutral (47).
SLC26A6 and the Pancreas
Physiological Role of SLC26A6 in the Pancreas
The secretion of and fluid is an essential function of pancreatic ductal epithelium and is critical for maintaining the integrality of the tissue. The pancreas of human secretes 1–2 L isotonic alkaline fluid every day, in which the concentration of bicarbonate may exceed 140 mM under stimulation (48). Under stimulating conditions, secretion depends critically upon the activity of CFTR anion channel (49) which is a cAMP-dependent anion channel located on the apical membrane. This rich liquid removes digestive enzymes in the duct branches, facilitates solubilization of macromolecular substances, neutralizes the protons secretion of acinar cells, inhabits premature activation of trypsinogen, and neutralizes gastric acid in the duodenum to provide the optimal pH environment for digestive enzymes. Guinea pigs could secrete pancreatic juice containing 140 mM under stimulation by secretin or cAMP, which is similar to the concentration of human pancreatic juice (50). Interestingly, Steward et al. explored the secretion mechanism of pancreatic ducts by isolating the pancreatic interlobular ducts in guinea pigs, and concluded that only one third of is secreted via the apical Cl−/ exchangers, and the other two thirds by CFTR (50). The Cl−/ exchangers of ductal epithelium have three, two are members of the SLC26 family distributed on the apical membrane and one is AE2 of the SLC4 family which distributed on the basolateral membrane (51–53). Interestingly, when the concentration of in the lumen reached the highest value (140 mM), Cl−/ exchange mediated by SLC26A3 and AE2 would be reversed, which reabsorbs instead of secretion (50). The function of SLC26A6 to secrete into the lumen is inhibited and the continuing secretion of will be mediated almost entirely (90%) by CFTR (49) (Figure 2). Ko et al. demonstrated that binding the highly conserved STAS domains of SLC26A6 to the regulatory (R) domains of CFTR can enhance the activity of both SLC26A6 and CFTR (36). Therefore, SLC26A6 has synergistic interactions with CFTR in the secretion of pancreatic ductal epithelium, and SLC26A6 deletion results in dis-regulation of CFTR in the pancreatic duct (54). Most people may think that SLC26A6 deletion could upregulate the ability of CFTR in a compensatory way to secrete , but there is no evidence to confirm this hypothesis. The explanation given by Wang et al. is that the activity of CFTR is always inhibited by SLC26A6 in the resting state, and SLC26A6 regulates the activity of CFTR by reducing the rate of CFTR activation in the stimulated state at physiological stimulus intensity (54). Therefore, SLC26A6 deletion could remove tonic inhibition of CFTR by SLC26A6 in the resting ducts and by reducing activation of CFTR by SLC26A6 in the stimulated ducts. Conversely, Ishiguro et al. (55) revealed that there was no change of SLC26A6 deletion on neither spontaneous nor stimulated secretion in isolated ducts or in vivo. However, reverse transcription (RT)-PCR showed an apparent upregulation of SLC26A3 mRNA, which may be a compensatory upregulation of SLC26A6 deletion to maintain bicarbonate secretion and pancreatic juice volume. Since SLC26A6 is predominantly distributed in the proximal portion of pancreas duct and SLC26A3 is distributed in the distal portion (36, 40), they are activated by different ion gradients (Figure 3).
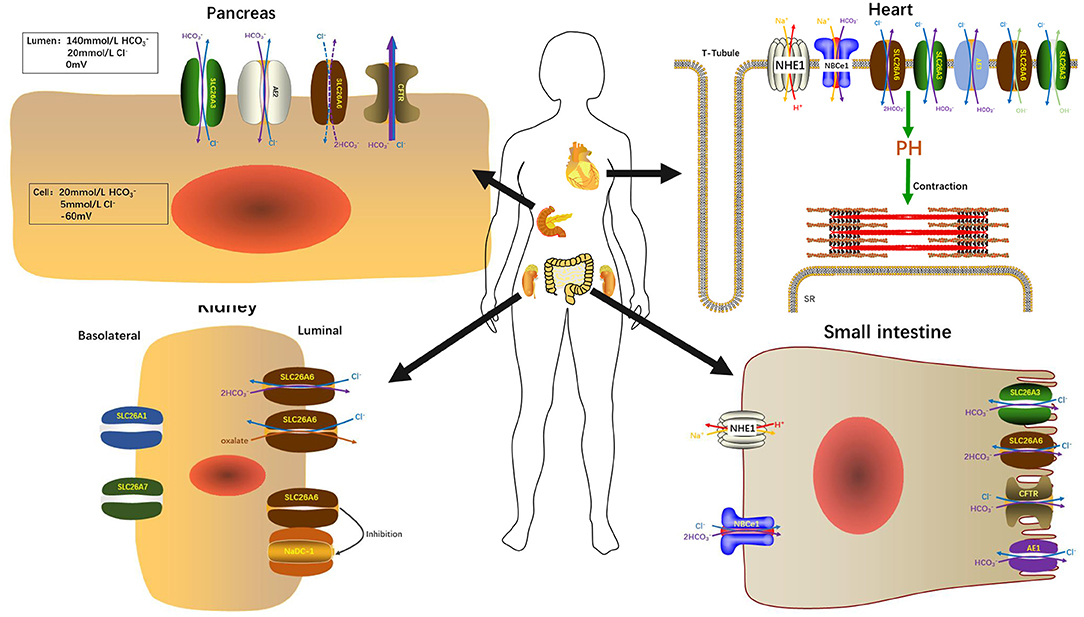
Figure 2. SLC26A6 participates in the maintenance of normal organ function in the pancreas, kidneys, heart, and small intestine.
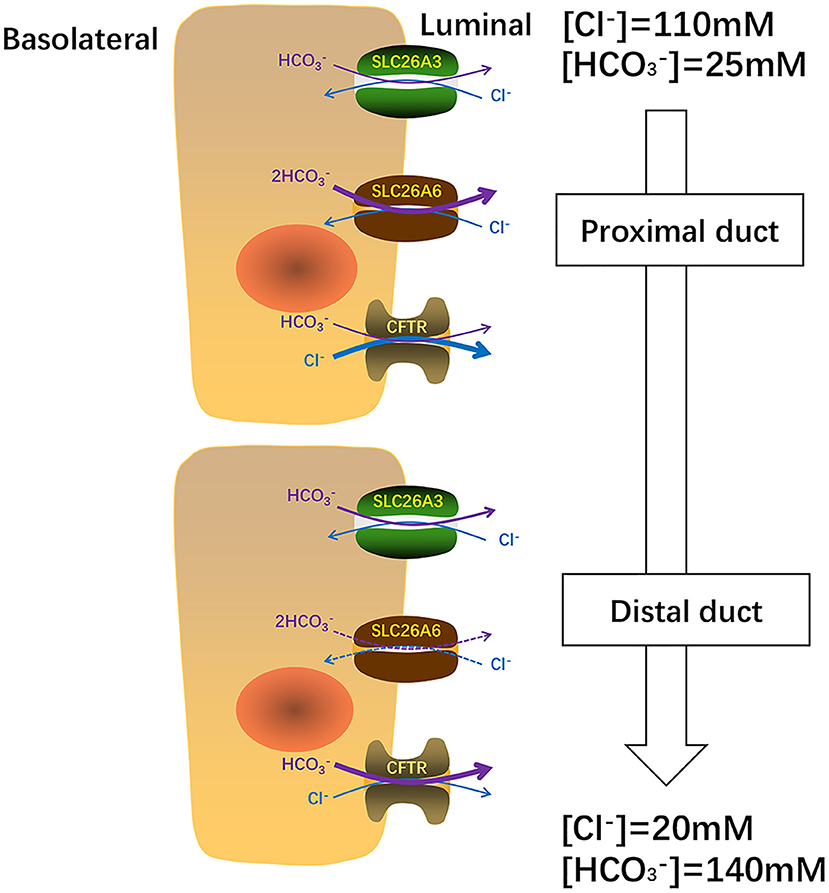
Figure 3. In pancreas, SLC26A6 and SLC26A3 perform their functions at different ion concentrations. SLC26A6 is predominantly responsible for the excretion of basal and CFTR is primarily responsible for Cl− secretion from the proximal duct. Close to the distal tubule, the concentration of increases, SLC26A3 plays a major role in the secretion of basal . When the concentration of in the distal tubule reaches a maximum of 140 uM, the secretion of mainly depends on CFTR.
SLC26A6 and Pancreatic Diseases
The distribution and role of SLC26A6 in human pancreatic ducts is still unclear. The pathological significance of SLC26A6 in the pancreas is little known. Deterioration of Pancreatic ductal secretion is observed in chronic pancreatitis. Therefore, SLC26A6 is a reasonable candidate for a chronic pancreatitis susceptibility gene. But the study from Balazs et al. verified the SLC26A6 is not related to mutations associated with chronic pancreatitis (56). Unfortunately, there is no more study on human pancreatic diseases. Further studies on the role of the anion exchanger in the human pancreas may clarify the role of secretion disorders in acute pancreatitis, chronic pancreatitis and related pancreatic diseases.
SLC26A6 and the Intestine
Physiologic Role of SLC26A6 in the Intestine
SLC26A6 and Intestinal Cl–/ Exchange
It is known that intestinal secretion is stimulated by cAMP (57–59). There are at least two secretion mechanisms of , one is Cl−-dependent and the other is Cl−-independent. The apical membrane Cl−-independent secretion stimulated by cAMP is considered to be mediated by the Cl− channel. CFTR is one of the Cl− channels responsible for secretion (60–62). Under stimulation, CFTR is still the most important conduction pathway for intestinal secretion of (63). On the country, the apical membrane Cl−-dependent secretion is mediated by the Cl−/ exchangers (60, 62). SLC26A6 and SLC26A3 are considered to play vital roles in Cl−-dependent secretion, primarily responsible for basal secretion related strongly to blood concentration (64). In addition, there are three mechanisms involved in intestinal acid-base transport (Figure 3), a DIDS-sensitive Na+/ co-transporter (NBC) and an amiloride-sensitive Na+/H+ exchanger (NHE1) present on the basolateral membranes and a Cl− / (AE4) exchanger located on the apical membranes of entrocyte (60, 65, 66). SLC26A3 and SLC26A6 are both expressed in the small intestine and large intestine. The difference is that SLC26A6 expression is very high in the small intestine (especially in the duodenum) but low in the colon (17) and SLC26A3 is expressed primarily in the colon and moderately in the small intestine (44). Therefore, SLC26A6 is particularly vital in mediating the increase of duodenal secretion with the raising blood (64). Also, in vitro studies have shown that its presence enhances the secretory rate of in the basal state (18, 67). The role of SLC26A6 in duodenal secretion crucially depends on the acid/base status under in vivo conditions, with very crucial contribution to basal secretory rates at high, but not at low, blood concentrations (64). In addition, SLC26A6 mediates secretion strongly dependent on carbonic anhydrase, while the Cl−-independent and cAMP-activated secretion are both largely independent of the enzyme (68). Based on SLC26A6-null mice, it was found that SLC26A6 is responsible for <30% of the basal secretion and had no respondence to cAMP and forskolin simulation, while prostaglandin E2-stimulated secretion is mediated via a Ca2+-dependent pathway (18, 67). The SLC26A3-mediated Cl−/ exchange contributes to the rest of ~60% of basal secretion (69). The mouse small intestine was further measured which revealed that the level of SLC26A6 and SLC26A3 expression reciprocates along the villus axis with SLC26A6 greatest in the upper villus in the lower villus/crypt and SLC26A3 greatest in the lower villus/crypt (Figure 4), but both exchangers are well-represented throughout the villus length (69, 70). And microfluorimetry studies of the upper villus epithelium found that SLC26A6 provides 70% of the total Cl−/ exchange, while SLC26A3 provides nearly all of the apical membrane Cl−/ exchange of the lower villus epithelium (71). SLC26A6 has been demonstrated to also participate in the transport of Cl−/ in the lower villi, but since SLC26A6 is an electrogenic transporter, cell acidification or membrane depolarization during glucose transport could mask the activity of SLC26A6 (72). Moreover, SLC26A6 may primarily serve other significant functions, such as the absorption of salt, nutrient-associated anion absorption (73) and regulating the pH of the intestinal lumen fluid. Studies demonstrated that compared with wild-type, SLC26A6-null mice show a decreased basal duodenal secretory rate (18). But the effects of SLC26A6-null mice on intestine and tubular functions are not profound at the steady state (18). This does not rule out that SLC26A6 deletion may lead to the compensatory upregulation of other Cl−-absorption transporters in the intestinal lumen to maintain homeostasis.
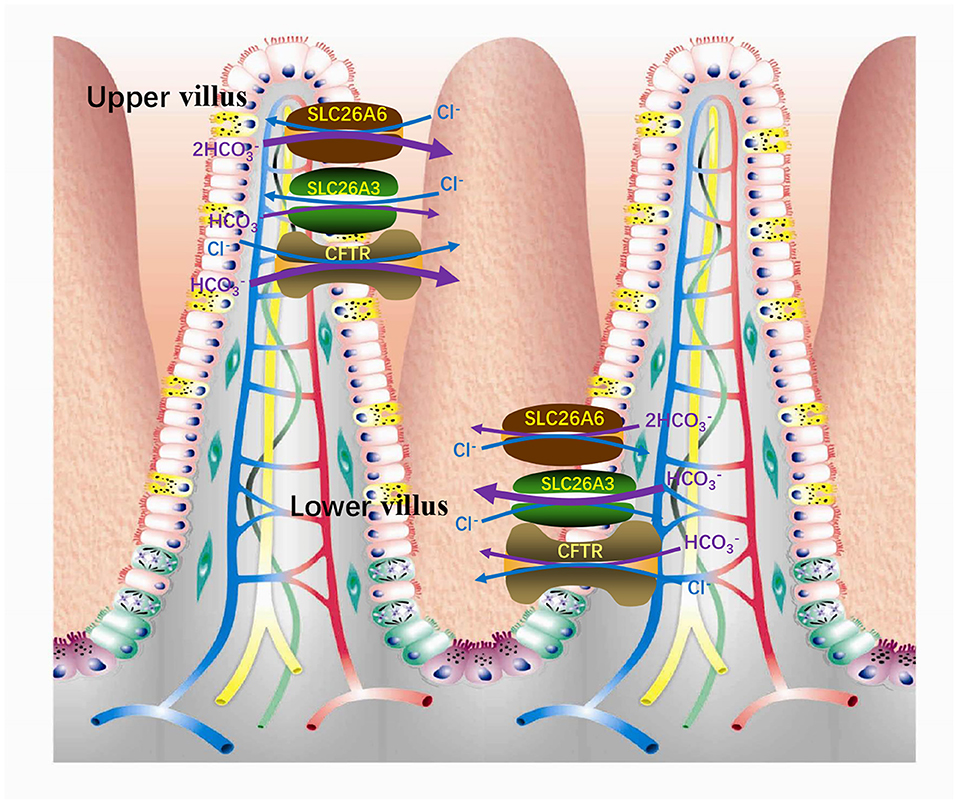
Figure 4. In the small intestinal villi epithelium, SLC26A6 is predominantly responsible for the basal secretion of the upper villi, while SLC26A3 is responsible for the lower villi.
SLC26A6 and Intestinal Cl–/Oxalate Exchange
Another function of SLC26A6 in the small intestine is to mediate oxalate excretion by the duodenum (9) and distal ileum (74), which plays an important role in oxalate homeostasis. Oxalate is indeed an obdurate anion and exists as a monovalent or divalent anion. It is not metabolized in the human body, but it is easily produced through enormous metabolic pathways (75). It is usually present in minute amounts related to other anions, yet small changes in its concentration in the existence of calcium can result in the deposition of calcium oxalate. The concentration of oxalate in plasma depends on dietary load, intestinal absorption, metabolic production and renal excretion. The transport of oxalate in intestine is bidirectional and net transport (76, 77). Transport methods include transcellular pathway and paracellular pathway. Studies demonstrated that intestinal oxalate secretion relies on a SLC26A6-dependent transcellular mechanism, while the oxalate absorption takes place by a paracellular channel (78). In general, in contrast to this phenomenon of basal net oxalate secretion in the small bowel and proximal colons, the distal colons of animals typically exhibit basal net absorption of oxalate (79, 80). SLC26A6 in the mouse ileum mediates apical secretion of oxalate in exchange for Cl− and is an important component of the transcellular serosal-to-mucosal unidirectional oxalate flux (74). The vectorial oxalate transport mediated by SLC26A6 appears to be more dependent on the direction and magnitude of counterion driver gradients than the intrinsic property of the protein (81). Although SLC26A6 is also expressed in the apical membranes of mouse colon (17), its role in colonic oxalate transport awaits further studies. In addition, SLC26A1, SLC26A2, and SLC26A3 also participate in the transport of intestinal oxalate (Figure 5). SLC26A1 is considered to a /oxalate transporter. It may mediate /oxalate exchange in parallel with the Cl−/oxalate exchange, again leading to the recycling of oxalate with minimal net oxalate transport (8, 74). SLC26A2 participates in the secretion of intestinal oxalate with trans-, Cl−, or oxalate itself (82). SLC26A3 mediates oxalate transport by absorbing oxalate and Cl− in the large and small intestines (83). AE1 is also detected to express in the apical membranes of ileum (84) and surface cells of the distal colon (85), which may also play a role in oxalate transport.
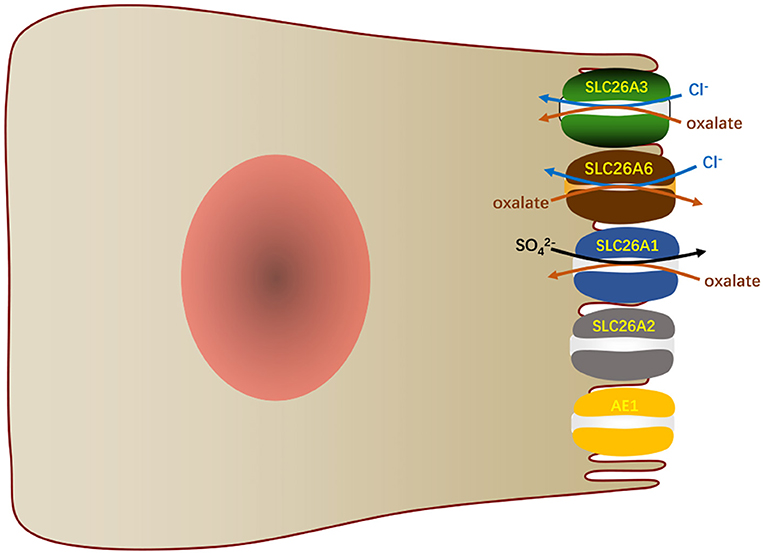
Figure 5. In the small intestinal villi epithelium, SLC26A6 and other transports participate in the oxalate transport.
SLC26A6 and Intestinal Diseases
Duodenal mucosal secretion plays important role in protecting duodenal mucosa against gastric-induced injure (86, 87). Clinical study found that duodenal mucosal secretion was found to be markedly diminished in Helicobacter pylori-associated duodenal ulcer patients (61). Helicobacter pylori infection impaired the expressions and functional activities of duodenal mucosal CFTR and SLC26A6 via TGFβ-mediated P38 MAPK signaling pathway, which contributed to the development of duodenal ulcer (88, 89). The studies showed that inflammatory bowel disease (IBD) patients usually accompanied with hyperoxaluria and kidney stones (90, 91), which might be due to the lack of oxalate transporters. Also, hyperoxaluria is a major complication after malabsorptive bariatric surgery of obesity (92, 93) and more than 50% of patients would be complicated. Previous studies demonstrated that SLC26A6-null mice have serious defects in the intestinal secretion of oxalate which could lead to enhanced net oxalate absorption and result in a high incidence of hyperoxemia, hyperuricemia and calcium oxalate urolithiasis (9, 74). And nephrolithiasis in the SLC26A6-null mouse is accompanied by 50–75% reduction in intestinal oxalate secretion with increased intestinal oxalate absorption (82). It has been shown that the activation of PKA signaling pathway can enhance SLC26A6 surface protein expression and increase the intrinsic activity of preexisting SLC26A6 surface membrane transporters to stimulate oxalate transport (94). Therefore, SLC26A6 may be used as one of the therapeutic targets for hyperoxaluria caused by various reasons.
SLC26A6 and the Kidney
Physiological Role of SLC26A6 in the Kidney
There are two ways for the body to excrete oxalate, one is via the intestinal tract as mentioned above, and the other is by the kidneys. The majority of oxalate excretion in the human body though the kidneys where 90–95% of circulating oxalate is removed via the urine (95, 96), and the rest is secreted into the intestine. When intestinal or kidney oxalate metabolism is defective, it can lead to hyperoxaluria which is the main risk factor for kidney stone formation (97). Calcium oxalate is the primary element in 70–80% of kidney stones (98), and the risk of stone formation was also affected by small changes in urinary oxalate concentration (99). The expression of SLC26A6 can be detected on the apical membrane of the proximal duct epithelium of the kidney (12), and the pivotal function of the proximal tubule is to retrieve the majority of NaCl and water filtered by the kidney. Previous studies have shown that oxalate cloud stimulate the reabsorption of Cl− in the proximal tubule, which means that the apical membrane Cl−/oxalate exchange mediates Cl− absorption as well as oxalate secretion (100–102). Moreover, microperfusion studies found that SLC26A6 was also the major apical Cl−/ exchangers in the proximal tubule straight segments. Wang et al. proposed that the NaCl reabsorption mediated by the Cl−/oxalate exchange in the proximal tubule was entirely contributed by SLC26A6, and the apical membrane Cl−/ exchange mediated by SLC26A6 was not a contributing factor to transtubular NaCl absorption (18). The Cl−/oxalate exchange mediated by SLC26A6 in the proximal tubule is essential for oxalate homeostasis (74). In addition, AE1 and SLC26A7 distributed on the basolateral membrane might participate in the transport of oxalate in the tubule epithelium (46, 103). SLC26A1 is considered to mediate basolateral oxalate-/ exchange in the proximal tubule (46). The function of SLC26A7 in the transport of oxalate is unclear. In addition to mediate oxalate secretion, SLC26A6 also can form a complex with the succinate transporter NaDC-1 and strongly inhibit NaDC-1 activity and interact with NaDC-1 to control absorption of citrate from the urinary lumen (104). Urinary citrate can chelate free Ca2+ to protect against Ca2+ oxalate crystallization. However, whether this mechanism can prevent the occurrence of kidney stones remains to be further studied.
SLC26A6 and Renal Diseases
Studies have indicated that SLC26A6-null mice have a 4-fold increase in urine oxalate excretion (9, 74), but serum oxalate levels were not apparently different between KO and WT mice, although there was a tendency toward hyperoxalemia in the KO mice (74). The high concentration of oxalate in the urine makes the kidney stones appear high frequency in SLC26A6-null mice, and the occurrence rate of male mice is higher than that of female mice (9). In SLC26A6-null mice, histological examination of the kidney demonstrated that the stones mainly comprise of calcium oxalate, and are primarily found in the lumen of cortical tubules and in the urinary space (9). Although a number of studies have shown a close relationship between the expression of SLC26A6 and kidney stone formation, but its precise role in the human diseases remain unknown (104–107). Studies have demonstrated that SLC26A6 has fundamental roles not only in proximal tubule NaCl transport but also in the prevention of hyperoxaluria as well as calcium oxalate nephrolithiasis. Therefore, further studies to explore the metabolism of oxalate, absorption and excretion in clinic is vital importance, and it may enhance oxalate excretion in pharmacology to provide new therapeutic targets.
SLC26A6 and the Heart
Physiological Role of SLC26A6 in the Heart
The homeostasis of cardiomyocytes depends on cytoplasmic buffer and membrane ion transporter, thus the regulation of pH in cardiomyocytes is extremely complicated. pH is a vital regulator of cardiac excitation and contraction (108), which is an adverse contributing factor of electric arrhythmia and cardiac hypertrophy (109). An uncompensated decrease in cytoplasmic pH leads to abnormal electrical activities, reducing Ca2+ transient and contraction in cardiomyocytes by potentially decreasing the binding of Ca2+ to troponin C as well as by affecting cross-bridges action resulting in maximal force reduction (108, 110), which would trigger arrhythmia (111). Inversely, twitch tension, resting tonic tension, voltage dependent tonic tension, and after-contraction contractile parameters can be enhanced with intracellular alkalosis in sheep cardiac fibers (112). H+ equivalent transporters Na+/H+ exchanger (NHE) and Na+- co-transporter (NBC) mediate acid extrusion, while Cl−/ exchangers and Cl−/OH− exchangers extrude excess base (113, 114). Also, sarcolemmal lactic acid transporter is recruited in response to increased anaerobic metabolism (115). Under normal physiological state, the regulating system can maintain the pH at a steady state value of 7.2. When coupled with a Na+-dependent acid excretion mechanism, Na+ loading increased Cl−/ exchange which can affect myocardial contractility and promote cardiac hypertrophy (116). Another function of Cl−/ exchange in the heart is to counter the alkalinizing effects of Na+/H+ exchange (116, 117), which can reduce the occurrence of cardiac hypertrophy (118, 119). SLC26A6, SLC26A3, and AEs (especially AE3) have been found to mediate Cl−/ exchange in the myocardium (19, 120) (Figure 3). Immunohistochemistry showed that SLC26A6, SLC26A3, and AE3 exist in the plasma membrane of ventricular myocytes (19). The expression level of SLC26A6 is lower in the atrium than in the ventricle, while AE3 is only detected in the ventricle (19). SLC26A6 is the major anion exchanger in the heart because the heart-dependent acid load is primarily mediated by SLC26A6 and its expression in the myocardial cell membrane is much higher than that of AE and SLC26A3 (19, 121). Furthermore, recovery from alkalinization induced by acetate is severely impaired in SLC26A6-null mice cardiomyocytes (121). Of note that SLC26A6 is a dual Cl−/, Cl−/OH− exchanger with unique implications for myocardial intracellular pH regulation (19). Also, SLC26A6 is the most important Cl−/ and Cl−/OH− exchanger for the myocardium maintaining normal activity. However, SLC26A6 mediates the physiological relevance of the Cl−/OH− exchange in the heart is not clear yet. In addition, SLC26A6 and SLC26A3 are the only Cl−/ exchangers of the SLC26 family that are expressed in the heart (19). The cardiac SLC26A6 not only regulates the pH, but also plays a unique role in regulating cardiac excitability and function of heart. The study of Sirish et al. demonstrated that SLC26A6 deletion resulted in the shortening of cardiac action potential (AP), cardiomyocyte Ca2+ transient and sarcoplasmic reticulum Ca2+ loading decrease, cardiomyocyte diminution of sarcomeric shortening, and cardiomyocyte pH elevation (121). In SLC26A6-null mice, these factors lead to a decrease of cardiac fractional shortening and cardiac contractility responses and alter cardiac conduction system, as seen in sinus bradycardia and fragmentation of the QRS electrocardiographic-recorded complex (121).
SLC26A6 and Heart Diseases
SLC26A6 is considered to be a primary transporter in the heart ventricle. Although the regulation mechanism of various ion transporters is complicated (19), studies have demonstrated that the expression of SLC26A6 and Cl− transporting activity are upregulated in the type 2 diabetic heart model, which reveals that effective SLC26A6 blockers may be efficient in regulating pH of type 2 diabetic hearts (122). Additionally, Cl− influx and efflux mediated by SLC26A6 may be beneficial to intracellular acidification in diabetic myocardium during cardioplegia-induced arrest (122). Evidences reveal that protein kinase C (PKC) can inhibit the transport activity of SLC26A6 (19, 123). The inhibitory effect of PKC is attributed to PKC-mediated displacement owing to the combination of carbonic anhydrase II and SLC26A6, and thus destroys the transport metabolites (123). Therefore, in order to prevent myocardial hypertrophy, attention should be paid to regulate the function of myocardial SLC26A6 to ensure the acid-base homeostasis of myocardial tissue. In the state of anesthesia and intensive care, patients with basic heart diseases should minimize the use of vasoactive drugs that stimulate α1-adrenergic receptors to avoid insufficient myocardial contraction.
SLC26A6 and the Placenta
Sulfate is significant for human growth and development and human usually get it from metabolism of sulfur-containing amino acids and the diet in the body. Also, sulfate is an essential nutrient for the growth and development of fetus (124). However, the developing fetus have negligible capacity to generate sulfate from methionine and cysteine (125, 126) and depends on sulfate supplied from maternal circulation via placental sulfate transporters (127). For human, several transporters have been detected in the placenta, which include SLC26A6 (21). During the period of pregnancy, the level of maternal circulating sulfate increases by nearly 2-fold (128), and the increased plasma sulfate levels are associated with elevated sulfate reabsorption of kidney (129). In the kidney, sulfate is filtered in the glomerulus and then reabsorbed via epithelial cells of the proximal tubule, firstly across the apical membrane where SLC13A1, SLC26A2, and SLC26A6 are expressed and secondly via SLC26A1 on the basolateral membrane (31, 46, 130). Therefore, maintaining the normal function of transporters, such as SLC26A6 in the placenta is essential for fetal growth and development. In future clinical applications, monitoring the function of placental transporters may be used to predict and evaluate neonatal cartilage development.
Conclusion
In summary, SLC26A6 transporter mediates the exchange of anions in mammalian cells, thereby participating in the maintenance of normal physiological functions of various organs. However, there are still many problems on SLC26A6 need to be solved. Firstly, the transport base of SLC26A6 has not yet been illuminated at the molecular level. Secondly, the cellular mechanism that regulates and fine-tune the activity of SLC26A6 transporter has not been elucidated. Finally, it is about the influence of the normal function and dysfunction of SLC26A6 on human related diseases. The connection and role of SLC26A6 with the body may become a research hotspot and may become a new molecular marker for the diagnosis and treatment of human-related diseases. The development of drugs targeting SLC26A6 will provide new treatment directions for human-related diseases.
Author Contributions
JW and WW wrote the manuscript. HW participated in information collection, analysis, and organization. BT revised the manuscript for clarity and style. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82073087).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Atala A. Re: missense mutations in SLC26A8, encoding a sperm-specific activator of CFTR, are associated with human asthenozoospermia. J Urol. (2014) 191:554–5. doi: 10.1016/j.juro.2013.10.082
2. Blackman SM, Commander CW, Watson C, Arcara KM, Strug LJ, Stonebraker JR, et al. Genetic modifiers of cystic fibrosis-related diabetes. Diabetes. (2013) 62:3627–35. doi: 10.2337/db13-0510
3. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet. (1997) 17:411–22. doi: 10.1038/ng1297-411
4. Hastbacka J, de la Chapelle A, Mahtani MM, Clines G, Reeve-Daly MP, Daly M, et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. (1994) 78:1073–87. doi: 10.1016/0092-8674(94)90281-X
5. Hoglund P, Haila S, Socha J, Tomaszewski L, Saarialho-Kere U, Karjalainen-Lindsberg ML, et al. Mutations of the down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nat Genet. (1996) 14:316–9. doi: 10.1038/ng1196-316
6. Liss HP, Bernstein J. Ribavirin aerosol in the elderly. Chest. (1988) 93:1239–41. doi: 10.1378/chest.93.6.1239
7. Rungta RL, Choi HB, Tyson JR, Malik A, Dissing-Olesen L, Lin PJC, et al. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. (2015) 161:610–21. doi: 10.1016/j.cell.2015.03.029
8. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, et al. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest. (2010) 120:706–12. doi: 10.1172/JCI31474
9. Jiang Z, Asplin JR, Evan AP, Rajendran VM, Velazquez H, Nottoli TP, et al. Calcium oxalate urolithiasis in mice lacking anion transporter SLC26A6. Nat Genet. (2006) 38:474–8. doi: 10.1038/ng1762
10. Xu J, Song P, Miller ML, Borgese F, Barone S, Riederer B, et al. Deletion of the chloride transporter SLC26A9 causes loss of tubulovesicles in parietal cells and impairs acid secretion in the stomach. Proc Natl Acad Sci USA. (2008) 105:17955–60. doi: 10.1073/pnas.0800616105
11. Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, et al. Deletion of the chloride transporter SLC26A7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem. (2009) 284:29470–9. doi: 10.1074/jbc.M109.044396
12. Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc Natl Acad Sci USA. (2001) 98:9425–30. doi: 10.1073/pnas.141241098
13. Xie Q, Welch R, Mercado A, Romero MF, Mount DB. Molecular characterization of the murine SLC26A6 anion exchanger: functional comparison with SLC26A1. Am J Physiol Renal Physiol. (2002) 283:F826–38. doi: 10.1152/ajprenal.00079.2002
14. Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med. (2013) 34:494–515. doi: 10.1016/j.mam.2012.07.009
15. Stewart AK, Shmukler BE, Vandorpe DH, Reimold F, Heneghan JF, Nakakuki M, et al. SLC26 anion exchangers of guinea pig pancreatic duct: molecular cloning and functional characterization. Am J Physiol Cell Physiol. (2011) 301:C289–303. doi: 10.1152/ajpcell.00089.2011
16. Stewart AK, Yamamoto A, Nakakuki M, Kondo T, Alper SL, Ishiguro H. Functional coupling of apical Cl−/ exchange with CFTR in stimulated secretion by guinea pig interlobular pancreatic duct. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G1307–17. doi: 10.1152/ajpgi.90697.2008
17. Wang Z, Petrovic S, Mann E, Soleimani M. Identification of an apical Cl−/ exchanger in the small intestine. Am J Physiol Gastrointest Liver Physiol. (2002) 282:G573–9. doi: 10.1152/ajpgi.00338.2001
18. Wang Z, Wang T, Petrovic S, Tuo B, Riederer B, Barone S, et al. Renal and intestinal transport defects in SLC26A6-null mice. Am J Physiol Cell Physiol. (2005) 288:C957–65. doi: 10.1152/ajpcell.00505.2004
19. Alvarez BV, Kieller DM, Quon AL, Markovich D, Casey JR. SLC26A6: a cardiac chloride-hydroxyl exchanger and predominant chloride-bicarbonate exchanger of the mouse heart. J Physiol. (2004) 561:721–34. doi: 10.1113/jphysiol.2004.077339
20. Pierucci-Alves F, Akoyev V, Stewart JC III, Wang LH, Janardhan KS, Schultz BD. Swine models of cystic fibrosis reveal male reproductive tract phenotype at birth. Biol Reprod. (2011) 85:442–51. doi: 10.1095/biolreprod.111.090860
21. Dawson PA, Rakoczy J, Simmons DG. Placental, renal, and ileal sulfate transporter gene expression in mouse gestation. Biol Reprod. (2012) 87:43. doi: 10.1095/biolreprod.111.098749
22. Shcheynikov N, Yang D, Wang Y, Zeng W, Karniski LP, So I, et al. The SLC26A4 transporter functions as an electroneutral Cl−/I−/ exchanger: role of SLC26A4 and SLC26A6 in I− and secretion and in regulation of CFTR in the parotid duct. J Physiol. (2008) 586:3813–24. doi: 10.1113/jphysiol.2008.154468
23. Abdulnour-Nakhoul S, Nakhoul HN, Kalliny MI, Gyftopoulos A, Rabon E, Doetjes R, et al. Ion transport mechanisms linked to bicarbonate secretion in the esophageal submucosal glands. Am J Physiol Regul Integr Comp Physiol. (2011) 301:R83–96. doi: 10.1152/ajpregu.00648.2010
24. Petrovic S, Wang Z, Ma L, Seidler U, Forte JG, Shull GE, et al. Colocalization of the apical Cl−/ exchanger PAT1 and gastric H-K-ATPase in stomach parietal cells. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G1207–16. doi: 10.1152/ajpgi.00137.2002
25. Jalali R, Zandieh-Doulabi B, DenBesten PK, Seidler U, Riederer B, Wedenoja S, et al. SLC26A3/Dra and SSLC26A6 in murine ameloblasts. J Dent Res. (2015) 94:1732–9. doi: 10.1177/0022034515606873
26. Yin K, Lei Y, Wen X, Lacruz RS, Soleimani M, Kurtz I, et al. SLC26A gene family participate in pH regulation during enamel maturation. PLoS ONE. (2015) 10:e0144703. doi: 10.1371/journal.pone.0144703
27. Thomson RB, Thomson CL, Aronson PS. N-glycosylation critically regulates function of oxalate transporter SLC26A6. Am J Physiol Cell Physiol. (2016) 311:C866–73. doi: 10.1152/ajpcell.00171.2016
28. Lohi H, Kujala M, Kerkela E, Saarialho-Kere U, Kestila M, Kere J. Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics. (2000) 70:102–12. doi: 10.1006/geno.2000.6355
29. Waldegger S, Moschen I, Ramirez A, Smith RJ, Ayadi H, Lang F, et al. Cloning and characterization of SLC26A6, a novel member of the solute carrier 26 gene family. Genomics. (2001) 72:43–50. doi: 10.1006/geno.2000.6445
30. Jin H, Wen G, Deng S, Wan S, Xu J, Liu X, et al. Oestrogen upregulates the expression levels and functional activities of duodenal mucosal CFTR and SLC26A6. Exp Physiol. (2016) 101:1371–82. doi: 10.1113/EP085803
31. Kujala M, Tienari J, Lohi H, Elomaa O, Sariola H, Lehtonen E, et al. SLC26A6 and SLC26A7 anion exchangers have a distinct distribution in human kidney. Nephron Exp Nephrol. (2005) 101:e50–8. doi: 10.1159/000086345
32. Simmons DG, Rakoczy J, Jefferis J, Lourie R, McIntyre HD, Dawson PA. Human placental sulfate transporter mRNA profiling from term pregnancies identifies abundant SLC13A4 in syncytiotrophoblasts and SLC26A2 in cytotrophoblasts. Placenta. (2013) 34:381–4. doi: 10.1016/j.placenta.2013.01.017
33. Geertsma ER, Chang YN, Shaik FR, Neldner Y, Pardon E, Steyaert J, et al. Structure of a prokaryotic fumarate transporter reveals the architecture of the SLC26 family. Nat Struct Mol Biol. (2015) 22:803–8. doi: 10.1038/nsmb.3091
34. Shibagaki N, Grossman AR. Probing the function of STAS domains of the Arabidopsis sulfate transporters. J Biol Chem. (2004) 279:30791–9. doi: 10.1074/jbc.M403248200
35. Shibagaki N, Grossman AR. The role of the STAS domain in the function and biogenesis of a sulfate transporter as probed by random mutagenesis. J Biol Chem. (2006) 281:22964–73. doi: 10.1074/jbc.M603462200
36. Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, et al. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. (2004) 6:343–50. doi: 10.1038/ncb1115
37. Rouached H, Berthomieu P, El Kassis E, Cathala N, Catherinot V, Labesse G, et al. Structural and functional analysis of the C-terminal STAS (sulfate transporter and anti-sigma antagonist) domain of the Arabidopsis thaliana sulfate transporter SULTR1.2. J Biol Chem. (2005) 280:15976–83. doi: 10.1074/jbc.M501635200
38. Lohi H, Lamprecht G, Markovich D, Heil A, Kujala M, Seidler U, et al. Isoforms of SLC26A6 mediate anion transport and have functional PDZ interaction domains. Am J Physiol Cell Physiol. (2003) 284:C769–79. doi: 10.1152/ajpcell.00270.2002
39. Fanning AS, Anderson JM. PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J Clin Invest. (1999) 103:767–72. doi: 10.1172/JCI6509
40. Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, et al. A molecular mechanism for aberrant CFTR-dependent transport in cystic fibrosis. EMBO J. (2002) 21:5662–72. doi: 10.1093/emboj/cdf580
41. Shcheynikov N, Wang Y, Park M, Ko SB, Dorwart M, Naruse S, et al. Coupling modes and stoichiometry of Cl−/ exchange by SLC26A6 and SLC26A3. J Gen Physiol. (2006) 127:511–24. doi: 10.1085/jgp.200509392
42. Chernova MN, Jiang L, Friedman DJ, Darman RB, Lohi H, Kere J, et al. Functional comparison of mouse SLC26A6 anion exchanger with human SLC26A6 polypeptide variants: differences in anion selectivity, regulation, and electrogenicity. J Biol Chem. (2005) 280:8564–80. doi: 10.1074/jbc.M411703200
43. Lamprecht G, Baisch S, Schoenleber E, Gregor M. Transport properties of the human intestinal anion exchanger DRA (down-regulated in adenoma) in transfected HEK293 cells. Pflugers Arch. (2005) 449:479–90. doi: 10.1007/s00424-004-1342-x
44. Melvin JE, Park K, Richardson L, Schultheis PJ, Shull GE. Mouse down-regulated in adenoma (DRA) is an intestinal Cl−/ exchanger and is up-regulated in colon of mice lacking the NHE3 Na+/H+ exchanger. J Biol Chem. (1999) 274:22855–61. doi: 10.1074/jbc.274.32.22855
45. Walker NM, Simpson JE, Yen PF, Gill RK, Rigsby EV, Brazill JM, et al. Down-regulated in adenoma Cl−/ exchanger couples with Na+/H+ exchanger 3 for NaCl absorption in murine small intestine. Gastroenterology. (2008) 135:1645–53 e1643. doi: 10.1053/j.gastro.2008.07.083
46. Karniski LP, Lotscher M, Fucentese M, Hilfiker H, Biber J, Murer H. Immunolocalization of sat-1 sulfate/oxalate/bicarbonate anion exchanger in the rat kidney. Am J Physiol. (1998) 275:F79–87. doi: 10.1152/ajprenal.1998.275.1.F79
47. Ohana E, Shcheynikov N, Yang D, So I, Muallem S. Determinants of coupled transport and uncoupled current by the electrogenic SLC26 transporters. J Gen Physiol. (2011) 137:239–51. doi: 10.1085/jgp.201010531
48. Domschke S, Domschke W, Rosch W, Konturek SJ, Sprugel W, Mitznegg P, et al. Inhibition by somatostatin of secretin-stimulated pancreatic secretion in man: a study with pure pancreatic juice. Scand J Gastroenterol. (1977) 12:59–63. doi: 10.1080/00365521.1977.12031112
49. Yamaguchi M, Steward MC, Smallbone K, Sohma Y, Yamamoto A, Ko SB, et al. Bicarbonate-rich fluid secretion predicted by a computational model of guinea-pig pancreatic duct epithelium. J Physiol. (2017) 595:1947–72. doi: 10.1113/JP273306
50. Steward MC, Ishiguro H, Case RM. Mechanisms of bicarbonate secretion in the pancreatic duct. Annu Rev Physiol. (2005) 67:377–409. doi: 10.1146/annurev.physiol.67.031103.153247
51. Greeley T, Shumaker H, Wang Z, Schweinfest CW, Soleimani M. Downregulated in adenoma and putative anion transporter are regulated by CFTR in cultured pancreatic duct cells. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G1301–1308. doi: 10.1152/ajpgi.2001.281.5.G1301
52. Hyde K, Harrison D, Hollingsworth MA, Harris A. Chloride-bicarbonate exchangers in the human fetal pancreas. Biochem Biophys Res Commun. (1999) 263:315–21. doi: 10.1006/bbrc.1999.1367
53. Romero MF, Fulton CM, Boron WF. The SLC4 family of transporters. Pflugers Arch. (2004) 447:495–509. doi: 10.1007/s00424-003-1180-2
54. Wang Y, Soyombo AA, Shcheynikov N, Zeng W, Dorwart M, Marino CR, et al. SLC26A6 regulates CFTR activity in vivo to determine pancreatic duct secretion: relevance to cystic fibrosis. EMBO J. (2006) 25:5049–57. doi: 10.1038/sj.emboj.7601387
55. Ishiguro H, Namkung W, Yamamoto A, Wang Z, Worrell RT, Xu J, et al. Effect of SLC26A6 deletion on apical Cl−/ exchanger activity and cAMP-stimulated bicarbonate secretion in pancreatic duct. Am J Physiol Gastrointest Liver Physiol. (2007) 292:G447–55. doi: 10.1152/ajpgi.00286.2006
56. Balazs A, Ruffert C, Hegyi E, Hritz I, Czako L, Takacs T, et al. Genetic analysis of the bicarbonate secreting anion exchanger SLC26A6 in chronic pancreatitis. Pancreatology. (2015) 15:508–13. doi: 10.1016/j.pan.2015.08.008
57. Minhas BS, Sullivan SK, Field M. Bicarbonate secretion in rabbit ileum: electrogenicity, ion dependence, and effects of cyclic nucleotides. Gastroenterology. (1993) 105:1617–29. doi: 10.1016/0016-5085(93)91056-N
58. Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, et al. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca2+-dependent secretion. J Physiol. (1997) 505:411–23. doi: 10.1111/j.1469-7793.1997.411bb.x
59. Vidyasagar S, Rajendran VM, Binder HJ. Three distinct mechanisms of secretion in rat distal colon. Am J Physiol Cell Physiol. (2004) 287:C612–21. doi: 10.1152/ajpcell.00474.2003
60. Clarke LL, Harline MC. Dual role of CFTR in cAMP-stimulated secretion across murine duodenum. Am J Physiol. (1998) 274:G718–26. doi: 10.1152/ajpgi.1998.274.4.G718
61. Hogan DL, Crombie DL, Isenberg JI, Svendsen P, Schaffalitzky de Muckadell OB, Ainsworth MA. CFTR mediates cAMP- and Ca2+-activated duodenal epithelial secretion. Am J Physiol. (1997) 272:G872–8. doi: 10.1152/ajpgi.1997.272.4.G872
62. Spiegel S, Phillipper M, Rossmann H, Riederer B, Gregor M, Seidler U. Independence of apical Cl−/ exchange and anion conductance in duodenal secretion. Am J Physiol Gastrointest Liver Physiol. (2003) 285:G887–97. doi: 10.1152/ajpgi.00083.2003
63. Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. (1999) 79:S23–45. doi: 10.1152/physrev.1999.79.1.S23
64. Singh AK, Sjoblom M, Zheng W, Krabbenhoft A, Riederer B, Rausch B, et al. CFTR and its key role in in vivo resting and luminal acid-induced duodenal secretion. Acta Physiol. (2008) 193:357–65. doi: 10.1111/j.1748-1716.2008.01854.x
65. Brown CD, Dunk CR, Turnberg LA. Cl−/ exchange and anion conductance in rat duodenal apical membrane vesicles. Am J Physiol. (1989) 257:G661–7. doi: 10.1152/ajpgi.1989.257.4.G661
66. Isenberg JI, Ljungstrom M, Safsten B, Flemstrom G. Proximal duodenal enterocyte transport: evidence for Na+-H+ and Cl−/ exchange and NaHCO3 cotransport. Am J Physiol. (1993) 265:G677–85. doi: 10.1152/ajpgi.1993.265.4.G677
67. Tuo B, Riederer B, Wang Z, Colledge WH, Soleimani M, Seidler U. Involvement of the anion exchanger SLC26A6 in prostaglandin E2− but not forskolin-stimulated duodenal secretion. Gastroenterology. (2006) 130:349–58. doi: 10.1053/j.gastro.2005.10.017
68. Uchiyama H, Hayashi H, Tanji K, Sugimoto O, Suzuki Y. pH stat studies on bicarbonate secretion in the isolated mouse ileum. Biomed Res. (2007) 28:239–46. doi: 10.2220/biomedres.28.239
69. Walker NM, Simpson JE, Brazill JM, Gill RK, Dudeja PK, Schweinfest CW, et al. Role of down-regulated in adenoma anion exchanger in secretion across murine duodenum. Gastroenterology. (2009) 136:893–901. doi: 10.1053/j.gastro.2008.11.016
70. Jacob P, Rossmann H, Lamprecht G, Kretz A, Neff C, Lin-Wu E, et al. Down-regulated in adenoma mediates apical Cl−/ exchange in rabbit, rat, and human duodenum. Gastroenterology. (2002) 122:709–24. doi: 10.1053/gast.2002.31875
71. Simpson JE, Schweinfest CW, Shull GE, Gawenis LR, Walker NM, Boyle KT, et al. PAT-1 (SLC26A6) is the predominant apical membrane Cl−/ exchanger in the upper villous epithelium of the murine duodenum. Am J Physiol Gastrointest Liver Physiol. (2007) 292:G1079–88. doi: 10.1152/ajpgi.00354.2006
72. Walker NM, Simpson JE, Hoover EE, Brazill JM, Schweinfest CW, Soleimani M, et al. Functional activity of Pat-1 (SLC26A6) Cl−/ exchange in the lower villus epithelium of murine duodenum. Acta Physiol. (2011) 201:21–31. doi: 10.1111/j.1748-1716.2010.02210.x
73. Seidler U, Rottinghaus I, Hillesheim J, Chen M, Riederer B, Krabbenhoft A, et al. Sodium and chloride absorptive defects in the small intestine in SLC26A6 null mice. Pflugers Arch. (2008) 455:757–66. doi: 10.1007/s00424-007-0318-z
74. Freel RW, Hatch M, Green M, Soleimani M. Ileal oxalate absorption and urinary oxalate excretion are enhanced in SLC26A6 null mice. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G719–728. doi: 10.1152/ajpgi.00481.2005
75. Marengo SR, Romani AM. Oxalate in renal stone disease: the terminal metabolite that just won't go away. Nat Clin Pract Nephrol. (2008) 4:368–77. doi: 10.1038/ncpneph0845
76. Hatch M, Freel RW. Intestinal transport of an obdurate anion: oxalate. Urol Res. (2005) 33:1–16. doi: 10.1007/s00240-004-0445-3
77. Hatch M, Freel RW. The roles and mechanisms of intestinal oxalate transport in oxalate homeostasis. Semin Nephrol. (2008) 28:143–51. doi: 10.1016/j.semnephrol.2008.01.007
78. Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. (2004) 447:710–21. doi: 10.1007/s00424-003-1090-3
79. Hatch M, Freel RW, Goldner AM, Earnest DL. Oxalate and chloride absorption by the rabbit colon: sensitivity to metabolic and anion transport inhibitors. Gut. (1984) 25:232–7. doi: 10.1136/gut.25.3.232
80. Hatch M, Freel RW, Vaziri ND. Mechanisms of oxalate absorption and secretion across the rabbit distal colon. Pflugers Arch. (1994) 426:101–9. doi: 10.1007/BF00374677
81. Freel RW, Morozumi M, Hatch M. Parsing apical oxalate exchange in Caco-2BBe1 monolayers: siRNA knockdown of SLC26A6 reveals the role and properties of PAT-1. Am J Physiol Gastrointest Liver Physiol. (2009) 297:G918–29. doi: 10.1152/ajpgi.00251.2009
82. Heneghan JF, Akhavein A, Salas MJ, Shmukler BE, Karniski LP, Vandorpe DH, et al. Regulated transport of sulfate and oxalate by SLC26A2/DTDST. Am J Physiol Cell Physiol. (2010) 298:C1363–75. doi: 10.1152/ajpcell.00004.2010
83. Freel RW, Whittamore JM, Hatch M. Transcellular oxalate and Cl− absorption in mouse intestine is mediated by the DRA anion exchanger SLC26A3, and DRA deletion decreases urinary oxalate. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G520–7. doi: 10.1152/ajpgi.00167.2013
84. Charney AN, Egnor RW, Henner D, Rashid H, Cassai N, Sidhu GS. Acid-base effects on intestinal Cl− absorption and vesicular trafficking. Am J Physiol Cell Physiol. (2004) 286:C1062–70. doi: 10.1152/ajpcell.00454.2003
85. Rajendran VM, Black J, Ardito TA, Sangan P, Alper SL, Schweinfest C, et al. Regulation of DRA and AE1 in rat colon by dietary Na depletion. Am J Physiol Gastrointest Liver Physiol. (2000) 279:G931–42. doi: 10.1152/ajpgi.2000.279.5.G931
86. Allen A, Flemstrom G. Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am J Physiol Cell Physiol. (2005) 288:C1–19. doi: 10.1152/ajpcell.00102.2004
87. Said H, Kaji I, Kaunitz JD. Gastroduodenal mucosal defense mechanisms. Curr Opin Gastroenterol. (2015) 31:486–91. doi: 10.1097/MOG.0000000000000211
88. Wen G, Jin H, Deng S, Xu J, Liu X, Xie R, et al. Effects of Helicobacter pylori infection on the expressions and functional activities of human duodenal mucosal bicarbonate transport proteins. Helicobacter. (2016) 21:536–47. doi: 10.1111/hel.12309
89. Wen G, Deng S, Song W, Jin H, Xu J, Liu X, et al. Helicobacter pylori infection downregulates duodenal CFTR and SLC26A6 expressions through TGFβ signaling pathway. BMC Microbiol. (2018) 18:87. doi: 10.1186/s12866-018-1230-8
90. Caudarella R, Rizzoli E, Pironi L, Malavolta N, Martelli G, Poggioli G, et al. Renal stone formation in patients with inflammatory bowel disease. Scanning Microsc. (1993) 7:371–9; discussion 379–80.
91. Pardi DS, Tremaine WJ, Sandborn WJ, McCarthy JT. Renal and urologic complications of inflammatory bowel disease. Am J Gastroenterol. (1998) 93:504–14. doi: 10.1111/j.1572-0241.1998.156_b.x
92. Duffey BG, Alanee S, Pedro RN, Hinck B, Kriedberg C, Ikramuddin S, et al. Hyperoxaluria is a long-term consequence of Roux-en-Y Gastric bypass: a 2-year prospective longitudinal study. J Am Coll Surg. (2010) 211:8–15. doi: 10.1016/j.jamcollsurg.2010.03.007
93. Nelson WK, Houghton SG, Milliner DS, Lieske JC, Sarr MG. Enteric hyperoxaluria, nephrolithiasis, and oxalate nephropathy: potentially serious and unappreciated complications of Roux-en-Y gastric bypass. Surg Obes Relat Dis. (2005) 1:481–5. doi: 10.1016/j.soard.2005.07.002
94. Arvans D, Alshaikh A, Bashir M, Weber C, Hassan H. Activation of the PKA signaling pathway stimulates oxalate transport by human intestinal Caco2-BBE cells. Am J Physiol Cell Physiol. (2020) 318:C372–9. doi: 10.1152/ajpcell.00135.2019
95. Costello JF, Smith M, Stolarski C, Sadovnic MJ. Extrarenal clearance of oxalate increases with progression of renal failure in the rat. J Am Soc Nephrol. (1992) 3:1098–104.
96. Osswald H, Hautmann R. Renal elimination kinetics and plasma half-life of oxalate in man. Urol Int. (1979) 34:440–50. doi: 10.1159/000280294
97. Noori N, Honarkar E, Goldfarb DS, Kalantar-Zadeh K, Taheri M, Shakhssalim N, et al. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (dietary approaches to stop hypertension)-style and low-oxalate diets. Am J Kidney Dis. (2014) 63:456–63. doi: 10.1053/j.ajkd.2013.11.022
98. Asplin JR. Hyperoxaluric calcium nephrolithiasis. Endocrinol Metab Clin North Am. (2002) 31:927–49. doi: 10.1016/S0889-8529(02)00030-0
99. Robertson WG, Peacock M. The cause of idiopathic calcium stone disease: hypercalciuria or hyperoxaluria? Nephron. (1980) 26:105–10. doi: 10.1159/000181963
100. Wang T, Egbert ALJr, Abbiati T, Aronson PS, Giebisch G. Mechanisms of stimulation of proximal tubule chloride transport by formate and oxalate. Am J Physiol. (1996) 271:F446–50. doi: 10.1152/ajprenal.1996.271.2.F446
101. Wang T, Giebisch G, Aronson PS. Effects of formate and oxalate on volume absorption in rat proximal tubule. Am J Physiol. (1992) 263:F37–42. doi: 10.1152/ajprenal.1992.263.1.F37
102. Wang T, Segal AS, Giebisch G, Aronson PS. Stimulation of chloride transport by cAMP in rat proximal tubules. Am J Physiol. (1995) 268:F204–10. doi: 10.1152/ajprenal.1995.268.2.F204
103. Ko SB, Luo X, Hager H, Rojek A, Choi JY, Licht C, et al. AE4 is a DIDS-sensitive Cl−/ exchanger in the basolateral membrane of the renal CCD and the SMG duct. Am J Physiol Cell Physiol. (2002) 283:C1206–18. doi: 10.1152/ajpcell.00512.2001
104. Ohana E, Shcheynikov N, Moe OW, Muallem S. SLC26A6 and NaDC-1 transporters interact to regulate oxalate and citrate homeostasis. J Am Soc Nephrol. (2013) 24:1617–26. doi: 10.1681/ASN.2013010080
105. Hirata T, Cabrero P, Berkholz DS, Bondeson DP, Ritman EL, Thompson JR, et al. In vivo Drosophilia genetic model for calcium oxalate nephrolithiasis. Am J Physiol Renal Physiol. (2012) 303:F1555–62. doi: 10.1152/ajprenal.00074.2012
106. Landry GM, Hirata T, Anderson JB, Cabrero P, Gallo CJ, Dow JA, et al. Sulfate and thiosulfate inhibit oxalate transport via a dPrestin (SLC26A6)-dependent mechanism in an insect model of calcium oxalate nephrolithiasis. Am J Physiol Renal Physiol. (2016) 310:F152–9. doi: 10.1152/ajprenal.00406.2015
107. Lu X, Sun D, Xu B, Pan J, Wei Y, Mao X, et al. In silico screening and molecular dynamic study of nonsynonymous single nucleotide polymorphisms associated with kidney stones in the SLC26A6 gene. J Urol. (2016) 196:118–23. doi: 10.1016/j.juro.2016.01.093
108. Orchard CH, Kentish JC. Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol. (1990) 258:C967–81. doi: 10.1152/ajpcell.1990.258.6.C967
109. Orchard CH, Cingolani HE. Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res. (1994) 28:1312–9. doi: 10.1093/cvr/28.9.1312
110. Fabiato A, Fabiato F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J Physiol. (1978) 276:233–55. doi: 10.1113/jphysiol.1978.sp012231
111. Crampin EJ, Smith NP, Langham AE, Clayton RH, Orchard CH. Acidosis in models of cardiac ventricular myocytes. Philos Trans A Math Phys Eng Sci. (2006) 364:1171–86. doi: 10.1098/rsta.2006.1763
112. Vaughan-Jones RD, Eisner DA, Lederer WJ. Effects of changes of intracellular pH on contraction in sheep cardiac Purkinje fibers. J Gen Physiol. (1987) 89:1015–32. doi: 10.1085/jgp.89.6.1015
113. Leem CH, Lagadic-Gossmann D, Vaughan-Jones RD. Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J Physiol. (1999) 517:159–80. doi: 10.1111/j.1469-7793.1999.0159z.x
114. Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. (2009) 46:318–31. doi: 10.1016/j.yjmcc.2008.10.024
115. Poole RC, Halestrap AP. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am J Physiol. (1993) 264:C761–82. doi: 10.1152/ajpcell.1993.264.4.C761
117. Perez NG, Alvarez BV, Camilion de Hurtado MC, Cingolani HE. pHi regulation in myocardium of the spontaneously hypertensive rat. Compensated enhanced activity of the Na+-H+ exchanger. Circ Res. (1995) 77:1192–200. doi: 10.1161/01.RES.77.6.1192
118. Karmazyn M, Kilic A, Javadov S. The role of NHE-1 in myocardial hypertrophy and remodelling. J Mol Cell Cardiol. (2008) 44:647–53. doi: 10.1016/j.yjmcc.2008.01.005
119. Yoshida H, Karmazyn M. Na+/H+ exchange inhibition attenuates hypertrophy and heart failure in 1-wk postinfarction rat myocardium. Am J Physiol Heart Circ Physiol. (2000) 278:H300–4. doi: 10.1152/ajpheart.2000.278.1.H300
120. Alvarez BV, Fujinaga J, Casey JR. Molecular basis for angiotensin II-induced increase of chloride/bicarbonate exchange in the myocardium. Circ Res. (2001) 89:1246–53. doi: 10.1161/hh2401.101907
121. Sirish P, Ledford HA, Timofeyev V, Thai PN, Ren L, Kim HJ, et al. Action potential shortening and impairment of cardiac function by ablation of SLC26A6. Circ Arrhythm Electrophysiol. (2017) 10:e005267. doi: 10.1161/CIRCEP.117.005267
122. Ji M, In Lee S, Lee SA, Son KH, Hong JH. Enhanced activity by NKCC1 and Slc26a6 mediates acidic pH and Cl− movement after cardioplegia-induced arrest of db/db diabetic heart. Mediators Inflamm. (2019) 2019:7583760. doi: 10.1155/2019/7583760
123. Alvarez BV, Vilas GL, Casey JR. Metabolon disruption: a mechanism that regulates bicarbonate transport. EMBO J. (2005) 24:2499–511. doi: 10.1038/sj.emboj.7600736
124. Dawson PA. Sulfate in fetal development. Semin Cell Dev Biol. (2011) 22:653–9. doi: 10.1016/j.semcdb.2011.03.004
125. Gaull G, Sturman JA, Raiha NC. Development of mammalian sulfur metabolism: absence of cystathionase in human fetal tissues. Pediatr Res. (1972) 6:538–47. doi: 10.1203/00006450-197206000-00002
126. Loriette C, Chatagner F. Cysteine oxidase and cysteine sulfinic acid decarboxylase in developing rat liver. Experientia. (1978) 34:981–2. doi: 10.1007/BF01915299
127. Barnes SK, Eiby YA, Lee S, Lingwood BE, Dawson PA. Structure, organization and tissue expression of the pig SLC13A1 and SLC13A4 sulfate transporter genes. Biochem Biophys Rep. (2017) 10:215–23. doi: 10.1016/j.bbrep.2017.04.005
128. Cole DE, Baldwin LS, Stirk LJ. Increased serum sulfate in pregnancy: relationship to gestational age. Clin Chem. (1985) 31:866–7. doi: 10.1093/clinchem/31.6.866
129. Cole DE, Baldwin LS, Stirk LJ. Increased renal reabsorption of inorganic sulfate in third-trimester high-risk pregnancies. Obstet Gynecol. (1985) 66:485–90.
Keywords: SLC26A6, pancreas, intestine, kidney, heart, placenta
Citation: Wang J, Wang W, Wang H and Tuo B (2021) Physiological and Pathological Functions of SLC26A6. Front. Med. 7:618256. doi: 10.3389/fmed.2020.618256
Received: 16 October 2020; Accepted: 30 December 2020;
Published: 21 January 2021.
Edited by:
Ihsan Ullah, Khyber Medical University, PakistanReviewed by:
Serenella Anzilotti, Institute of Research and Medical Care (IRCCS) SDN, ItalyDeepthi Rao, University of Missouri, United States
Copyright © 2021 Wang, Wang, Wang and Tuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Biguang Tuo, dHVvYmlndWFuZ0BhbGl5dW4uY29t
†These authors have contributed equally to this work